

Abstract
Transcriptional responses to extracellular stimuli involve tuning the rates of transcript production and degradation. Here, we show that the time-dependent profiles of these rates can be inferred from simultaneous measurements of precursor mRNA (pre-mRNA) and mature mRNA profiles. Transcriptome-wide measurements demonstrate that genes with similar mRNA profiles often exhibit marked differences in the amplitude and onset of their production rate. The latter is characterized by a large dynamic range, with a group of genes exhibiting an unexpectedly strong transient production overshoot, thereby accelerating their induction and, when combined with time-dependent degradation, shaping transient responses with precise timing and amplitude.
Keywords: half-life, operational strategy, pre-mRNA, transcriptional response
Introduction
A major component of cellular response to changing conditions is a shift of the transcriptome to a new state, which is more adequate for facing the new environment. In eukaryotes, this shift is governed by a highly dynamic interplay between epigenetic, co-transcriptional and post-transcriptional processes, which determine the temporal concentration profiles of RNAs by controlling their production and degradation (Orphanides and Reinberg, 2002; Garneau et al, 2007). The most commonly used mathematical description of the transcriptional response is expressed by the following differential equation (Gorini and Maas, 1957):
 |
Here, M is the mRNA concentration, dM/dt is its rate of change with respect to time t, β denotes the rate of transcript production and α is the transcript degradation coefficient. Response of a gene to stimulus is commonly described as an abrupt shift of its β and/or α to new values, which then remain constant (see Figure 1A, B, D and E). mRNA concentration then approaches its new asymptotic value, β/α, with kinetics determined solely by the mRNA degradation coefficient α (Alon, 2007). Thus, a small steady-state value of α implies slow dynamics (a long mRNA half-life, T1/2=ln2/α) and also supports an economically favorable low production rate (Shalem et al, 2008; Elkon et al, 2010).
Figure 1.

Pre-mRNA and mRNA response profiles for stimulus-induced production and degradation changes. Different time-dependent production rates (green) and degradation coefficients (gold) are shown in the left column. Resulting pre-mRNA (red) and mRNA (blue) profiles, obtained by numerically solving Equations (4) and (5), are presented for mRNAs with initial half-lives of 30 and 120 min in the middle and right columns, respectively. All strategies yield a 5-fold change in the steady-state mRNA level, pre-mRNA profiles closely mimic production, while mRNA FC profiles are delayed and may be qualitatively different. (A) Five-fold step increase of production rate at time t=0. A rapid increase of pre-mRNA levels is followed by slower mRNA response. Dashed black line indicates the rise time to half of the aimed level, determined by the mRNA half-life. (B) Signal-induced 5-fold step decrease of degradation rate. Pre-mRNA levels reflect unchanged production; mRNA accumulates now more slowly compared with (A). (C) Signal-induced transient overshoot in production rate accelerates mRNA induction, particularly for mRNAs with long initial half-lives. (D) Five-fold step decrease in production rate at t=0. (E) Degradation driven downregulation: response time is shorter due to reduction of the mRNA half-life. (F) A transient overshoot in degradation accelerates response time of downregulated genes.
Notably, many organisms across the phylogenetic tree exhibit rapid rise times of long-lived mRNAs, in contradiction to this simple model. In bacteria, accelerated production can be achieved by time-delayed negative autoregulation (Rosenfeld et al, 2002) and in yeast through transcriptional control by an incoherent feed-forward loop (Mangan and Alon, 2003; Mangan et al, 2003). In mammalian systems, however, the operational strategies which govern transcript production and degradation profiles remain less well characterized.
Important case studies for dynamics of transcript production and degradation are exposure to serum, pathogen components such as lipopolysaccharide (LPS), or growth factors like the epidermal growth factor (EGF). These stimuli initiate well-characterized signaling cascades that culminate in orchestrated transcriptional responses involving primary and secondary response genes (PRGs and SRGs; Cochran et al, 1983; Lau and Nathans, 1987; Iyer et al, 1999; Amit et al, 2007; Medzhitov and Horng, 2009). PRGs include immediate-early genes (IEGs), whose mRNA expression often peaks during the first hour and delayed early genes (DEGs), which mostly peak during the second hour and mainly comprise delayed PRGs (Amit et al, 2007; Tullai et al, 2007; Hao and Baltimore, 2009). While PRGs are rapidly induced by pre-existing transcription factors, without de novo protein synthesis (Herschman, 1991; Byun et al, 2009; Hargreaves et al, 2009; Ramirez-Carrozzi et al, 2009; Wang et al, 2009), induction of SRGs depends upon newly synthesized activators. Correct identification of transcripts as PRG or SRG is of central importance in order to understand the networks that govern the response to stimulus. According to currently accepted views, IEGs primarily comprise transcriptional regulators and are mostly encoded by short, intron-poor and short-lived transcripts. Their rapid induction is facilitated by permissive chromatin structures and by swift attenuation of pre-existing negative regulators (Hargreaves et al, 2009; Avraham et al, 2010). Expression of IEGs is controlled by activation-dependent feedback (e.g., receptor endocytosis), by negative autoregulation and by DEGs (Sassone-Corsi et al, 1988; Carballo et al, 1998; Amit et al, 2007). The latter regulate IEGs by transcriptional repression, destabilization of IEG transcripts or by attenuating the signaling pathways that lead to IEG induction. Regulation of DEGs remains less understood. The lag in their expression has been attributed to their distinct promoter properties, and to delays in transcription initiation and elongation (Tullai et al, 2007; Ramirez-Carrozzi et al, 2009). Intriguingly, many DEGs encode for long-lived mRNAs, yet they are rapidly induced (Tullai et al, 2007).
The aim of our study is to uncover and quantify the dynamics of transcriptional responses to stimuli and to elucidate the operational strategies that govern them. To this end, we introduced a simple method that allows, for the first time, genome-wide simultaneous measurement of pre-mRNA and mRNA expression. When combined with a new mathematical model for transcription dynamics, production and degradation profiles (i.e., time-dependent functions) can be reliably inferred from these measured pre-mRNA and mRNA profiles. Our transcriptome-wide study reveals that transcript production rates, reflected by pre-mRNA profiles, exhibit a high dynamic range, and identifies a subset of genes that exhibit novel relationships between transcript production and mRNA abundance profiles. In particular, we identify genes that exhibit pre-mRNA fold changes (FCs) that exceed by an unexpectedly high margin those of the associated mRNA. Indeed, such temporally confined production overshoot is used to solve the ‘conflict’ between long mRNA half-life and the need for rapid response. Taken together, our findings reveal complex dynamics of both pre-mRNA production and mRNA degradation rates, which shape the expression profiles of mRNAs in response to extracellular stimuli.
Results
Inference of production and degradation profiles requires measurement of pre-mRNA and mRNA dynamics
Cellular response to stimuli involves time-dependent variation of the production rate β and the degradation coefficient α of Equation (1). Exact inference of two unknown functions (α(t) and β(t)) from the time-dependent mRNA abundance M(t) alone is impossible. Furthermore, the expression profiles of transcripts with typical mRNA half-lives track very poorly the profile β(t) of RNA production (Barenco et al, 2009; Hao and Baltimore, 2009). Thus, independent assessments of production or degradation rates (Fan et al, 2002; Shalem et al, 2008; Hao and Baltimore, 2009) are necessary.
