

SUMMARY
Several families have been reported with autosomal dominant frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS), genetically linked to chromosome 9p21. Here we report an expansion of a non-coding GGGGCC hexanucleotide repeat in the gene C9ORF72 that is strongly associated with disease in a large FTD/ALS kindred, previously reported to be conclusively linked to chromosome 9p. This same repeat expansion was identified in the majority of our families with a combined FTD/ALS phenotype and TDP-43 based pathology. Analysis of extended clinical series found the C9ORF72 repeat expansion to be the most common genetic abnormality in both familial FTD (11.7%) and familial ALS (22.5%). The repeat expansion leads to the loss of one alternatively spliced C9ORF72 transcript and to formation of nuclear RNA foci, suggesting multiple disease mechanisms. Our findings indicate that repeat expansion in C9ORF72 is a major cause of both FTD and ALS.
INTRODUCTION
Frontotemporal dementia (FTD) and amyotrophic lateral sclerosis (ALS) are both devastating neurological diseases. FTD is the second most common cause of pre-senile dementia in which degeneration of the frontal and temporal lobes of the brain results in progressive changes in personality, behavior, and language with relative preservation of perception and memory (Graff-Radford and Woodruff, 2007). ALS affects 2 in 100,000 people and has traditionally been considered a disorder in which degeneration of upper and lower motor neurons gives rise to progressive spasticity, muscle wasting, and weakness. However, ALS is increasingly recognized to be a multisystem disorder with impairment of frontotemporal functions such as cognition and behavior in up to 50% of patients (Giordana et al., 2011; Lomen-Hoerth et al., 2003; Phukan et al., 2007). Similarly, as many as half of FTD patients develop clinical symptoms of motor neuron dysfunction (Lomen-Hoerth et al., 2002). The concept that FTD and ALS represent a clinicopathological spectrum of disease is strongly supported by the recent discovery of the transactive response DNA binding protein with Mr 43 kD (TDP-43) as the pathological protein in the vast majority of ALS cases and in the most common pathological subtype of FTD (Neumann et al., 2006) (now referred to as frontotemporal lobar degeneration with TDP-43 pathology, FTLD-TDP) (Mackenzie et al., 2009).
A positive family history is observed in ~10% of ALS patients (Gros-Louis et al., 2006), while up to 50% of FTD patients report family members with FTD or related cognitive and behavioral changes (Graff-Radford and Woodruff, 2007), supporting the important contribution of genetic factors to these diseases. The most common currently known cause of familial FTLD-TDP involves loss-of-function mutations in the gene for the secreted growth factor progranulin (GRN) (Baker et al., 2006; Cruts et al., 2006). Although GRN deficiency has been directly linked to TDP-43 dysfunction and aggregation in a neuronal culture model of disease and in GRN knock-out mice, the exact relationship between GRN insufficiency and TDP-43 dysfunction remains unknown (Ahmed et al., 2010; Guo et al., 2010; Yin et al., 2010). In familial ALS, ~15-20% of patients are found to have mutations in the Cu/Zn superoxide dismutase gene (SOD1) (Rosen et al., 1993). Treatments shown to be effective in SOD1 mouse models, however, have generally not been effective in ALS clinical trials, and the absence of TDP-43 pathology in cases with SOD1 mutations suggests that motor neuron degeneration in these cases may result from a different mechanism (Mackenzie et al., 2007). For these reasons, the recent identification of mutations in TDP-43 (encoded by TARDBP) (Kabashi et al., 2008; Sreedharan et al., 2008) and the related RNA-binding protein fused in sarcoma (FUS) (Kwiatkowski et al., 2009; Vance et al., 2009) in ~5% of familial ALS patients has significantly shifted the focus of ALS research and implicated abnormal RNA processing as a critical process in ALS pathogenesis (Lagier-Tourenne et al., 2010).
