

Abstract
Pathogen recognition is the first and crucial step in innate immunity. Molecular families involved in the recognition of pathogens and activation of the innate immune responses in immunoreactive cells include the Toll-like receptor family in mammals and the peptidoglycan recognition protein (PGRP) family in Drosophila, which sense microorganisms in an extracellular or luminal compartment. Other emerging families are the intracellular recognition molecules for bacteria, such as nucleotide binding and oligomerization domain-like receptors in mammals and PGRP–LE in Drosophila, several of which have been shown to detect structures of bacterial peptidoglycan in the host cell cytosol. Exciting advances in recent studies on autophagy indicate that macroautophagy (referred to here as autophagy) is selectively induced by intracellular recognition molecules and has a crucial role in the elimination of intracellular pathogens, including bacteria, viruses and parasites. This review discusses recent studies related to intracellular recognition molecules and innate immune responses to intracellular pathogens, and highlights the role of autophagy in innate immunity.
Keywords: autophagy, inflammatory disease, innate immunity, intracellular pathogens, pattern recognition molecules
The first important step in innate immunity is the recognition of various infectious microbes as distinct from self, which leads to the induction of the appropriate innate immune response. Activation of innate immune responses in response to pathogens, therefore, relies on the detection of conserved microbial motifs. The recognition of infectious microbes in innate immunity is achieved though the detection of pathogen-associated molecular patterns, the conserved microbial components, including lipopolysaccharide (LPS), peptidoglycan (PGN), flagellin and nucleic acids from bacteria, fungi or viruses, respectively, which are essential for the microbial survival, but are not found in higher eukaryotes (1). This initial recognition is mediated by a set of genome-encoded pattern recognition molecules, which sense the conserved pathogen-associated molecular patterns. The study of innate immune recognition molecules originated with the identification of Drosophila Toll, a plasma membrane-associated receptor, which itself does not have a direct role in the recognition of microbes, but is essential for the innate immune responses against fungi and Gram-positive bacteria by receiving the immune signalling on the cellular membrane to activate downstream signalling (2, 3). Cloning of the mammalian homologues of Drosophila Toll, Toll-like receptors (TLRs), and their functional studies have led to great advances in understanding how mammalian cells sense microbe infection (4). TLR-2, 4 and 5 recognize PGN, LPS and flagellin, respectively, on the surface of immune responding cells to detect bacteria or fungi, whereas TLR3, 7 and 9 recognize poly IC, ssRNA, CpG DNA, respectively, inside the endosome to detect invasive viruses (5). These specificities of the recognition of these family members of TLRs enable the host cells to sense wide ranges of microbes. In Drosophila, different from mammals, the recognition of bacteria or fungi depends on peptidoglycan recognition proteins (PGRPs). Some PGRP members, PGRP–SA, SD, LC and LE, are essential for the recognition of peptidoglycan or LPS in bacteria, similar to TLR-2 or TLR-4, respectively, in mammals, and activate immune signalling pathways that are conserved from flies to mammals (5). PGRP–SA and SD function in the haemolymph, a circulatory fluid in body cavity of insects, whereas PGRP–LC is on the surface of immune responding cells facing the haemolymph to sense the infectious microbes outside or on the surface of the host cells (6). The different molecular families used for pathogen recognition, TLRs in mammals and PGRPs in Drosophila, might be related to the differences in the body structures of each species and the methods of detecting pathogen invasion of the bodies. In mammals, microbes that penetrate the epithelial cells of the body are first detected in the local lymph by TLRs on macrophages, leading to inflammatory responses that cause the accumulation of macrophages and neutrophils at the site of the infection. In contrast, insect bodies usually comprise one body cavity that contains tissues floating in haemolymph in which invaded microbes easily move, making them easily detected by circulating PGRPs, which provoke the induction and secretion of antimicrobial peptides to the haemolymph.
Intracellular recognition of bacteria via NOD-like receptors
Intracellular bacteria, such as Salmonella, Listeria and Mycobacterium, invade the host cells and grow inside the cells, thereby escaping from these humoral or cellular luminal defences. Recent studies revealed that mammalian cells have another family of detection molecules other than TLRs, called nucleotide binding and oligomerization domain (NOD)-like receptors (NLRs), which are present in the cytoplasm of the host cells and detect fragments of bacterial peptidoglycan (7). The discovery of these intracellular pathogen-associated molecular patterns receptors suggests that host cells use these receptors in addition to the extracellular receptors as an intracellular defence line against invasive bacteria.