Direct measurements of α(t) and β(t) involve interference with the system: prototypical methods induce transcription arrest (e.g., by actinomycin D, ActD) followed by measurements of mRNA decay to obtain mRNA half-lives. More recently introduced methods involve biosynthetic tagging by averaging the incorporation of labeled nucleotides (e.g., 4-thiouracil, 4-sU) over a certain time interval to measure newly synthesized RNA (Miller et al, 2011; Rabani et al, 2011; Schwanhausser et al, 2011). When combined with measurements of mRNA abundance, either method can be used to infer production and degradation rates. Transcription arrest may alter transcript stability and thereby the measurement may affect the properties one wishes to measure. Methods based on incorporation of labeled nucleotides are more promising, and the underlying assumptions, that cellular uptake and incorporation of tagged nucleotides is constant over stimulus, and that labeled transcripts are spliced, exported from the nucleus and degraded at the same rate as their unlabeled counterparts, may be valid. By contrast, our method involving simultaneous and direct measurements of mRNA and pre-mRNA abundance profiles, M(t) and P(t), is not only much simpler, but also free of any ‘interference’ with either transcription or mRNA degradation. Moreover, since no time interval for labeling is required, our method allows for tracking of pre-mRNA and mRNA profiles at high temporal resolution.
Mathematical modeling of the transcription process
We extended the ‘minimal model’ of Equation (1) and describe production of a particular transcript in terms of P(t) and M(t). Dynamics of these variables are described by two coupled linear differential equations of the form
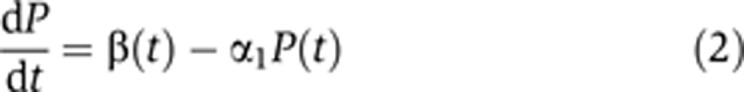 |
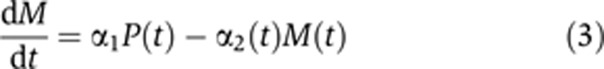 |
Here, β is the (time-dependent) production rate of pre-mRNA, α1 denotes the conversion (splicing) coefficient of pre-mRNA to mRNA and α2(t) represents the degradation coefficient of mRNA. This simple model relies on the following main assumptions:
(i) Time-dependent production rate: in principle, the production rate of a particular transcript may depend on the activity of various proteins (e.g., transcription factors, Pol II and so on), and may occur monotonically or in transcriptional bursts (Suter et al, 2011). We assume that all these can be absorbed in an effective (gene-specific) time-dependent production rate β(t) (Larson et al, 2011; Suter et al, 2011). We do not introduce explicit dependence of β on either P or M, because Pol II most likely elongates across the distal transcription units irrespective either of the number of polymerases recruited or of previous rounds of transcription (Darzacq et al, 2007; Hager et al, 2009; Singh and Padgett, 2009; Wada et al, 2009; Larson et al, 2011; Suter et al, 2011).
(ii) For most genes, the measured data can be explained and fitted well without incorporating in the model processes involving storage or transport of mRNA to specific cellular compartments (e.g., export to the cytoplasm). The main approximation introduced by this assumption is that even though mRNA is degraded only in the cytoplasm, in our model degradation is proportional to M(t), the total mRNA (rather than to the cytoplasmic fraction). Since the vast majority of the mRNA transcripts we study do localize to the cytoplasm (data not shown) and export of newly synthesized mRNA to the cytoplasm occurs much faster than mRNA degradation, using α2M as the degradation rate is a good approximation.
(iii) Degradation of pre-mRNA in the nucleus, beyond its conversion to mRNA, is minimal. Hence, all pre-mRNA lost (in Equation (2)) is converted and reappears as mRNA (in Equation (3)).
(iv) The conversion coefficient α1 is independent of time and is not affected by the stimulus.
As described below, assumptions (iii) and (iv) were confirmed experimentally. Finally, our model conforms with the fact that pre-mRNA splicing temporally coincides with other processing events such as capping, RNA editing and poly(A)-tail addition (Hirose and Manley, 1998; Orphanides and Reinberg, 2002; Proudfoot et al, 2002).
In both quantitative PCR (qPCR) and microarray measurements, the output signal for different nucleotide sequences is characterized by different amplification and hybridization efficiencies. Hence, these measurements yield reliably only relative concentrations, for example, FC, measured, for each transcript, with respect to its concentration in some reference condition. Throughout this study, we used the pre-stimulus state, t=0, as our reference, and all relative concentrations shown are the FC with respect to this reference. Hence, we used a trivial transformation of variables to cast Equations (2) and (3) in a form that presents the dynamics of normalized FC variables, 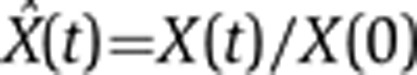 , where X stands for any of the quantities P, M, β, α:
, where X stands for any of the quantities P, M, β, α:
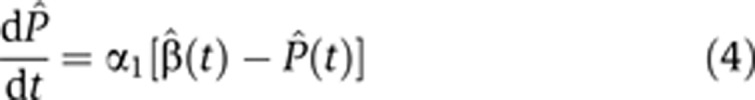 |
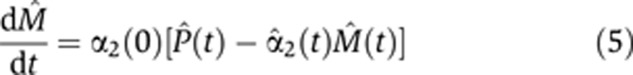 |
These are the actual dynamic equations we used to infer for each transcript its normalized, time-dependent production and degradation rates 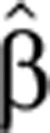 (t) and
(t) and 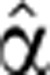 2(t). These rates were inferred (see Materials and methods) from measurements of the corresponding Pˆ(t) and Mˆ(t). Note that for fast conversion, that is, α1≫1, Equation (4) can be rewritten (using first order Taylor expansion) as
2(t). These rates were inferred (see Materials and methods) from measurements of the corresponding Pˆ(t) and Mˆ(t). Note that for fast conversion, that is, α1≫1, Equation (4) can be rewritten (using first order Taylor expansion) as 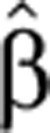 (t)≈Pˆ(t+1/α1), demonstrating that the production FC profile is given, to good approximation, by the time-shifted pre-mRNA FC.
(t)≈Pˆ(t+1/α1), demonstrating that the production FC profile is given, to good approximation, by the time-shifted pre-mRNA FC.
Pre- and post-stimulus values of conversion coefficients α1(t) were determined by direct measurements (see below in subsection Conversion with a constant coefficient dominates pre-mRNA outflux) while degradation coefficients α2(0) were derived from the transcript FC profiles by optimization of the fit to the data (see Materials and methods).
The predicted transcript responses to different production and degradation profiles
To demonstrate the time-dependent FC of pre-mRNA and mRNA generated by our model, we solved numerically Equations (4) and (5) for different time-varying forms of transcript production 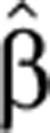 (t) and degradation
(t) and degradation 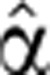 2(t). We find that the temporal pre-mRNA profile closely resembles the time-dependent transcript production rate
2(t). We find that the temporal pre-mRNA profile closely resembles the time-dependent transcript production rate 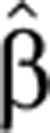 (t) for both upregulated and downregulated transcripts (Figure 1).
(t) for both upregulated and downregulated transcripts (Figure 1).
In contrast, the profile of mRNA is affected also by degradation and exhibits much slower kinetics, particularly for mRNAs with long half-lives (Elkon et al, 2010). In terms of end point steady-state levels, a 5-fold increase in production (Figure 1A) is equivalent to a 5-fold reduction in degradation (Figure 1B). However, the response time, that is, the time until half the end point change is reached, is much longer in the latter case. Our simulations of upregulation also demonstrate that a simple step to new production or degradation rates cannot generate the rapid dynamics of transcriptional responses that were in fact observed across a series of experimental conditions (Iyer et al, 1999; Amit et al, 2007, 2009), especially for medium to long-lived mRNAs. In contrast, as seen in Figure 1C, marked acceleration of the response of even long-lived mRNAs can be achieved by a strategy of production overshoot (defined as a transient increase of the pre-mRNA FC to values that exceed at least twice the maximal mRNA FC). Thus, unlike the response time to simple step changes in β and/or α2, which is governed by α2 alone (Alon, 2007; Shalem et al, 2008; Elkon et al, 2010), our results emphasize the importance of short pulses of production to achieve rapid transcriptional induction.