Further support for the concept that FTD and ALS are closely related conditions is the recognition that both clinical syndromes may occur within the same family, often with an autosomal dominant pattern of inheritance. This familial association is not well explained by the currently recognized genetic defects; GRN mutations are not associated with significant motor neuron deficits, while patients carrying mutations in SOD1, TARDBP or FUS are rarely affected by FTD. Linkage analysis in several autosomal dominant families in which affected members develop either ALS or FTD or both, and where the pathology is consistently TDP-positive, have suggested a major locus for FTD/ALS on chromosome 9p21. Combined data defined a minimum linkage region of 3.7Mb, containing only five known genes (Boxer et al., 2011; Gijselinck et al., 2010; Le Ber et al., 2009; Luty et al., 2008; Morita et al., 2006; Pearson et al., 2011; Valdmanis et al., 2007; Vance et al., 2006). Importantly, the same chromosomal region has been identified in several large independent genome-wide association studies (GWAS) of both ALS and FTD, implicating the genetic defect at chromosome 9p in sporadic forms of both diseases (Laaksovirta et al., 2010; Shatunov et al., 2010; Van Deerlin et al., 2010; van Es et al., 2009). Furthermore, the associated ‘risk’ haplotype has been the same in all ALS and FTD populations studied and has also recently been shown to be present in all affected members of several 9p-linked FTD/ALS families (Mok et al., 2011).
Our collaborative group from the University of British Columbia (UBC), the University of California, San Francisco (UCSF) and the Mayo Clinic Rochester (MCR) previously reported a large autosomal dominant FTD/ALS kindred named VSM-20 for ‘Vancouver, San Francisco and Mayo family 20’, with conclusive linkage to chromosome 9p (maximum two-point LOD-score, 3.01) (Boxer et al., 2011). Post mortem evaluation of three affected members showed a combination of FTLD-TDP and ALS with TDP-immunoreactive pathology (Figure 1). Previous extensive sequencing of all exons and exon-intron boundaries of the genes within the candidate region did not identify the disease causing mutation in this family. Here we provide evidence that disease in family VSM-20 is caused by an expanded hexanucleotide repeat in a non-coding region of chromosome 9 open reading frame 72 (C9ORF72) and that this repeat expansion is the most common cause of familial FTD and ALS identified to date.
Figure 1. Neuropathology in familial FTD/ALS linked to chromosome 9p (family VSM-20).

(A, B) FTLD-TDP characterized by TDP-43 immunoreactive neuronal cytoplasmic inclusions and neurites in (A) neocortex and (B) hippocampal dentate granule cell layer. (C) TDP-34 immunoreactive neuronal cytoplasmic inclusions in spinal cord lower motor neurons, typical of ALS. (D) Numerous neuronal cytoplasmic inclusions and neurites in cerebellar granular layer immunoreactive for ubiquitin but not TDP-43. Scale bar: (A) 15 μm, (B) 30 μm, (C) 100 μm, (D) 12 μm.
RESULTS
Expanded GGGGCC hexanucleotide repeat in C9ORF72 is the cause of chromosome 9p21-linked FTD/ALS in family VSM-20
In the process of sequencing the non-coding region of C9ORF72, we detected a polymorphic GGGGCC hexanucleotide repeat (g.26724GGGGCC(3_23) in the reverse complement of AL451123.12 starting at nt 1), located between non-coding C9ORF72 exons 1a and 1b. Fluorescent fragment-length analysis of this region in samples from members of family VSM-20 resulted in an aberrant segregation pattern. All affected individuals appeared homozygous in this assay, and affected children appeared not to inherit an allele from the affected parent (Figure 2A-B). To determine whether the lack of segregation was the result of single allele amplification due to the presence of an unamplifiable repeat expansion, we used a repeat-primed PCR method specifically designed to the observed GGGGCC hexanucleotide repeat. This method suggested the presence of repeat expansions in all affected members of family VSM-20, but not in unaffected relatives (Figure 2C). Subsequent analysis of 909 healthy controls by fluorescent fragment-length analysis identified 315 who were homozygous, however no repeat expansions were observed by repeat-primed PCR. The maximum size of the repeat in controls was 23 units. These findings suggested the presence of a unique repeat expansion in family VSM-20 and prompted us to perform Southern blot analysis on DNA from four different affected and one unaffected member of VSM-20. In addition to the expected normal allele, we detected a variably sized expanded allele, too large to be amplified by PCR, which was found only in the affected individuals (Figure 2D). In all but one patient, the expanded alleles appeared as single discrete bands; however, in patient 20-17 (Figure 2D, lane 5) two discrete high molecular weight bands were observed, suggesting somatic instability of the repeat. Based on this small number of patients we estimated the number of GGGGCC repeat units to range from approximately 700 to 1600.