The first description of intracellular receptors in mammals for bacterial detection was Nod1 (nucleotide-binding oligomerization domain 1; also referred to as Card4), which mediates NF-κB and JNK activation following invasive Shigella flexneri infection (8). Nod1 was originally identified as a novel member of caspase-recruitment domain (CARD)-containing protein, which is capable of activating NF-κB when it is overexpressed (9, 10). The NLR family comprises more than 20 members that contain the molecular feature of an N-terminal CARD domain, which is responsible for protein-protein interaction; a C-terminal leucine-rich repeat domain; and a central nucleotide-biding and oligomerization domain (NOD or NACHT domain). Leucine-rich repeats domains generally comprise 20–30 amino acids and are responsible for the detection of bacterial components.
Following biochemical studies on the molecules recognized by NLRs, many studies on the functions of NLRs in the susceptibility to intracellular bacteria were reported. NOD1 and NOD2 are required for resistance against Shigella flexneri (8, 11); NOD1, NOD2, Nlrc4 (Ipaf) and Nlrp3 (NALP3) against Listeria monocytogenes (12–14); Nlrc4 and NOD2 against Salmonella typhimurium (15); and Naip5 (Birc 1e) against Legionella pneumophila (16, 17).
Consistent with the functional importance of NLRs in the resistance against intracellular bacteria in vivo, NLRs activate immune signalling pathways (18). CARD9, an adaptor protein required for innate immune responses to L. monocytogenes interacts with NOD2 and activates p38 and JNK to produce proinflammatory cytokines (19). Despite the evidence of NLRs and signalling molecules, which are essential for innate immune responses such as cytokine production and inflammatory responses after infection of intracellular bacteria, it remains unclear how host animals combat invasive pathogens that grow within the host cells. One prominent innate immune response mechanism that effectively eliminates bacteria inside the cells is autophagy, a fundamental non-selective degradation system for proteins and organelles. The relationship between the intracellular sensors and autophagy is discussed below.
Intracellular receptors in insects
Genetic screenings using Drosophila melanogaster and studies on family proteins have revealed many pattern recognition proteins that function in the recognition of extracellular pathogens. PGRP–SA recognizes Gram-positive bacteria and activates the Toll pathway, one of the two major immune signalling pathways for the activation of NF-κB-like transcription factors, together with Gram-negative-binding protein (GNBP)-1 (20, 21). PGRP–SD functions in the resistance against Gram-positive bacteria and has some redundancy with PGRP–SA and GNBP-1 (22). GNBP-3 is essential for activation of the Toll pathway in response to fungal infection (23). For resistance against Gram-negative bacteria, PGRP–LC and PGRP–LE redundantly function as pattern recognition receptors and activate the imd pathway, another immune signalling pathway (24).
In contrast to these extracellular sensors for microbes, the intracellular sensors in insects for intracellular bacteria, viruses, and parasites are mostly unknown. Only one sensor has been shown to detect pathogens within the cells and it has a crucial role in the resistance against the pathogen: PGRP–LE. PGRP–LE was originally identified as an extracellular pattern recognition receptor that detects the DAP-type peptidoglycan, the type of peptidoglycan, the peptide stem of which has meso-diaminopimelic acid (DAP) at the third residue, and possessed by Gram-negative and some species of Gram-positive bacteria (25), although the molecular nature of PGRP–LE is different from that of the other PGRPs; PGRP–LE has no signal peptides for secretion nor a transmembrane domain.
The intracellular role of PGRP–LE was revealed by the mosaic analysis of clonal expression of the PGRP–LE gene. PGRP acts in a non-cell autonomous manner in the fat body to activate the imd pathway whereas in the malphigian tubules PGRP–LE activates the imd pathway in cell-autonomous manner, suggesting that in some situations or in some tissues, PGRP–LE acts inside the cells (24). Following the evidence that the Drosophila culture cell line S2 cells react to transfected tracheal cytotoxin (TCT), a partial structure of DAP-type peptidoglycan to induce the antimicrobial peptides expression, PGRP–LE was shown to have a crucial role in the resistance against Listeria monocytogenes, an intracellular Gram-positive bacterium that has a DAP-type peptidoglycan (26, 27). Inhibition of the intracellular growth of L. monocytogenes after infection in vitro in cultured haemocytes, phagocytotic macrophage-like cells of Drosophila, requires PGRP–LE, but not PGRP–LC. Interestingly this PGRP–LE dependent inhibition of bacterial growth does not require factors working in the Toll pathway or imd pathway, but does require Atg5 or Atg1, factors functioning in autophagy (27). PGRP–LE co-localizes with the invaded L. monocytogenes to induce autophagy, and this induction at the site of the bacteria is totally dependent on PGRP–LE.