Simulations of responses to different strategies that lead to downregulation are shown in Figure 1D–F. In order to accelerate downregulation, cells have to transiently increase degradation rates; otherwise even complete arrest of production will result in decay at a rate not faster than the initial half-life of the transcript.
Another point to note is the insensitivity of the mRNA profiles to the precise value of the conversion coefficient α1. The reason is that typically conversion times are much shorter than mRNA half-lives (Supplementary Figure S1A).
Measuring pre-mRNA and mRNA expression using intronic and exonic probe sets
As a model system for studying strategies of mammalian transcriptional responses to extracellular signals, we used the human mammary epithelial MCF10A cell line. Within 8 h of stimulation with EGF, MCF10A cells develop a migratory and invasive phenotype that requires de novo transcription of pro-migratory genes (Amit et al, 2007; Katz et al, 2007). Importantly, under our experimental conditions MCF10A cells remain viable for over 10 days, but do not proliferate, thus precluding the confounding effect of mRNA dilution due to cell division (see Materials and methods).
We extracted total RNA from biological triplicates at 7 time points following stimulation, hybridized these samples to 21 Affymetrix GeneChip Human Exon 1.0 ST Arrays, and measured, for the first time, transcriptome-wide dynamics of mRNA and pre-mRNA. The key to this advance was using signals from exonic and intronic probe sets (PS), both present on these arrays (Figure 2A). Owing to the instantaneous degradation of introns that were spliced out of pre-mRNA (Singh and Padgett, 2009), the signal of intronic PS reflects the amount of the respective pre-mRNA (validated for example for the VCL gene by comparing Figures 2D and 3C). Exons, on the other hand, are common to both pre-mRNA and mature mRNA. Because mature mRNA abundance vastly exceeds that of pre-mRNA (the ratio of their steady-state levels equals the ratio of their half-lives), the signals of exonic PS are dominated by mRNA.
Figure 2.

EGF induces distinct transcriptional dynamics of pre-mRNAs and mRNAs in MCF10A cells. (A) Experimental outline: MCF10A mammary epithelial cells were stimulated with EGF for the indicated intervals and RNA was hybridized to Affymetrix Exon Arrays. Annotation and signal intensity-based filtering was performed to define probe sets (PS) interrogating exons (blue marks) and introns (red). Fold changes (FCs) of intronic and exonic PS were weighed separately to define the pre-mRNA and mRNA FC for each gene. (B) Pre-mRNA and mRNA profiles of 441 transcriptionally induced genes. The left (right) heatmap displays mRNA (pre-mRNA) expression, as defined by the gene-level exon (intron) FC profiles; genes are grouped according to the peak time of their mRNA expression and within each such group ordered according to the peak time of their pre-mRNA FC. Finally, within each subgroup the transcripts were ordered according to the correlation between the pre-mRNA and mRNA profiles (rightmost panel). The order of genes is the same in both heatmaps. Note that the log-transformed expression values of each row were centered and normalized (separately for pre-mRNA and mRNA, owing to their different dynamic range). Hence, the log-FC values at t=0 are not uniformly 0. Green lines on the bar on the left indicate genes with production overshoot (i.e., the maximal FC of intronic PS exceeded that of exonic PS by >2-fold). Genes chosen for detailed validation (Figure 3) are indicated. (C) Scatter plots comparing gene-level intron (pre-mRNA) and exon (mRNA) FC during EGF stimulation. Genes with mRNA FC⩽1.5 over the whole time course (grey dots), transcriptionally induced genes with production overshoot (green dots) or without (red dots) are shown. Note the similarity of mRNA FC of overshooting and non-overshooting genes. (D) Space-time description of probe-level FC of an overshooting gene. Upper panel: Genomic organization of the vinculin (VCL) gene (122 kb). Arrow length corresponds to 50 kb. Positions of exonic (blue) and intronic (red) PS are indicated. Lower panels: FC (log2 scale) of each PS with respect to its baseline value is shown for the time course outlined in (A). Only PS with present calls in all replicates of the respective time points compared are shown. Error bars (in gold) represent standard deviations. Note that intronic and exonic PS display different behavior and dynamic range.
Figure 3.

Genes with distinct mRNA expression profiles exhibit production overshoot. Real-time quantitative PCR (qPCR) measurements of expression FC profiles of selected genes with production overshoot (A–F) or no production overshoot (G, H), in a high-resolution time course following stimulation of MCF10A cells with EGF. Dots represent averages of technical triplicates, solid lines represent best fit (see Supplementary information) of pre-mRNA (red) and mature mRNA (blue). Production profiles (green lines) and degradation (gold) were inferred from these measurements. Half-lives (t1/2) of pre-mRNAs were measured as described in the text, whereas those of mRNA were estimated from fit to the data (see Materials and methods).
We classified each PS as interrogating exons or introns, by combining annotation-based criteria with constraints on signal quality and intensity (see Supplementary information). Properly weighted intronic readings were used to assess, at each time point, the gene-level pre-mRNA expression, while gene-level mRNA expression levels were computed by combining signals from the gene's exonic PS. Of note, our strategy of using signals from intronic PS to measure changes in pre-mRNA expression may be applicable to most multicellular organisms: Analysis of the number of intron-containing genes and the size of their introns revealed that the majority of genes, from C. elegans to human, contain introns that are large enough to be interrogated by one or more PS (Supplementary Figure S2). The main limitation in this methodology may reside in (i) the paucity of intronic PS in existing microarray platforms and (ii) difficulty to detect gene-level intronic FC above noise level in less abundant transcripts. Analysis of three biological replicates of each time point permitted us to reliably detect gene-level exonic FC for about 8000 genes. In nearly half of those, a sufficient number of intronic PS (see Materials and methods) were present and exhibited a signal clearly exceeding noise level, thus allowing definition of gene-level intronic FC values (see Supplementary information and Supplementary Figure S3).
Genome-wide time-dependent pre-mRNA and mRNA transcriptional responses to EGF stimulation
By measuring the FC of pre-mRNAs (introns) and mRNAs (exons) at 7 time points, we identified 441 transcriptionally induced genes (with maximum FC exceeding 2.1 for pre-mRNA and 1.5 for mRNA, see Supplementary information for choice of cutoffs). Figure 2B displays the time-dependent FC profiles of these genes. Genes were first grouped according to the peak time of their mRNA FC; members of each group were then internally ordered according to the peak time of their pre-mRNA. Finally, each subgroup of transcripts that shared both mRNA and pre-mRNA peak times was sorted by the correlation between the two profiles (see right bar on Figure 2B and Supplementary information). As expected, the onset and peak expression time of introns typically preceded that of exons (Figure 2B; Supplementary Figure S1B–E). Surprisingly, most genes exhibited markedly different temporal profiles of mRNA and pre-mRNA expression (Figure 2B, right bar). For instance, the mRNA of LAMC2 peaked 8 h after EGF stimulation, while its pre-mRNA had reached its maximum FC already after 20 to 30 min (Figures 2B and 3E). Strikingly, genes with similar peak times of mRNA expression exhibited clearly distinct pre-mRNA dynamics. Intuitively, genes with similar pre-mRNA profiles would be more likely to share common cis-regulatory elements compared with genes exhibiting similar mRNA profiles. Our analyses of genes sharing either similar pre-mRNA or mRNA profiles, however, did not reveal a significant enrichment in known transcription factor-binding DNA sequence motifs (data not shown). Importantly, by 20 min after EGF stimulus, pre-mRNA levels of most upregulated mRNAs had already increased (see heatmaps in Figure 2B), suggesting that the initial regulation of these genes occurs via the primary transcriptional response, whereas the amplitude and duration of induction of these genes may be differentially shaped by newly synthesized transcriptional and post-transcriptional regulators.