Figure 2. Expanded GGGGCC hexanucleotide repeat in C9ORF72 causes FTD and ALS linked to chromosome 9p in family VSM-20.

(A) Segregation of GGGGCC repeat in C9ORF72 and flanking genetic markers in disguised linkage pedigree of family VSM-20. The arrowhead denotes the proband. For the GGGGCC repeat, numbers indicate hexanucleotide repeat units and the X denotes that the allele could not be detected. Black symbols represent patients affected with frontotemporal dementia (left side filled), amyotrophic lateral sclerosis (right side filled) or both. White symbols represent unaffected individuals or at-risk individuals with unknown phenotype. Haplotypes for individuals 20-1, 20-2 and 20-3 are inferred from genotype data of siblings and offspring. (B) Fluorescent fragment length analyses of a PCR fragment containing the GGGGCC repeat in C9ORF72. PCR products from the unaffected father (20-9), affected mother (2-10) and their offspring (20-16, 20-17 and 20-18) are shown illustrating the lack of transmission from the affected parent to affected offspring. Numbers under the peaks indicate number of GGGGCC hexanucleotide repeats. (C) PCR products of repeat-primed PCR reactions separated on an ABI3730 DNA Analyzer and visualized by GENEMAPPER software. Electropherograms are zoomed to 2000 relative fluorescence units to show stutter amplification. Two expanded repeat carriers (20-8 and 20-15) and one non-carrier (20-5) from family VSM-20 are shown. (D) Southern blotting of four expanded repeat carriers and one non-carrier from family member of VSM-20 using genomic DNA extracted from lymphoblast cell lines. Lane 1 shows DIG-labeled DNA Molecular Weight Marker II (Roche) with fragments of 2027, 2322, 4361, 6557, 9416, 23130 bp, lane 2 shows DIG-labeled DNA Molecular Weight Marker VII (Roche) with fragments of 1882, 1953, 2799, 3639, 4899, 6106, 7427, and 8576 bp. Patients with expanded repeats (lanes 3-6) show an additional allele from 6-12kb, while a normal relative (lane 7) only shows the expected 2.3kb wild-type allele.
Expanded GGGGCC hexanucleotide repeat in C9ORF72 is a frequent cause of disease in FTD and ALS patient populations
The proband of family VSM-20 (20-6) is part of a highly selected series of 26 probands ascertained at UBC, Vancouver, Canada, with a confirmed pathological diagnosis of FTLD-TDP and a positive family history of FTD and/or ALS. We previously identified GRN mutations in seven probands (26.9%) from this series, all from families with a clinically pure FTD phenotype; however, the genetic basis for the disease in the other families remained unknown. Using a combination of fluorescent fragment-length and repeat-primed PCR analyses, we now found that 16 of the 26 FTLD-TDP families in this series (61.5%) carry expanded alleles of the GGGGCC hexanucleotide repeat; nine with a combined FTD/ALS phenotype and seven with clinically pure FTD. In five of these families, DNA was available from multiple affected members and in all cases, the repeat expansion was found to segregate with disease (Figure 2 and Figure S1). These findings suggest that GGGGCC expansions in C9ORF72 are the most common cause of familial FTLD-TDP.
To further determine the frequency of GGGGCC hexanucleotide expansions in C9ORF72 in patients with FTLD-TDP pathology and to assess the importance of this genetic defect in the etiology of patients clinically diagnosed with FTD and ALS, we analyzed 696 patients (93 pathologically diagnosed FTLD-TDP, 374 clinical FTD, and 229 clinical ALS) derived from three well-characterized patient series ascertained at the Mayo Clinic Florida (MCF) and MCR (Table S1). This resulted in the identification of 59 additional unrelated patients carrying GGGGCC repeat expansions, including 22 patients without a known family history (Table 1, Figure S1). In a subset of these patients the sporadic nature of the disease could potentially be explained by the early death of one or both parents (3/22), adoption (1/22) or a lack of sufficient information (8/22); however, in 10 patients the clinical records suggested a true sporadic nature of the disease. The GGGGCC repeat was found in 18.3% of all patients with FTLD-TDP pathology from the MCF brain bank, and explained 22.5% of familial cases in this series. It should be noted however, that this is a dementia-focused series with an under-representation of ALS. The frequency in our clinical FTD patient series was 3.0% of sporadic cases and 11.7% of familial patients. In our clinical ALS series, 4.1% of the sporadic and 23.5% of patients with a positive family history carried repeat expansions. Importantly, a direct comparison of the frequency of repeat expansions in C9ORF72 with mutations in SOD1, TARDBP and FUS revealed GGGGCC expansions to be the most common genetic cause of sporadic and familial ALS in our clinical series (Table 1). In clinical FTD, GGGGCC repeat expansions were found to be more common than either GRN or microtubule associated protein tau (MAPT) mutations in familial cases, and of equal frequency to GRN mutations in sporadic FTD.