Detection of L. monocytogenes by PGRP–LE inside the cells provokes other immune responses than autophagy. Strategic microarray analysis to search for PGRP–LE dependent gene expression in response to bacterial infection revealed that the novel antibacterial peptide Listericin is expressed in a JAK-STAT pathway-dependent manner, and is secreted from the cells (28). Thus, intracellular recognition of the bacteria by PGRP–LE induces more than one immune response to defend the body from the infection at different stages of the infection.
Although autophagy is induced via PGRP–LE when DAP-type peptidoglycan is transfected to Drosophila cells, transfection of Lys-type peptidoglycan, the peptidoglycan in most Gram-positive bacteria, induces autophagy in a PGRP–LE independent manner, suggesting that a different and unknown intracellular recognition molecule(s) for the Lys-type peptidoglycan is present in the cells (27). An intracellular recognition protein(s) with such binding properties has not been found in mammalian cells, despite the clear induction of autophagy of the invading Group A Streptococcus (29).
Role of autophagy in elucidation of microbes
Autophagy is a fundamental lysosome-dependent process well conserved in eukaryotes from yeast to humans involved in the turnover of molecules and organelles in the cytoplasm to maintain cellular homeostasis. In addition to this function, its induction to a higher level for supplying nutrients under nutrient deprived conditions is linked to a broad range of biological processes; i.e. development, aging, cancer, neurodegenerative diseases and inflammatory disorders (30). At the beginning of the process of autophagy, undesirable or recyclable proteins and organelles are sequestered in a structure known as the isolation membrane, and this membrane elongates to form the autophagosome, a structure distinct from the phagosome by the presence of a double membrane. Autophagosomes then mature by fusion with lysosomes to become autolysosomes, in which the captured substances are eventually degraded. These processes along with the drastic membrane traffic are mediated by evolutionally conserved factors known as autophagy-related (ATG) proteins. Many of the ATG proteins were originally identified in the screenings using yeast to isolate genes involved in the responses to nutrient limitation (31). The identification of these factors has led to numerous studies in mammals to elucidate the mechanism of autophagy and how each factor functions in these processes (32). The molecules involved in autophagy are well conserved even in Drosophila, which allows for analyses of the physiological role of autophagy in whole animals and of its mechanism using powerful genetic methods in insects (33).
Invasive pathogens, including intracellular bacteria, viruses and parasites, are degraded by autophagy when they invade the host cell, and this autophagic degradation acts as an innate immune defence mechanism to eliminate intracellular microbes (34). Because autophagosomes are capable of engulfing whole organelles such as mitochondria, it is not surprising that autophagosomes can surround whole pathogens and degrade them inside the cell. When microbes invade the host cells, autophagosome formation is usually detected in a couple of hours or less, and the autophagic processes begin within hours. Therefore, autophagy works as a front line innate immune response against invasive microbes.
Induction of autophagy by pathogen sensors
Various intracellular microbes are eliminated by autophagy, and this elimination is in many cases crucial for host survival against the microbe (Table 1). Although autophagy is a non-selective degradation process, autophagosomes do not form randomly in the cytoplasm, but rather sequester the bacteria selectively (29). Furthermore, autophagosomes that engulf the microbe are sometimes much larger than those formed during nutrient starvation, suggesting that the elongation step of the autophagosome membrane is involved in bacteria-surrounding autophagy. The mechanism underlying selective induction of autophagy at the site of microbe invasion is largely unknown. The intracellular pattern recognition proteins against microbes, which detect microbes inside the cells and enable autophagosomes to form at the site of the invasion, are clues to clarify the molecular mechanisms. In the first report of the recognition protein-dependent activation of autophagy during microbe infection, the authors performed experiments using Drosophila, in which autophagy induction against L. monocytogenes in Drosophila haemocytes is dependent on PGRP–LE (27, Fig. 1). Importantly, autophagic induction via the recognition protein is independent from the known and well-studied immune signalling pathways, the Toll pathway and the imd pathway, the activation of which results in the activation of NF-κB like transcription factors, suggesting that a novel mechanism of induction exists in the cells (27). In parallel, in mammalian cells, NOD1 and NOD2, the intracellular sensors described above, have a critical role in autophagy induction upon entry of S. flexneri or L. monocytogenes into the cells (35). The NOD-dependent activation of autophagy is independent from RIP2, an adaptor protein for the activation of NF-κB to trigger proinflammatory signals; and NEMO, an NF-κB essential modulator, suggesting a striking similarity in mechanism of the activation of autophagy between these species. The binding of pattern recognition receptors to invading bacteria for targeting adaptor protein(s) to recruit factors essential for autophagosomes is a possible mechanism of autophagy activation.
Table I.
Microbes eliminated by autophagy.