Complementary time-dependent amplitude information revealed that most of the induced genes exhibited much higher and narrower peaks of pre-mRNA FC compared with mRNA FC (Figures 2C and 3). Whereas the early FC of many pre-mRNAs tremendously exceeded that of their respective mRNAs, their FCs at later time points were comparable. In particular, the peak pre-mRNA FC of 18% (79/441) of the induced genes exceeded the peak FC of their corresponding mRNAs by >2-fold (green lines on the bar in Figure 2B and green dots in Figure 2C). We refer to these as genes exhibiting production overshoot. The role of a brief pulse of production which significantly exceeds the eventual mRNA FC is to accelerate the rise of mRNA abundance and thereby shorten response time. Production overshoot is the strategy of choice to overcome the ‘dynamic barrier’ imposed by long mRNA life times on the ‘classical’ step rise in production described above (Figure 1A). Production overshoot is not synonymous (see Figure 2C) to having either higher long-time mRNA FC or to a transient peak of mRNA abundance.
Genes exhibiting production overshoot are mostly PRGs
Production overshoot was typically found in genes exhibiting significantly earlier onset and peak of production, as well as earlier mRNA peak time compared with genes without overshooting introns. For example, within the first 40 min of EGF stimulation pre-mRNA levels had increased by >2-fold in 96% of the overshooting versus 45% of the non-overshooting genes (Supplementary Figure S1B–E). Such rapid kinetic characteristics identify genes with production overshoot as PRGs. To examine this notion, we considered a data set of 98 genes identified as PRGs in glioblastoma cells stimulated with the platelet-derived growth factor in the presence of cycloheximide (Tullai et al, 2007). Thirteen (13%) of these PRGs were also transcriptionally induced by EGF in our MCF10A cells. Nine out of these thirteen PRGs (69%) exhibited production overshoot in MCF10A cells indicating highly significant overlap (P<4.0E−06, hypergeometric test). Functional annotation analysis (Huang da et al, 2009) of the overshooting genes revealed their significant enrichment by functional categories associated with cell adhesion and motility (Supplementary File 1).
A typical example of production overshoot is shown in Figure 2D for the vinculin (VCL) gene, which encodes for a tension sensor localized to focal adhesions. Here, EGF stimulation leads to a transient, 16-FC in expression of intronic PS, reached 40–60 min after stimulation and rapidly fading thereafter. In contrast, exonic PS display delayed and much more subtle changes, exhibiting a peak FC of 2 between 120 and 240 min followed by a slow decrease. Note that in large genes such as VCL (122 kb) or ITGA2 (105 kb, Supplementary Figure S4), the space-time dependence of intronic PS reflects the propagation of the initial wave of polymerases sweeping along the gene (Singh and Padgett, 2009; Wada et al, 2009; Figure 2D).
Inferring transcript production and degradation dynamics from pre-mRNA and mRNA profiles
To obtain time-dependent transcript production and mRNA degradation rates, we measured pre-mRNA and mRNA expression profiles by qPCR (see Materials and methods and Supplementary Figure S5) in a frequently sampled time course experiment. We selected 12 upregulated genes (indicated in Figure 2B), 9 with production overshoot and, for comparison, 3 without production overshoot. Additionally, we profiled MYC as well as three dramatically induced IEGs: HBEGF, NR4A1 and PTGS2 (IEGs would otherwise have been underrepresented in our analyses due to lack of a sufficient number of intronic PS, see Materials and methods). Results for downregulated genes are described later (Figure 4).
Figure 4.

EGF driven downregulation of pre-mRNAs and mRNAs in MCF10A cells. (A) Pre-mRNA and mRNA profiles of 364 transcriptionally repressed genes. The left heatmap displays mRNA expression as defined by the gene-level exon FC profiles; genes are grouped first according to the time of their lowest mRNA FC, and within each such group, they are ordered according to the time of their lowest pre-mRNA FC, as reflected by intronic FC (right heatmap). The order of the genes is the same in both heatmaps. Names of genes that were chosen for detailed validation are indicated. Note that mRNA profiles are very poor indicators of shutdown of pre-mRNA production. Heatmaps were normalized as in Figure 2B. (B) qPCR measurements of expression FC profiles of selected downregulated genes in a high-resolution time course following stimulation of MCF10A cells with EGF. Dots represent averages of technical triplicates, solid lines represent best fit (see Materials and methods) of pre-mRNA (red) and mature mRNA (blue). Production profiles (green lines), mRNA half-lives and degradation (gold) were inferred from these measurements as described in Materials and methods.
Overshooting pre-mRNA levels occurred in genes with diverse mRNA profiles, such as exhibiting transient or sustained induction, high or low level fold change, and early or late peaks (Figure 3; Supplementary Figure S5). Genes, for which exon arrays did not reveal an overshoot in intron profiles, did not exhibit overshoot in qPCR measurements either (Figure 3G and H; Supplementary Figure S5), validating the array results. Transcript production and degradation dynamics (Figure 3, 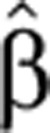 (t) and
(t) and 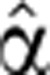 2(t) are shown in green and gold curves, respectively) were inferred from measured pre-mRNA and mRNA time courses (as described in Materials and methods).
2(t) are shown in green and gold curves, respectively) were inferred from measured pre-mRNA and mRNA time courses (as described in Materials and methods).
Importantly, these functions were obtained without any interference with the transcriptional response.
In an independent experiment, we measured the values of the pre-mRNA conversion coefficient α1, in the pre-stimulus steady state as well as following stimulation, using transcription arrest during short temporal intervals (see below, Supplementary Figure S6A). These values were found to be highly similar across all transcripts analyzed. Since the relevant time scale of conversion is a few minutes, the problems associated with the severe disruption of the cells (caused by transcription arrest) have not yet taken effect and the results are reliable. By contrast, the pre-stimulus mRNA degradation coefficients α2(0) could reliably be determined without transcription arrest, by fitting our data (see Materials and methods), and exhibited wide variation between transcripts. Analysis of the statistical error of the inferred profiles (described in Supplementary information) shows that the observed temporal variations are statistically significant.
Profiles shown in Figure 3 reveal that the overshoot in pre-mRNA levels reflects an overshoot in the production rate. In contrast to pre-mRNA levels, mRNA levels often peaked while production had already decreased much below its peak level, or even returned to its initial level (e.g., see NR4A1 and VCL in Figure 3B and C). Some of the genes also exhibited a second, albeit much smaller, peak of production (e.g., TUFT1 in Figure 3D and CD55 in Supplementary Figure S5).
mRNA degradation coefficients of several genes also exhibited non-monotonic behavior, including stabilization at long times (e.g., AREG and HBEGF; Figure 3G and H), demonstrating that cells delicately balance degradation with time-varying production to establish the desired temporal mRNA profiles. The inferred EGF-induced changes of mRNA stability at long times were qualitatively confirmed by experiments employing transcription arrest (Supplementary Figure S6B). The inferred (slightly <2-fold) stabilization in AREG and HBEGF (see Figure 3G and H) was confirmed, while the actual measured degradation times were significantly longer than the ones inferred without transcription arrest (pre-stimulus for AREG 69 versus 26 min, for HBEGF 28 versus 18 min), indicating the extent to which such methods are comparable.