Table 1.
Frequency of chromosome 9p repeat expansion in FTLD and ALS
| Cohort | N | Number of mutation carriers (%) |
|||||
|---|---|---|---|---|---|---|---|
| c9FTD/ALS | GRN | MAPT | SOD1 | TARDBP | FUS | ||
| UBC FTLD-TDP | |||||||
| Familial | 26 | 16 (61.5) | 7 (26.9) | n/a | n/a | n/a | n/a |
| MCF FTLD-TDP | |||||||
| Familial | 40 | 9 (22.5) | 6 (15.0) | n/a | n/a | n/a | n/a |
| Sporadic a | 53 | 8 (15.1) | 8 (15.1) | n/a | n/a | n/a | n/a |
| MC Clinical FTD | |||||||
| Familial | 171 | 20 (11.7) | 13 (7.6) | 12 (6.3) | n/a | n/a | n/a |
| Sporadic | 203 | 6 (3.0) | 6 (3.0) | 3 (1.5) | n/a | n/a | n/a |
| MCF Clinical ALS | |||||||
| Familial | 34 | 8 (23.5) | n/a | n/a | 4 (11.8) | 1 (2.9) | 1 (2.9) |
| Sporadic | 195 | 8 (4.1) | n/a | n/a | 0 (0.0) | 2 (1.0) | 3 (1.5) |
Includes 22 individuals for which no information on family history was available.
ALS=Amyotrophic lateral sclerosis; c9FTD/ALS= (GGGGCC)n repeat expansion at chromosome 9p identified in this study; FTD = frontotemporal dementia; FTLD-TDP=Frontotemporal lobar degeneration with TDP-43 pathology; FUS=fused in sarcoma gene; GRN=Progranulin gene; MAPT=Microtubule associated protein tau gene; MC=Mayo Clinic; MCF=Mayo Clinic Florida; n/a = not assessed; SOD1=superoxide dismutase 1 gene; TARDBP=TAR DNA-binding protein 43 gene UBC=University of British Columbia.
Clinical and pathological characteristics of expanded GGGGCC repeat carriers
Clinical data was obtained for the 26 unrelated expanded repeat carriers from the clinical FTD series and the 16 unrelated carriers from the ALS series. The median age of onset was comparable in the two series (FTD: 56.2 years, range 34-72 years; ALS: 54.5 years, range 41-72 years), with a slightly shorter mean disease duration in the ALS patients (FTD: 5.1 ± 3.1 years, range 1-12 years, N=18; ALS: 3.6 ± 1.6 years, range 1-6 years, N=7). The FTD phenotype was predominantly behavioral variant FTD (bvFTD) (25/26). Importantly, seven patients from the FTD series (26.9%) had concomitant ALS and eight patients (30.7%) had relatives affected with ALS. In comparison, the frequency of a family history of ALS in the remainder of our FTD population (those without repeat expansions) was only 5/348 (1.4%). In the ALS series, all mutation carriers presented with classical ALS with the exception of one patient diagnosed with progressive muscular atrophy without upper motor neuron signs. Three patients (18.8%) were diagnosed with a combined ALS/FTD phenotype. In the ALS patients with expanded repeats, 11/16 (68.8%) reported relatives with FTD or dementia, compared to only 61/213 (28.6%) of ALS patients without repeat expansions. Finally, autopsy was subsequently performed on 11 FTD and three ALS expanded repeat carriers from the clinical series, and in all cases, TDP-43 based pathology was confirmed.