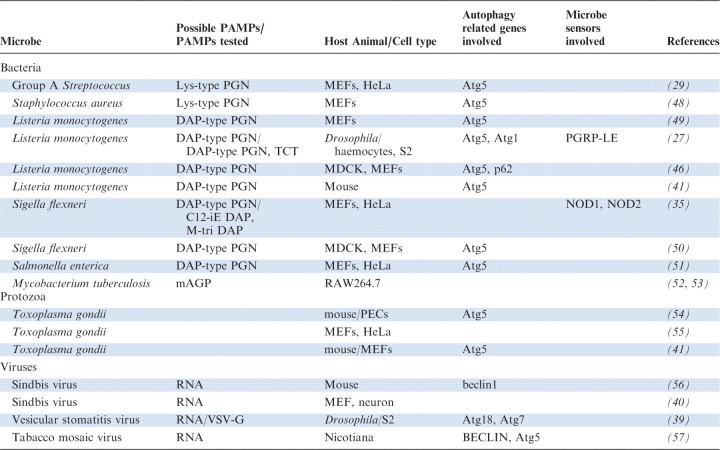 |
PAMPs, Pathogen associated molecular patterns; mAGP, mycolyl–arabinogalactan–peptidoglycan; MEFs, mouse embrionic fibroblasts; PECs, primed peritoneal exudate cells.
Fig. 1.

Pathogens and innate immune signalling for autophagy induction in flies and mammals. Autophagy is induced by the infection of intracellular bacteria and some kind of viruses. In flies, PGN of invasive bacteria is detected by PGRPs inside cells, which provokes autophagy independent from Toll pathway and Imd pathway. Viral glycoprotein (VSV-G) causes autophagy induction via unidentified sensor. In mammals, NLRs detect bacterial PGN in the cytosol of infected cells to induce autophagy independent from RIP2, while TLRs on the cell plasma membrane also involved in the activation of autophagy via NF-κB signalling pathway. Sindbis virus infection causes autophagy in mice, although the pathogen sensors involved for the induction has yet not been clarified.
TLRs, well-characterized pattern recognition proteins in mammals, are also involved in the activation of autophagy (36, 37). The TLR4 ligand LPS is able to induce autophagy in RAW264.7 cells in a TLR4-dependent manner, and TLR7 induces autophagy after single-strand RNA stimulation. The activation of autophagy via TLRs is dependent on the signalling pathway that leads to NF-κB activation; TLR4-mediated autophagy depends on TRIF, and TLR7 on MyD88 (36, 37). It is therefore likely that the mechanism for the activation of autophagy via TLRs is distinct from the activation mechanism via PGRP–LE and NODs, which is consistent with the distinct location of pathogen recognition between these sensors; i.e. TLR4 on the surface of the plasma membrane, TLR7 in the endosome, and PGRP–LE and NODs in the cytoplasm.
It was recently demonstrated that autophagy also functions in antiviral immunity (Table I, 38), but the involvement of the pattern recognition receptors for viruses in the induction of autophagy is not clear. In the case of vesicular stomatitis virus infection in Drosophila cells, induction of autophagy might not be the result of recognition of the viral genome RNA, because transfection of viral pattern recognition molecules, such as viral RNA or RNP isolated from the virus, does not induce autophagy, but rather transfection of the viral glycoprotein VSV-G particles cause autophagy induction (39). In mouse cells infected with sindbis virus, autophagy induction requires replication of the virus in the cytoplasm of the host cells; the viral capsid proteins are targeted to the autophagosomes by making a complex with p62, a known adaptor of selective autophagy, and functions in the clearance of protein aggregates in neurons (40). Therefore, the major role of autophagy in the resistance against sindbis virus might not be elimination of the virus itself, but clearance of the virus protein containing aggregates, which may cause neural toxicity, might contribute to animal survival. The involvement of a cytoplasmic virus sensor, retinoic acid-inducible gene I, in autophagy induction has not been clarified.
Infection of Toxoplasma gondii in mouse macrophages also induces autophagy, and Atg5, a key molecule for autophagy is required for the resistance (41). Although autophagy genes are clearly involved in the resistance to T. gondii, classical autophagosomal membranes are not observed to surround T. gondii, suggesting that Atg5 plays a novel role in the elimination of the microbe rather than classical autophagy, with intracellular membrane dynamics causing damage to the parasitiphorous vacuole. For that process, it remains an open question if there is any intracellular sensor(s) present in the cytoplasm of the host cells to activate the signalling pathway for Atg5-dependent pathogen elimination.