Downregulated genes
An important aspect of transcriptional response involves downregulation of many transcripts. Due to the typically long mRNA half-lives, the timing of transcript production shut-down is hard to determine from mRNA data. We identified 364 downregulated genes characterized by exon FC<0.7 and/or intron FC<0.5. Time-dependent mRNA and pre-mRNA temporal profiles of these genes emphasize again the mismatch between mRNA and production profiles (Figure 4A). Transcription dynamics of six genes, measured at high temporal resolution (Figure 4B), revealed non-trivial production dynamics, mostly involving pronounced early shut-down followed by partial restoration of production. Importantly, in our experimental system, mRNA downregulation always involved a decrease of production (versus relying on mRNA degradation only, e.g., Figure 1F).
Conversion with a constant coefficient dominates pre-mRNA outflux
Inference of production and degradation profiles from our model was derived assuming that the pre-mRNA to mRNA conversion coefficient α1 does not vary with time. An alternative scenario, of conversion slowing down (due to either saturation of the pre-mRNA processing machinery (Patel et al, 2002; Pessa et al, 2006; Singh and Padgett, 2009) or to prolonged nuclear retention of incompletely processed transcripts (Prasanth et al, 2005)), could lead to pile-up of pre-mRNA and interfere with correct estimation of production rates. To exclude this possibility, we directly calculated the pre-mRNA conversion coefficients (α1) from measurements of pre-mRNA decay following transcription arrest. This was done for both unstimulated MCF10A cells and EGF-stimulated cells, yielding very similar decay rates of pre-mRNA under the two conditions (Figure 5A; Supplementary Figure S6A), confirming our modeling assumptions: indeed, α1 remains unchanged over a large range of pre-mRNA concentrations.
Figure 5.

Validation of model assumptions. (A) Pre-mRNA decay in unstimulated (green curves) and EGF-stimulated (red curves) MCF10A cells: transcription was arrested by applying ActD (10 μM) to mock-stimulated MCF10A cells or to cells near the peak of pre-mRNA induction after pre-incubation with EGF (20 ng/ml) for 20 min (AREG, HBEGF, PTGS2) or 40 min (TGFA), respectively. RNA was isolated at the indicated time points and qPCR was used to measure pre-mRNA decay. Note that the pre-mRNA conversion coefficient is unaffected by EGF stimulation. (B) Dynamics of pre-, exon and mature mRNA after transcription arrest at the peak of pre-mRNA induction: MCF10A cells were stimulated with EGF for 20 min before applying ActD as described in (A). Pre-mRNA (red), exonic (black) and mature mRNA (blue) were measured by qPCR. Note that pre-mRNA outflux is predominantly determined by conversion to mRNA. All symbols and error bars denote mean values and standard deviations of technical triplicates from one out of three independently repeated experiments. RNA half-lives (t1/2) and asymptotic constants (k) were calculated and curves were fit as described in Materials and methods.
Another explicit assumption of our model is that the half-life of pre-mRNA is mainly determined by conversion to mRNA, rather than by degradation. Conceivably, a significant level of pre-mRNA degradation by the nuclear RNAi machinery, that changes in the course of the stimulus (Bousquet-Antonelli et al, 2000; Hargreaves et al, 2009; Guang et al, 2010), could also contribute to the increased pre-mRNA levels, which we attributed to sharply increased production. Because for most genes the ratio of pre-mRNA to mRNA is very low—even close to the peak of pre-mRNA—assessing the quantitative impact of pre-mRNA degradative processes is quite challenging. To accomplish this, we analyzed two highly induced genes (PTGS2 and NR4A1), both exhibiting overshooting pre-mRNA. We arrested transcription near the point of maximum pre-mRNA accumulation (at which the pre-mRNA to mRNA ratio was high enough), and measured pre-mRNA, mRNA and exon FCs by qPCR. Since the copy number of exons is preserved by pre-mRNA conversion, but not preserved when pre-mRNA is degraded, loss of exons would be indicative of non-negligible pre-mRNA degradation. We found that conversion was the predominant process of pre-mRNA depletion; the rapid decay of pre-mRNA was accompanied by nearly constant exon abundance and an increase of mRNA (Figure 5B).
Production overshoot accelerates the induction of mRNAs
Our results demonstrate that the production dynamics of many induced genes do not exhibit a simple step increase (Figure 1A); rather, production overshoot is the strategy of choice (Figure 1C) and is most likely employed in order to accelerate mRNA response. Therefore, we compared the temporal profiles of mRNAs of induced transcripts with and without pre-mRNA overshoot. To eliminate the confounding effect of mRNA half-life, we first grouped genes into sets of similar half-lives, and then compared the dynamics of transcripts within each set. mRNA half-life was estimated for all induced genes using the fitting procedure as described in Materials and methods.
Beyond revealing the expected faster response and earlier peak times for short-lived versus long-lived mRNAs (Shalem et al, 2008; Hao and Baltimore, 2009; Elkon et al, 2010), our analyses demonstrated clearly shorter response times for genes exhibiting production overshoot across the entire range of mRNA half-lives (Figure 6). Similar results were obtained, when information on mRNA half-lives from another data source, which employed biosynthetic labeling (genomic run-on) methods to calculate mRNA half-lives in different cell lines, was used (Friedel et al, 2009; Supplementary Figure S7).
Figure 6.

Pre-mRNA production overshoot accelerates the response time of mRNAs. Genes that were transcriptionally induced by EGF stimulation of MCF10A cells (as described in legend to Figure 2) were grouped according to their mRNA half-lives (extracted from our measured expression profiles, as described in Materials and methods). Results are shown for three representative groups with half-lives between (A) 0–90 min (B) 90–240 min and (C) 240–480 min, respectively. The two columns on the left depict the percentage of genes, whose exons and introns peaked at the indicated time points, green for overshooting and brown for non-overshooting genes. The two columns on the right present the mean temporal expression FC profiles for exons and introns, for the overshooting (green) and non-overshooting (brown) genes. Note that within each group of genes with similar half-lives we observe earlier mRNA peak times for the genes with pre-mRNA production overshoot.
Production overshoot is a generic operational strategy enabling accelerated response
To evaluate the generality of production overshoot in transcriptional responses, we studied two additional very disparate cell types subjected to different types of stimuli, namely LPS-stimulated murine bone marrow-derived primary dendritic cells (DCs), and human embryonic stem cells (hESCs) exposed to retinoic acid (RA). Binding of LPS to Toll-like receptor-4 at the plasma membrane instigates signaling cascades that culminate in the activation of transcription factors such as NFκB, which induce an inflammatory response and maturation of DCs (Medzhitov and Horng, 2009; Supplementary Figure S8A). By contrast, RA diffuses through the cell membrane and forms a transcription regulatory complex with the RA receptor, which promotes hESC differentiation toward ectodermal (i.e., neuronal) fates within several days (Supplementary Figure S8B; Boyer et al, 2005).
In both systems, the temporal profiles of pre-mRNAs and mRNAs during the very initial phase of the transcriptional response to stimulation revealed transcripts exhibiting production overshoot, along with several known changes in mRNA stability accompanying transcriptional induction (Hao and Baltimore, 2009; Figure 7; Supplementary Figure S9). The observed occurrence of production overshoot in the three very different stimulated systems described herein demonstrates that this operational strategy is a general characteristic of mammalian transcriptional responses to extracellular cues.
Figure 7.