Comparison of haplotypes carrying expanded GGGGCC repeats with previously reported chromosome 9p ‘risk’ haplotype
We previously described a ~140kb risk haplotype on chromosome 9p21, that was shared by four chromosome 9p-linked families and showed significant association with FTD and ALS in at least eight populations (Mok et al., 2011). To determine whether all GGGGCC expanded repeat carriers identified in this study also carried this ‘risk’ haplotype, and to further study the significance of this finding, we selected the variant rs3849942 as a surrogate marker for the ‘risk’ haplotype for genotyping in our patient and control populations. All 75 unrelated expanded repeat carriers had at least one copy of the ‘risk’ haplotype (100%) compared to only 23.1% of our control population. In order to associate the repeat sizes with the presence or absence of the ‘risk’ haplotype, we further focused on controls homozygous for rs3849942 (505 GG and 49 AA) and determined the distribution of the repeat sizes in both groups (Figure 3). We found a striking difference in the number of GGGGCC repeats, with significantly longer repeats on the ‘risk’ haplotype tagged by allele ‘A’ compared to the wild-type haplotype tagged by allele ‘G’ (median repeat length: risk haplotype = 8, wild-type haplotype = 2; average repeat length: risk haplotype = 9.5, wild-type haplotype = 3.0; p<0.0001). Sequencing analysis of 48 controls in which the repeat length was the same on both alleles (range = 2-13 repeat units) further showed that the GGGGCC repeat was uninterrupted in all individuals.
Figure 3. Correlation of GGGGCC hexanucleotide repeat length with rs3849942, a surrogate marker for the previously published chromosome 9p ‘risk’ haplotype.
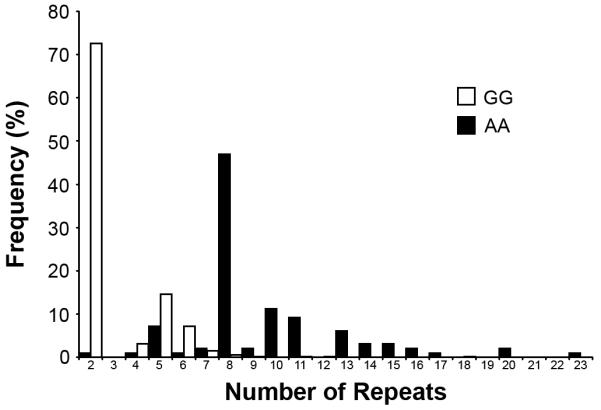
Histograms of number of GGGGCC repeats in 505 controls homozygous for the rs3849942 G-allele and 49 controls homozygous for the rs3849942 A-allele.
Expanded GGGGCC repeat affects C9ORF72 expression in a transcript-specific manner
One potential mechanism by which expansion of a non-coding repeat region might lead to disease is by interfering with normal expression of the encoded protein. Through a complex process of alternative splicing, three C9ORF72 transcripts are produced which are predicted to lead to the expression of two alternative isoforms of the uncharacterized protein C9ORF72 (Figure 4A). Transcript variants 1 and 3 are predicted to encode for a 481 amino acid long protein encoded by C9ORF72 exons 2-11 (NP_060795.1; isoform a), whereas variant 2 is predicted to encode a shorter 222 amino acid protein encoded by exons 2-5 (NP_659442.2; isoform b) (Figure 4A). RT-PCR analysis showed that all C9ORF72 transcripts were present in a variety of tissues, and immunohistochemical analysis in brain further showed that C9ORF72 was largely a cytoplasmic protein in neurons (Figure S2).
Figure 4. Effect of expanded hexanucleotide repeat on C9ORF72 expression.