Autophagy genes, intracellular sensors and inflammatory disease
One notable aspect of autophagy genes and intracellular pathogen sensors in innate immunity is their role in tissue homeostasis (42). Crohn’s disease, a chronic inflammatory disorder of the intestine, has a strong genetic association with autophagy; genome-wide scans have identified single-nucleotide polymorphisms (SNPs) of Crohn’s disease patients in the autophagy gene ATG16L1, autophagy-stimulatory GTPase IRGM and intracellular bacterial sensor NOD2. Furthermore, involvement of ATG16L1 in Crohn’s disease is supported by studies using ATG16L1 knockout (ATG16L1–/–) or hypomorphic mutant mice (ATG16L1HM) (43). ATG16L1HM mice show abnormalities in the Paneth cells, specialized intestinal epithelial cells that secrete antimicrobial proteins, with disorganized granules in the cells and defects in the granule exocytosis pathway (44). In foetal liver-derived macrophages of ATG16L1–/– mice, aberrantly enhanced production of an inflammatory cytokine, interleukin 1β (IL-1β), is observed in response to LPS stimulation (45). The finding that stimulation by non-invasive Escherichia coli or Enterobactor aerogenes causes abnormal cytokine secretion from ATG16L1–/– cells, but Salmonella typhimurium, an invasive bacterium, does not, suggests that it is not selective autophagy provoked by the detection of pathogen-related molecules in the cells, but rather it is a pathogen sensor(s) for the extracellular bacteria that is involved in activating the immune signalling pathway for secretion, which is affected by the loss of basal autophagy (45). Signalling from the intracellular sensor to enhance cytokine signalling is not excluded, however, because macrophages expressing the disease-associated NOD2 mutant also show enhanced expression of IL-1β when stimulated with a NOD2 ligand· Identification of the in vivo stimulants of the intestinal macrophages may help to address this issue.
Conclusion and future perspectives
Extensive studies of the role of autophagy reveal crucial functions of autophagy in immunity. Studies of autophagy as an innate immune response began with experiments using cultured cells, which revealed the molecular and cellular mechanisms of autophagic elimination of microbes. To elucidate the physiological role of autophagy in tissues or in the whole body, animals with conditional knockout of autophagy-related genes and in vivo infection models are indispensable (32).
The central issue of selective autophagy in innate immunity is the mechanism of the induction of autophagy at the site of the microbe. In case of bacterial infection, detection of the bacteria by recognition proteins triggers the whole process as described in this review, but downstream mechanisms including the recruitment of factors and the membrane involvement in autophagy at the target site require further elucidation. Targeting of L. monocytogenes to the autophagosome requires p62, which binds to poly-ubiquitin and LC3, an essential factor for autophagy, by associating with the autophagosome membrane (46). Another ubiquitin-associated factor that requires for the restriction of the intracellular bacterial growth is NDP52, which co-localizes with ubiquitin-coated Salmonella enterica, and binds to LC3 (47). The relationship between the pattern recognition protein-mediated induction of autophagy and these ubiquitin associated proteins-mediated pathway remains unclear, but further and precise analyses of the nucleation and elongation of the autophagosome may shed light on how bacteria are selectively degraded.
This review focuses on the elimination of microbes by their recognition and autophagy. Although in some cases autophagy is a powerful and indispensable process for host survival against microbes, microbes often escape from or even utilize autophagy in various strategies (38). Therefore, investigation of their mechanism and manipulation could lead to more targeted therapeutic approaches.
Funding
This work was supported by Grants-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan; the Japan Society for the Promotion of Science; the Program for the Promotion of Basic Research Activities for Innovative Biosciences (PROBRAIN); the Strategic International Cooperative program from Japan Science and Technology Agency; the National Institutes of Health (AI07495); the Takeda Science Foundation; the Mitsubishi Foundation; the Naito Foundation; and the Cabinet Office, Government of Japan through its “Funding Program for Next Generation World-Leading Researchers”.
Conflict of interest
None declared.