Pre-mRNA production overshoot is a general operational strategy in mammalian transcriptional networks. The plots in (A–F) show qPCR measurements of pre-mRNA (red) and mature mRNA (blue) expression profiles for selected genes exhibiting production overshoot. Dots represent averages of technical triplicate measurements and solid lines represent best fit (see Supplementary information). Production and degradation profiles were inferred from these measurements as described in Materials and methods, and their time-dependent profiles are indicated by green and gold curves, respectively. (A–C) Production overshoot in retinoic acid (RA)-stimulated human embryonic stem cells. (D–F) Production overshoot in lipopolysaccharide (LPS)-stimulated primary murine dendritic cells.
Discussion
Transcriptional responses of cells to external signals involve orchestrated changes in transcript production and degradation rates. These changes are often assumed to be simple shifts of production and degradation to new constant values. By combining mathematical modeling with measured temporal profiles of pre-mRNA and mRNA abundance in response to extracellular stimuli, we obtained, with unprecedented resolution, the time-dependent behavior of the processes that control transcript induction, that is, production and degradation. We discovered and quantified a most prominent feature of the pre-mRNA profiles of many genes, reflecting a transient pulse of production of previously unanticipated high dynamic range. Thus, production FC can exhibit a large overshoot over eventual mRNA FC. Moreover, genes with similar mRNA peak times exhibit a wide variation in production peak times, suggesting that the expression of such genes may be governed by different regulatory elements. Most EGF-induced genes initiate their production within the first hour after stimulation (Supplementary Figure S1B and C, blue curves). Two recently published studies addressed related issues. The first used global run-on and sequencing (Gro-seq) in a breast cancer cell line after estradiol stimulation (Hah et al, 2011), while the second used pulse labeling by 4sU in LPS-stimulated DCs. In agreement with our results, the first study reported that a large fraction of the transcriptional response was executed very rapidly, while the ensuing change of mRNA abundance was delayed by intervals that varied between 1 and 3 h. In contrast, the second study reported that changes in total mRNA lagged behind the corresponding changes in newly synthesized RNA by a fairly uniform interval of 15–30 min (Rabani et al, 2011).
Beyond production, the temporal profiles of mRNA induction are shaped also by degradation (Barenco et al, 2009; Hao and Baltimore, 2009). Our quantitative assessment properly weighs the relative contributions of production and degradation to the dynamics of transcriptional responses.
Production overshoot is instrumental, together with time-dependent degradation, in shaping precisely transient expression profiles, to bring a transcript to the right level at the right time and for the right duration. We found that most genes exhibiting production overshoot are bona fide PRGs and are enriched by executors of the phenotypic response to stimulation. These genes encode for relatively long-lived mRNAs, whose levels may be maintained at an economically favorable low production rate in the absence of stimuli.
Whether the overshoot is a digital all-or-none phenomenon, tuned only by the fraction of cells responding to stimulation (Podtschaske et al, 2007) or by the duration of production (Lahav et al, 2004; Suter et al, 2011), is an open question. Alternatively, it may comprise a graded transcriptional response in individual cells, demonstrated, for example, by independent NFκB binding to adjacent regulatory sites (Giorgetti et al, 2010). The underlying molecular mechanisms may also include cooperative action of transcription factors, regulation of the number of polymerases traveling across the gene, and regulation of polymerase processivity (Baugh et al, 2009; Wada et al, 2009). The very rapid offset kinetics of overshooting pre-mRNA production, which often precedes significant changes in mRNA abundance, suggests that if cells use feedback to induce this decrease, its mechanism likely relies on sensing the levels of pre-mRNA or nuclear mRNA, rather than of cytoplasmic mRNA or protein. An attractive and likely alternative to feedback is a mechanism of ‘prewired control’—production is designed to have a transient pulse-like profile. In different cellular model systems, we found that different genes exhibit production overshoot at different times, suggesting that the molecular mechanisms governing production overshoot may be gene and context dependent, and will require additional studies. Our findings are an essential prerequisite for such studies.
We believe that our demonstration of how similar mRNA profiles can be generated by very different production profiles constitutes an important conceptual advance. The insights gained by our modeling approach and experiments provide a consistent framework toward quantitative elucidation of operational and molecular strategies used by cells to regulate transcriptional responses to extracellular signals.
Materials and methods
Cell culture and stimulation
MCF10A cells were cultured as described in Katz et al (2007) and stimulated with EGF (20 ng/ml) for the indicated intervals. Bone marrow-derived murine DCs from C57BL/6 mice were prepared as previously described (Amit et al, 2009) and stimulated with LPS (100 ng/ml). H9 human ESCs were cultured as described in Supplementary information and stimulated with all-trans RA (1 μM). For RNA decay experiments, ActD was used as indicated to arrest transcription.
RNA isolation and microarray hybridization
RNA was isolated using the PerfectPure RNA Cultured Cell Kit (5 Prime, Hamburg, Germany) including DNAse 1 digestion and rRNA depleted. Samples were processed as recommended by the microarray manufacturer and hybridized to Affymetrix GeneChip Human Exon 1.0 ST arrays. Microarray data are deposited in Gene Expression Omnibus (accession number GSE24391).
Real-time qPCR
For qPCR of pre-mRNA and mRNA, respectively, forward primers were positioned in the second intron and exon, respectively, and a shared reverse primer was positioned in the third constitutive exon. For amplification of exon mRNA, primer pairs were positioned in the third exon. All qPCRs were performed using Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) on a 7900HT Fast Real Time PCR System platform (Applied Biosystems) along with non-template controls, melt curve analysis and cDNA dilution series. Detailed methods and curve fitting are described in Supplementary information and primer sequences are listed in Supplementary File 1.
Microarray data analyses
Affymetrix Expression Console (parameters: Annotation confidence—full, Summarization method—iterPLIER include DABG, Background—PM-GCBG, Normalization method—none) was used, followed by normalization of all arrays together using a Lowess multi-array algorithm and signal-dependent noise estimation, as described in Zeisel et al (2010). Annotation and signal-based information was used to define exonic and intronic PS. Intronic and exonic PS were used to calculate the gene-level FC of pre-mRNAs and mRNAs. A detailed description of microarray data processing is given in Supplementary information and Supplementary Figures S3 and S10.
Inference of transcript production and degradation profiles
Pre-mRNA, mRNA and exon FC were measured for selected genes using qPCR at up to 27 time points. For all measurements, the average of three technical replicates was plotted versus time. In order to infer the production profile, we used Equation (4). A particular 5-parameter functional form Pˆfit (t) (see Supplementary information) was used and the parameters were determined by best fit to the data. The value of the pre-mRNA conversion coefficient was determined for each transcript (see Figure 5A and Supplementary Figure S6A). The time derivative of the fitted function was taken (analytically) and Equation (4) was inverted to yield an analytic form of the production profile,
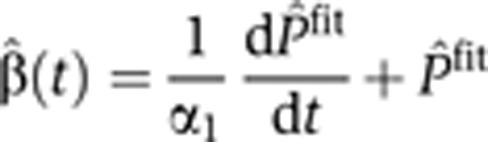 |
Next, for each gene, the pre-stimulus mRNA degradation rate α2(0) and degradation profile 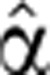 2(t) were inferred in a nested iterative procedure. A particular 7-parameter form was assumed for this function (see Supplementary information). The main iterative procedure is the following: start with an initial guess
2(t) were inferred in a nested iterative procedure. A particular 7-parameter form was assumed for this function (see Supplementary information). The main iterative procedure is the following: start with an initial guess 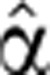 2(t)=1, optimize (as described below) the pre-stimulus coefficient to get α2(1)(0); using this value, optimize the degradation profile to get a new
2(t)=1, optimize (as described below) the pre-stimulus coefficient to get α2(1)(0); using this value, optimize the degradation profile to get a new 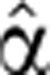 2(t), optimize again α2(0) to obtain the next optimal α2(0) and iterate until convergence.