(A) Overview of the genomic structure of the C9ORF72 locus (top panel) and the C9ORF72 transcripts produced by alternative pre-mRNA splicing (bottom panels). Boxes represent coding (white) and non-coding (grey) exons and the positions of the start codon (ATG) and stop codon (TAA) are indicated. The GGGGCC repeat is indicated with a red diamond. The position of rs10757668 is indicated with a green star. (B) Sequence traces of C9ORF72 exon 2 spanning rs10757668 in gDNA (top panel) and cDNA (bottom panels) prepared from frontal cortex of an FTLD-TDP patient carrying an expanded GGGGCC repeat. The arrow indicates the presence of the wild-type (G) and mutant (A) alleles of rs10757668 in gDNA. Transcript specific cDNAs were amplified using primers spanning the exon 1b/exon 2 boundary (variant 1) or exon 1a/exon 2 boundary (variant 2 and 3). Sequenced traces derived from cDNA transcripts indicate the loss of variant 1 but not variant 2 or 3 mutant RNA. Similar results were obtained for two unrelated FTLD-TDP mutation carriers. The bottom panel shows a non-expanded repeat carrier heterozygous for rs10757668 to confirm the presence of both alleles of transcript variant 1 validating the method. (C) mRNA expression analysis of C9ORF72 transcript variant 1 using a custom-designed Taqman expression assay. Top panel shows lymphoblast cell lines derived from expanded repeat carriers from family VSM-20 (n=7) and controls (n=7) and bottom panel shows RNA extracted from frontal cortex brain samples from FTLD-TDP patients with (n=7) and without (n=7) the GGGGCC repeat expansion. Data indicate mean ± s.e.m. ** indicates P<0.01. (D) mRNA expression analysis of all C9ORF72 transcripts encoding for C9ORF72 isoform a (variant 1 and 3) using inventoried ABI Taqman expression assay Hs_00945132. Top panel shows RNA extracted from lymphoblast cell lines derived from expanded repeat carriers from family VSM-20 (n=7) and controls (n=7) and bottom panel shows RNA extracted from frontal cortex brain samples from FTLD-TDP patients with (n=7) and without (n=7) the GGGGCC repeat expansion. Data indicate mean ± s.e.m. * indicates P<0.05.
The GGGGCC hexanucleotide repeat is located between two alternatively-spliced non-coding first exons, and depending on their use, the expanded repeat is either located in the promoter region (for transcript variant 1) or in intron 1 (for transcript variants 2 and 3) of C9ORF72 (Figure 4A). This complexity raises the possibility that the expanded repeat affects C9ORF72 expression in a transcript-specific manner. To address this issue, we first determined whether each of the three C9ORF72 transcripts, carrying the expanded repeat, produce mRNA expression in brain. For this, we selected two GGGGCC repeat carriers for which frozen frontal cortex brain tissue was available and who were heterozygous for the rare sequence variant rs10757668 in C9ORF72 exon 2. Comparison of sequence traces of C9ORF72 exon 2 in gDNA and transcript-specific cDNAs amplified from these patients showed the absence of variant 1 transcribed from the mutant RNA (G-allele) but normal transcription of variant 2 and 3 (Figure 4B). The loss of variant 1 expression in the GGGGCC repeat carriers was further confirmed by real-time RT-PCR using a custom-designed Taqman assay specific to variant 1. In lymphoblast cell lines of patients from family VSM-20 and in frontal cortex samples from unrelated FTLD-TDP patients carrying expanded repeats, the level of C9ORF72 variant 1 was approximately 50% reduced compared to non-repeat carriers (Figure 4C). Since C9ORF72 variants 1 and 3, which each contain a different non-coding first exon, both encode C9ORF72 isoform a (NP_060795.1), we next determined the effect of the expanded repeats on the total levels of transcripts encoding this isoform (variants 1 and 3 combined) using an inventoried ABI Taqman assay (Hs_00945132). Significant mRNA reductions were observed in both lymphoblast cells (34% reduction) and frontal cortex samples (38% reduction) from expanded repeat carriers (Figure 4D). In contrast, no appreciable changes in total levels of C9ORF72 protein could be observed by western blot analysis of lymphoblast cell lysates or brain (Figure S2) or by immunohistochemical analysis of C9ORF72 in post-mortem brain or spinal cord tissue from expanded repeat carriers (Figure S2). These protein expression data should however be considered preliminary, since they are based on a limited number of samples using relatively uncharacterized commercially obtained C9ORF72 antibodies without detailed quantitative analyses.