Glossary
Abbreviations
- LPS
lipopolysaccharide
- PGN
peptidoglycan
References
- 1.Akira S, Uematsu S, Takeuchi O. Pattern recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 2.Lemaitre B, Hoffmann J. The host defense of Drosophila melanogaster. Ann. Rev. Immunol. 2007;25:697–743. doi: 10.1146/annurev.immunol.25.022106.141615. [DOI] [PubMed] [Google Scholar]
- 3.Lemaitre B, Nicolas E, Michaut L, Reichhart J M, Hoffmann JA. The dorsoventral regulatory gene cassette spätzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86:973–983. doi: 10.1016/s0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- 4.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-loke receprotes. Nat. Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 5.Dziaeski R. Peptidoglycan recognition proteins (PGRPs) Mol. Immnol. 2004;40:877–886. doi: 10.1016/j.molimm.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 6.Kurata S. Extracellular and intracellular pathogen recognition by Drosophila PGRP-LE and PGRP-LC. Int. Immunol. 2010;22:143–148. doi: 10.1093/intimm/dxp128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanneganti T-D, Lamkanfi M, Núñez Intracellular NOD-like receptors in host defense and disease. Immunity. 2007;27:549–559. doi: 10.1016/j.immuni.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 8.Girardin SE, Tournebize R, Mavris M, Page AL, Li X, Stark GR, Bertin J, SiStefano PS, Yaniv M, Sansonetti PJ, Philpott DJ. CARD/Nod1 mediates NF-kappaB and JNK activation by invasive Shigella flexneri. EMBO Rep. 2001;2:736–742. doi: 10.1093/embo-reports/kve155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bertin J, Nir W-J, Fischer CM, Tayber OV, Errada PR, Grant JR, Keilty JJ, Gosselin ML, Robison KE, Wong GHW, Glucksmann MA, DiStefano PS. Human CARD4 protein is a novel CED-4/Apaf-1 cell death family member that activates NF-κB. J. Biol. Chem. 1999;274:12955–12958. doi: 10.1074/jbc.274.19.12955. [DOI] [PubMed] [Google Scholar]
- 10.Inohara N, Koseki T, del Peso L, Hu Y, Yee C, Chen S, Carrio R, Merino J, Liu D, Ni J, Núñez G. Nod1, an Apaf-1-like Activator of Caspase-9 and Nuclear Factor-κB. J. Biol. Chem. 1999;274:14560–14567. doi: 10.1074/jbc.274.21.14560. [DOI] [PubMed] [Google Scholar]
- 11.Nigro G, Fazio LL, Martino MC, Rossi G, Tattoli I, Liparoti V, De Castro C, Molinaro A, Philpott DJ, Bernardini ML. Muramylpeptide shedding modulates cell sending of Shigella flexneri. Cell. Microbiol. 2008;10:682–695. doi: 10.1111/j.1462-5822.2007.01075.x. [DOI] [PubMed] [Google Scholar]
- 12.Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Núñez G, Flavell RA. Nod2-Dependent Regulation of Innate and Adaptive Immunity in the Intestinal Tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 13.Warren SE, Mao DP, Rodriguez AE, Miao EA, Aderem A. Multiple Nod-Like Receptors Activate Caspase 1 during Listeria monocytogenes Infection. J. Immunol. 2008;180:7558–7564. doi: 10.4049/jimmunol.180.11.7558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim Y-G, Park J-H, Shaw MH, Franchi L, Inohara N, Núñez G. The cytosolic sensors Nod1 and Nod2 are critical for bacterial recognition and host defense after exposure to toll-like receptor ligands. Immunity. 2008;28:246–257. doi: 10.1016/j.immuni.2007.12.012. [DOI] [PubMed] [Google Scholar]
- 15.Franchi L, Amer A, Body-Malapel M, Kanneganti T-D, Özören N, Jagirdar R, Inohara N, Vandenabeele P, Bertin J, Coyle A, Grant EP, Núñez G. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1β in salmonella-infected macrophages. Nat. Immunol. 2006;7:576–582. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 16.Wright EK, Goodart SA, Growney JD, Hadinoto V, Endrizzi MG, Long EM, Sadigh K, Abney AL, Bernstein-Hanley I, Dietrich WF. Naip5 Affects Host Susceptibility to the Intracellular Pathogen Legionella pneumophila. Curr. Biol. 2003;13:27–36. doi: 10.1016/s0960-9822(02)01359-3. [DOI] [PubMed] [Google Scholar]
- 17.Zamboni DS, Kobayashi KS, Kohlsdorf T, Ogura Y, Long EM, Vence RE, Kuida K, Mariathasan S, Dixit VM, Flavell RA, Dietrich WF, Roy CR. The Birc1e cytosolic pattern-recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat. Immunol. 2006;7:318–325. doi: 10.1038/ni1305. [DOI] [PubMed] [Google Scholar]
- 18.Fritz JH, Ferrero R, Philpott DJ, Girardin SE. Nod-like proteins in immunity, inflammation and disease. Nat. Immunol. 2006;7:1250–1257. doi: 10.1038/ni1412. [DOI] [PubMed] [Google Scholar]
- 19.Hsu Y-M, Zhang Y, You Y, Wang D, Li H, Duramad O, Qin X-F, Dong C, Lin X. The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat. Immunol. 2007;8:198–205. doi: 10.1038/ni1426. [DOI] [PubMed] [Google Scholar]