2(t), optimize again α2(0) to obtain the next optimal α2(0) and iterate until convergence.
We describe here the optimization procedure for the case of fixed α2(0). We took an initial guess 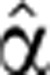 2(0)(t), substituted it for
2(0)(t), substituted it for 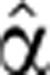 2(t) in the right hand side of Equation (5), together with the analytic fitted function Pˆfit(t). Next, we integrated Equation (5) numerically to yield an approximate Mˆ(0) (t). The least squares deviation of this function from the measured data was calculated, and new values for the parameters were set to define
2(t) in the right hand side of Equation (5), together with the analytic fitted function Pˆfit(t). Next, we integrated Equation (5) numerically to yield an approximate Mˆ(0) (t). The least squares deviation of this function from the measured data was calculated, and new values for the parameters were set to define 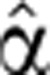 2(1)(t). The process was iterated until convergence to a function
2(1)(t). The process was iterated until convergence to a function 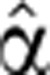 2(t) that gave the best fit to the measured Mˆ(t). A similar iterative process was used to optimize the pre-stimulus degradation coefficient for a given degradation profile (
2(t) that gave the best fit to the measured Mˆ(t). A similar iterative process was used to optimize the pre-stimulus degradation coefficient for a given degradation profile (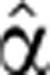 2(t)).
2(t)).
The error of the inferred functions, estimated on the basis of the qPCR measurement noise, is explained in Supplementary information and presented, for a single transcript, NR4A1, in Supplementary Figure S11.
Functions used for fit
The functions used to fit the pre-mRNA and degradation FC profiles, Pˆfit (t) and 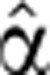 2(t) are described in detail in Supplementary information.
2(t) are described in detail in Supplementary information.
Supplementary Material
Excel File
Supplementary Figures S1–11
Acknowledgments
We thank Naama Barkai, Uri Alon, Tzachi Pilpel and Moshe Oren for fruitful discussions. This work was supported by the Leir Charitable Foundation, a Weizmann-Mario Negri collaborative research grant, the MD Moross Institute for Cancer Research, Arresto Biosciences, the European Commission, the Dr Miriam and Sheldon Adelson Medical Research Foundation, the Kekst Family Institute for Medical Genetics, the Kirk Center for Childhood Cancer and Immunological Disorders, the Women's Health Research Center funded by the Bennett-Pritzker Endowment Fund, the Marvelle Koffler Program for Breast Cancer Research, the German Research Foundation (DIP), National Cancer Institute (CA72981), and the Flight Attendants Medical Research Institute (FAMRI). ED is the incumbent of the Henri J Leir Professorial Chair, YY is the incumbent of the Harold and Zelda Goldenberg Professorial Chair, GR holds the Djerassi Chair in Oncology at the Tel-Aviv University and WJK is supported by a PhD Track for Specialist MDs fellowship of The Linda and Michael Jacobs Charitable Trust.
Author contributions: AZ, WJK, YY and ED designed and analyzed experiments and conceived mathematical modeling; AZ, WJK, NM, JMT, RK and JJ-H performed experiments; SJ, YS, GR, YY and ED provided funding and experimental platforms. AZ, WJK, YS, YY and ED wrote the manuscript.
Footnotes
The authors declare that they have no conflict of interest.
References
- Alon U (2007) An Introduction to Systems Biology—Designs Principles of Biological Circuits. Boca Raton, FL: Chapman and Hall/CRC [Google Scholar]
- Amit I, Citri A, Shay T, Lu Y, Katz M, Zhang F, Tarcic G, Siwak D, Lahad J, Jacob-Hirsch J, Amariglio N, Vaisman N, Segal E, Rechavi G, Alon U, Mills GB, Domany E, Yarden Y (2007) A module of negative feedback regulators defines growth factor signaling. Nat Genet 39: 503–512 [DOI] [PubMed] [Google Scholar]
- Amit I, Garber M, Chevrier N, Leite AP, Donner Y, Eisenhaure T, Guttman M, Grenier JK, Li W, Zuk O, Schubert LA, Birditt B, Shay T, Goren A, Zhang X, Smith Z, Deering R, McDonald RC, Cabili M, Bernstein BE et al. (2009) Unbiased reconstruction of a mammalian transcriptional network mediating pathogen responses. Science 326: 257–263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avraham R, Sas-Chen A, Manor O, Steinfeld I, Shalgi R, Tarcic G, Bossel N, Zeisel A, Amit I, Zwang Y, Enerly E, Russnes HG, Biagioni F, Mottolese M, Strano S, Blandino G, Borresen-Dale AL, Pilpel Y, Yakhini Z, Segal E et al. (2010) EGF decreases the abundance of microRNAs that restrain oncogenic transcription factors. Sci Signal 3: ra43. [DOI] [PubMed] [Google Scholar]
- Barenco M, Brewer D, Papouli E, Tomescu D, Callard R, Stark J, Hubank M (2009) Dissection of a complex transcriptional response using genome-wide transcriptional modelling. Mol Syst Biol 5: 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baugh LR, Demodena J, Sternberg PW (2009) RNA Pol II accumulates at promoters of growth genes during developmental arrest. Science 324: 92–94 [DOI] [PubMed] [Google Scholar]
- Bousquet-Antonelli C, Presutti C, Tollervey D (2000) Identification of a regulated pathway for nuclear pre-mRNA turnover. Cell 102: 765–775 [DOI] [PubMed] [Google Scholar]
- Boyer LA, Lee TI, Cole MF, Johnstone SE, Levine SS, Zucker JP, Guenther MG, Kumar RM, Murray HL, Jenner RG, Gifford DK, Melton DA, Jaenisch R, Young RA (2005) Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122: 947–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun JS, Wong MM, Cui W, Idelman G, Li Q, De Siervi A, Bilke S, Haggerty CM, Player A, Wang YH, Thirman MJ, Kaberlein JJ, Petrovas C, Koup RA, Longo D, Ozato K, Gardner K (2009) Dynamic bookmarking of primary response genes by p300 and RNA polymerase II complexes. Proc Natl Acad Sci USA 106: 19286–19291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carballo E, Lai WS, Blackshear PJ (1998) Feedback inhibition of macrophage tumor necrosis factor-alpha production by tristetraprolin. Science 281: 1001–1005 [DOI] [PubMed] [Google Scholar]
- Cochran BH, Reffel AC, Stiles CD (1983) Molecular cloning of gene sequences regulated by platelet-derived growth factor. Cell 33: 939–947 [DOI] [PubMed] [Google Scholar]
- Darzacq X, Shav-Tal Y, de Turris V, Brody Y, Shenoy SM, Phair RD, Singer RH (2007) In vivo dynamics of RNA polymerase II transcription. Nat Struct Mol Biol 14: 796–806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkon R, Zlotorynski E, Zeller KI, Agami R (2010) Major role for mRNA stability in shaping the kinetics of gene induction. BMC Genomics 11: 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Yang X, Wang W, Wood WH III, Becker KG, Gorospe M (2002) Global analysis of stress-regulated mRNA turnover by using cDNA arrays. Proc Natl Acad Sci USA 99: 10611–10616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedel CC, Dolken L, Ruzsics Z, Koszinowski UH, Zimmer R (2009) Conserved principles of mammalian transcriptional regulation revealed by RNA half-life. Nucleic Acids Res 37: e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau NL, Wilusz J, Wilusz CJ (2007) The highways and byways of mRNA decay. Nat Rev Mol Cell Biol 8: 113–126 [DOI] [PubMed] [Google Scholar]
- Giorgetti L, Siggers T, Tiana G, Caprara G, Notarbartolo S, Corona T, Pasparakis M, Milani P, Bulyk ML, Natoli G (2010) Noncooperative interactions between transcription factors and clustered DNA binding sites enable graded transcriptional responses to environmental inputs. Mol Cell 37: 418–428 [DOI] [PubMed] [Google Scholar]
- Gorini L, Maas WK (1957) The potential for the formation of a biosynthetic enzyme in Escherichia coli. Biochim Biophys Acta 25: 208–209 [DOI] [PubMed] [Google Scholar]
- Guang S, Bochner AF, Burkhart KB, Burton N, Pavelec DM, Kennedy S (2010) Small regulatory RNAs inhibit RNA polymerase II during the elongation phase of transcription. Nature 465: 1097–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hager GL, McNally JG, Misteli T (2009) Transcription dynamics. Mol Cell 35: 741–753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hah N, Danko CG, Core L, Waterfall JJ, Siepel A, Lis JT, Kraus WL (2011) A rapid, extensive, and transient transcriptional response to estrogen signaling in breast cancer cells. Cell 145: 622–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao S, Baltimore D (2009) The stability of mRNA influences the temporal order of the induction of genes encoding inflammatory molecules. Nat Immunol 10: 281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves DC, Horng T, Medzhitov R (2009) Control of inducible gene expression by signal-dependent transcriptional elongation. Cell 138: 129–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herschman HR (1991) Primary response genes induced by growth factors and tumor promoters. Annu Rev Biochem 60: 281–319 [DOI] [PubMed] [Google Scholar]
- Hirose Y, Manley JL (1998) RNA polymerase II is an essential mRNA polyadenylation factor. Nature 395: 93–96 [DOI] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4: 44–57 [DOI] [PubMed] [Google Scholar]
- Iyer VR, Eisen MB, Ross DT, Schuler G, Moore T, Lee JC, Trent JM, Staudt LM, Hudson J Jr, Boguski MS, Lashkari D, Shalon D, Botstein D, Brown PO (1999) The transcriptional program in the response of human fibroblasts to serum. Science 283: 83–87 [DOI] [PubMed] [Google Scholar]
- Katz M, Amit I, Citri A, Shay T, Carvalho S, Lavi S, Milanezi F, Lyass L, Amariglio N, Jacob-Hirsch J, Ben-Chetrit N, Tarcic G, Lindzen M, Avraham R, Liao YC, Trusk P, Lyass A, Rechavi G, Spector NL, Lo SH et al. (2007) A reciprocal tensin-3-cten switch mediates EGF-driven mammary cell migration. Nat Cell Biol 9: 961–969 [DOI] [PubMed] [Google Scholar]
- Lahav G, Rosenfeld N, Sigal A, Geva-Zatorsky N, Levine AJ, Elowitz MB, Alon U (2004) Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat Genet 36: 147–150 [DOI] [PubMed] [Google Scholar]
- Larson DR, Zenklusen D, Wu B, Chao JA, Singer RH (2011) Real-time observation of transcription initiation and elongation on an endogenous yeast gene. Science 332: 475–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau LF, Nathans D (1987) Expression of a set of growth-related immediate early genes in BALB/c 3T3 cells: coordinate regulation with c-fos or c-myc. Proc Natl Acad Sci USA 84: 1182–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan S, Alon U (2003) Structure and function of the feed-forward loop network motif. Proc Natl Acad Sci USA 100: 11980–11985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan S, Zaslaver A, Alon U (2003) The coherent feedforward loop serves as a sign-sensitive delay element in transcription networks. J Mol Biol 334: 197–204 [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Horng T (2009) Transcriptional control of the inflammatory response. Nat Rev Immunol 9: 692–703 [DOI] [PubMed] [Google Scholar]
- Miller C, Schwalb B, Maier K, Schulz D, Dumcke S, Zacher B, Mayer A, Sydow J, Marcinowski L, Dolken L, Martin DE, Tresch A, Cramer P (2011) Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol Syst Biol 7: 458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orphanides G, Reinberg D (2002) A unified theory of gene expression. Cell 108: 439–451 [DOI] [PubMed] [Google Scholar]
- Patel AA, McCarthy M, Steitz JA (2002) The splicing of U12-type introns can be a rate-limiting step in gene expression. EMBO J 21: 3804–3815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessa HK, Ruokolainen A, Frilander MJ (2006) The abundance of the spliceosomal snRNPs is not limiting the splicing of U12-type introns. RNA 12: 1883–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podtschaske M, Benary U, Zwinger S, Hofer T, Radbruch A, Baumgrass R (2007) Digital NFATc2 activation per cell transforms graded T cell receptor activation into an all-or-none IL-2 expression. PLoS ONE 2: e935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanth KV, Prasanth SG, Xuan Z, Hearn S, Freier SM, Bennett CF, Zhang MQ, Spector DL (2005) Regulating gene expression through RNA nuclear retention. Cell 123: 249–263 [DOI] [PubMed] [Google Scholar]
- Proudfoot NJ, Furger A, Dye MJ (2002) Integrating mRNA processing with transcription. Cell 108: 501–512 [DOI] [PubMed] [Google Scholar]
- Rabani M, Levin JZ, Fan L, Adiconis X, Raychowdhury R, Garber M, Gnirke A, Nusbaum C, Hacohen N, Friedman N, Amit I, Regev A (2011) Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat Biotechnol 29: 436–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Carrozzi VR, Braas D, Bhatt DM, Cheng CS, Hong C, Doty KR, Black JC, Hoffmann A, Carey M, Smale ST (2009) A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell 138: 114–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld N, Elowitz MB, Alon U (2002) Negative autoregulation speeds the response times of transcription networks. J Mol Biol 323: 785–793 [DOI] [PubMed] [Google Scholar]
- Sassone-Corsi P, Sisson JC, Verma IM (1988) Transcriptional autoregulation of the proto-oncogene fos. Nature 334: 314–319 [DOI] [PubMed] [Google Scholar]
- Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M (2011) Global quantification of mammalian gene expression control. Nature 473: 337–342 [DOI] [PubMed] [Google Scholar]
- Shalem O, Dahan O, Levo M, Martinez MR, Furman I, Segal E, Pilpel Y (2008) Transient transcriptional responses to stress are generated by opposing effects of mRNA production and degradation. Mol Syst Biol 4: 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, Padgett RA (2009) Rates of in situ transcription and splicing in large human genes. Nat Struct Mol Biol 16: 1128–1133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter DM, Molina N, Gatfield D, Schneider K, Schibler U, Naef F (2011) Mammalian genes are transcribed with widely different bursting kinetics. Science 332: 472–474 [DOI] [PubMed] [Google Scholar]
- Tullai JW, Schaffer ME, Mullenbrock S, Sholder G, Kasif S, Cooper GM (2007) Immediate-early and delayed primary response genes are distinct in function and genomic architecture. J Biol Chem 282: 23981–23995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada Y, Ohta Y, Xu M, Tsutsumi S, Minami T, Inoue K, Komura D, Kitakami J, Oshida N, Papantonis A, Izumi A, Kobayashi M, Meguro H, Kanki Y, Mimura I, Yamamoto K, Mataki C, Hamakubo T, Shirahige K, Aburatani H et al. (2009) A wave of nascent transcription on activated human genes. Proc Natl Acad Sci USA 106: 18357–18361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zang C, Cui K, Schones DE, Barski A, Peng W, Zhao K (2009) Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138: 1019–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Amir A, Kostler WJ, Domany E (2010) Intensity dependent estimation of noise in microarrays improves detection of differentially expressed genes. BMC Bioinformatics 11: 400. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Excel File
Supplementary Figures S1–11