- 20.Gottar M, Gobert V, Michel T, Belvin M, Duyk G, Hoffmann J A, Ferrandon D, Royet J. The Drosophila immune response against Gram-negative bacteria is mediated by a peptidoglycan recognition protein. Nature. 2002;416:640–644. doi: 10.1038/nature734. [DOI] [PubMed] [Google Scholar]
- 21.Gobert V, Gottar M, Matskevich AA, Rutschmann S, Royet J, Belvin M, Hoffmann JA, Ferrandon D. Dual activation of the Drosophila toll pathway by two pattern recognition receptors. Science. 2003;302:2126–2130. doi: 10.1126/science.1085432. [DOI] [PubMed] [Google Scholar]
- 22.Bischoff V, Vignal C, Boneca IG, Michel T, Hoffmann JA, Royet J. Function of the drosophila pattern-recognition receptor PGRP-SD in the detection of Gram-positive bacteria. Nat. Immunol. 2004;5:1175–1180. doi: 10.1038/ni1123. [DOI] [PubMed] [Google Scholar]
- 23.Gottar M, Gobert V, Matskevich AA, Reichhart J-M, Wang C, Butt TM, Belvin M, Hoffmann JA, Ferrandon D. Dual detection of fungal infections in Drosophila via recognition of glucans and sensing of virulence factors. Cell. 2006;127:1425–1437. doi: 10.1016/j.cell.2006.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takehana A, Yano T, Mita S, Kotani A, Oshima Y, Kurata S. Peptidoglycan recognition protein (PGRP)-LE and PGRP-LC act synergistically in Drosophila immunity. EMBO J. 2004;23:4690–4700. doi: 10.1038/sj.emboj.7600466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Takehana A, Katsuyama T, Yano T, Oshima Y, Takada H, Aigaki T, Kurata S. Overexpression of a pattern-recognition receptor, peptidoglycanrecognition protein-LE, activates imd/relish-mediated antibacterial defense and the prophenoloxidase cascade in Drosophila larvae. Proc. Natl Acad. Sci. USA. 2002;99:13705–13710. doi: 10.1073/pnas.212301199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaneko T, Yano T, Aggarwal K, Lim J-H, Ueda K, Oshima Y, Peach C, Erturk-Hasdemir D, Goldman WE, Oh B-H, Kurata S, Silverman N. PGRP-LC and PGRP-LE have essential yet distinct functions in the drosophila immune response to monomeric DAP-type peptidoglycan. Nat. Immunol. 2006;7:715–723. doi: 10.1038/ni1356. [DOI] [PubMed] [Google Scholar]
- 27.Yano T, Mita S, Ohmori H, Oshima Y, Fujimoto Y, Ueda R, Takada H, Goldman WE, Fukase K, Silverman N, Yoshimori T, Kurata S. Autophagic control of Listeria though intracellular innate immune recognition in drosophila. Nat. Immunol. 2008;9:908–916. doi: 10.1038/ni.1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goto A, Yano T, Terashima J, Iwashita S, Oshima Y, Kurata S. Cooperative Regulation of the Induction of the Novel Antibacterial Listericin by Peptidoglycan Recognition Protein LE and the JAK-STST Pathway. J. Biol. Chem. 2010;285:15731–15738. doi: 10.1074/jbc.M109.082115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakagawa I, Amano A, Mizushima N, Yamamoto A, Yamaguxhi H, Kamimoto T, Nara A, Funao J, Nakata M, Tsuda K, Hamada S, Yoshimori T. Autophagy defends cells against invading Group A Streptococcus. Science. 2004;306:1037–1040. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 30.Yang Z, Klionsky D. Eaten alive: a history of macroautophagy. Nat. Cell Biol. 2010;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakatogawa H, Suzuki K, Kamada Y, Ohsumi Y. Dynamics and diversity in autophagy mechanisms: lessons from yeast. Nat. Rev. Mol. Cell Biol. 2009;10:458–467. doi: 10.1038/nrm2708. [DOI] [PubMed] [Google Scholar]
- 32.Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010;12:823–830. doi: 10.1038/ncb0910-823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang Y-Y, Neufeld TP. Autophagy takes flight in Drosophila. FEBS Lett. 2010;584:1342–1349. doi: 10.1016/j.febslet.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 2007;7:767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Travasso LH, Carneiro LM, Ramjeet M, Hussey S, Kim Y-G, Magalhães J, Yuan L, Soares F, Chea E, Bourhis LL, Boneca IG, Allaoui A, Jones NL, Nuñez G, Girardin SE, Philpott DJ. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 2009;11:55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 36.Xu Y, Jagannath C, Liu X-D, Sharafkhaneh A, Kolodziejska KE, Eissa T. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity. 2007;27:135–144. doi: 10.1016/j.immuni.2007.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Delgado MA, Elmaoued RA, Davis AS, Kyei G, Deretic V. Toll-like receptors control autophagy. EMBO J. 2008;27:1110–1121. doi: 10.1038/emboj.2008.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deretic V, Levine B. Autophagy, Immunity, and Microbial Adaptations. Cell Host Micro. 2009;5:527–549. doi: 10.1016/j.chom.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shelly S, Lukinova N, Bambina S, Berman A, Cherry S. Autophagy is an essential component of drosophila immunity against vesicular stomatitis virus. Immunity. 2009;30:588–598. doi: 10.1016/j.immuni.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Orvrdahl A, MacPherson S, Sumpter R, Tallóczy Z, Zou Z, Levine B. autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Micro. 2010;7:115–127. doi: 10.1016/j.chom.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhao Z, Fux B, Goodwin M, Dunay IR, Strong D, Miller BC, Cadwell K, Delgado MA, Ponpuak M, Green KG, Schmidt RE, Mizushima N, Deretic V, Sibley LD, Virgin HW. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Micro. 2008;4:458–469. doi: 10.1016/j.chom.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Virgin HW, Levine B. Autophagy genes in immunity. Nat. Immunol. 2009;10:461–470. doi: 10.1038/ni.1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yano T, Kurata S. An unexpected twist for autophagy in Crohn’s disease. Nat. Immunol. 2009;10:134–135. doi: 10.1038/ni0209-134. [DOI] [PubMed] [Google Scholar]
- 44.Cadwell K, Liu JY, Broun SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, Stone CD, Brunt EM, Xavier RJ, Sleckman BP, Li E, Mizushima N, Stappenbeck T, Virgin HW. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–263. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saitoh T, Fujita N, Jang MH, Uematsu S, Yang BG, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T, Akira S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature. 2008;456:264–269. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 46.Yoshikawa Y, Ogawa M, Hain T, Yoshida M, Fukumatsu M, Kim M, Mimuro H, Nakagawa I, Yanagawa T, Ishii T, Kakizuka A, Sztul E, Chakraborty T, Sasakawa C. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat. Cell Biol. 2009;11:1233–1240. doi: 10.1038/ncb1967. [DOI] [PubMed] [Google Scholar]
- 47.Thurston TL, Ryzhakov G, Bloor S, von Muhlinen N, Randow F. The TBK1 adaptor and autophagy receptor NDP52 restricts the proliferation of ubiquitin-coated bacteria. Nat. Immunol. 2009;10:1215–1221. doi: 10.1038/ni.1800. [DOI] [PubMed] [Google Scholar]
- 48.Amano A, Nakagawa I, Yoshimori T. Autophagy in Innate Immunity against Intracellular Bacteria. J. Biochem. 2006;140:161–166. doi: 10.1093/jb/mvj162. [DOI] [PubMed] [Google Scholar]
- 49.Py B, Lipinsky MM, Yuan J. Autophagy Limits Listeria monocytogenes Intracellular Growth in the Early Phase of Primary Infection. Autophagy. 2007;3:117–125. doi: 10.4161/auto.3618. [DOI] [PubMed] [Google Scholar]
- 50.Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. Escape of Intracellular Shigella from Autophagy. Science. 2005;307:727–731. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- 51.Brimingham CL, Smith AC, Bakowski MA, Yoshimori T, Brumell JH. Autophagy Controls Salmonella Infection in Response to Damage to the Salmonella-containing Vacuole. J. Biol. Chem. 2006;281:11374–11383. doi: 10.1074/jbc.M509157200. [DOI] [PubMed] [Google Scholar]
- 52.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy Is a Defense Mechanism Inhibiting BCG and Mycobacterium tuberculosis Survival in Infected Macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 53.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular Mycobacteria. Science. 2006;313:1438–1441. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 54.Ling YM, Shaw MH, Ayala C, Coppens I, Taylor GA, Ferguson DJP, Yap GS. Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J. Exp. Med. 2006;203:2063–2071. doi: 10.1084/jem.20061318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Y, Weiss LM, Orlofsky A. Host cell autophagy is induced by Toxoplasma gondii and contributes to parasite growth. J. Biol. Chem. 2009;284:1694–1701. doi: 10.1074/jbc.M807890200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liang XH, Kleeman LK, Jiang HH, Gordon G, Goldman JE, Berry G, Herman B, Levine B. Protection against fatal Sindbis virus encephalitis by Beclin, a novel Bcl-2-interacting protein. J. Virol. 1998;72:8586–8596. doi: 10.1128/jvi.72.11.8586-8596.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu YL, Schiff M, Czymmek K, Tallóczy Z, Levine B, Dinesh-Kumar SP. Autophagy regulates programmed cell death during the plant innate immune response. Cell. 2005;121:567–577. doi: 10.1016/j.cell.2005.03.007. [DOI] [PubMed] [Google Scholar]