The transcribed GGGGCC repeat forms nuclear RNA foci in affected central nervous system regions of mutation carriers
In recent years, intracellular accumulation of expanded nucleotide repeats as RNA foci in the nucleus and/or cytoplasm of affected cells has emerged as an important disease mechanism for the growing class non-coding repeat expansion disorders (Todd and Paulson, 2010). To determine whether GGGGCC repeat expansions in C9ORF72 result in the formation of RNA foci, we performed RNA fluorescence in situ hybridization (FISH) in paraffin-embedded sections of post-mortem frontal cortex and spinal cord tissue from FTLD-TDP patients. For each neuroanatomical region, sections from two patients with expanded GGGGCC repeats and two affected patients with normal repeat lengths were analyzed. Using a probe targeting the GGGGCC repeat [probe (GGCCCC)4], multiple RNA foci were detected in the nuclei of 25% of cells in both the frontal cortex and the spinal cord from patients carrying the expansion, whereas a signal was observed in only 1% of cells in tissue sections from non-carriers (Figure 5A-C). Foci were never observed in any of the samples using a probe targeting the unrelated CCTG repeat [probe (CAGG)6], implicated in myotonic dystrophy type 2 (DM2) (Liquori et al., 2001), further supporting the specificity of the RNA foci composed of GGGGCC in these patients (Figure 5D).
Figure 5. Expanded GGGGCC hexanucleotide repeat forms nuclear RNA foci in human brain and spinal cord.

(A) Multiple RNA foci in the nucleus (stained with DAPI, blue) of a frontal cortex neuron of the proband of family 63 (63-1) using a Cy3-labeled (GGCCCC)4 oligonucleotide probe (red). (B) RNA foci observed in the nucleus of two lower motor neurons in FTD/ALS patient (13-7) carrying an expanded GGGGCC repeat using a Cy3-labeled (GGCCCC)4 oligonucleotide probe. (C) Absence of RNA foci in the nucleus of cortical neuron from FTLD-TDP patient (44-1) without an expanded GGGGCC repeat in C9ORF72. (D) spinal cord tissue sections from patient 13-7 that showed RNA foci with the (GGCCCC)4 oligonucleotide probe in (B) do not show any foci with a Cy3-labeled (CAGG)6 oligonucleotide probe (negative control probe). Scale bar: 10 μm (A and C), 20 μm (B and D).
DISCUSSION
The identification of an expanded non-coding GGGGCC repeat in C9ORF72 resolves an important question in the FTD and ALS fields, namely the genetic basis of FTD/ALS linked to chromosome 9p21. This finding adds FTD/ALS to the growing class of non-coding repeat expansion disorders, which includes the myotonic dystrophies (DM1 and DM2) (Brook et al., 1992; Liquori et al., 2001; Mahadevan et al., 1992), fragile-X associated tremor/ataxia syndrome (FXTAS) (Galloway and Nelson, 2009; Tassone et al., 2004), and several spinocerebellar ataxias (SCA8, SCA10, SCA31, SCA36) (Daughters et al., 2009; Kobayashi et al., 2011; Moseley et al., 2006; Sato et al., 2009).
We identified a total of 75 unrelated expanded GGGGCC repeat carriers in the 722 patients included in this study (10.4%). Patients presented with FTD, ALS or a combination of both. The highest frequency of C9ORF72 repeat expansions was observed in a selected series of pathologically confirmed FTLD-TDP probands with a strong family history of FTD and/or ALS ascertained at UBC (61.6%). A second pathologically confirmed FTLD-TDP series from the MCF brain bank showed a lower frequency of repeat expansion in familial cases (22.5%); the difference most likely reflecting the much smaller number of ALS patients and the fact that in most of the families, the proband had only a single relative with dementia of unspecified type. Expanded GGGGCC repeats in C9ORF72 also accounted for 11.7% of familial FTD and 23.5% of familial ALS patients from our sequential series of clinical patients ascertained at Mayo Clinic. A direct comparison with mutation frequencies of the previously identified common genes for FTD and ALS in our series showed that C9ORF72 repeat expansions are the most common cause of familial forms of FTD and ALS identified to date. C9ORF72 also explained the disease in a significant proportion of sporadic FTD and ALS patients and was the most common genetic cause of sporadic ALS in our series (4%). Therefore, the GGGGCC repeat expansion is the first genetic abnormality identified to be a common cause of both FTD and ALS phenotypes, is expected to be present in the majority of FTD/ALS families, and likely accounts for most of the risk associated with the recently reported FTLD-TDP and ALS GWAS hits in this region.
The expanded GGGGCC repeat is located in the non-coding region of C9ORF72, a gene that encodes an uncharacterized protein with no known domains or function, but which is highly conserved across species. We show that in normal individuals at least three alternatively spliced C9ORF72 transcripts (variants 1-3) are expressed in most tissues including brain. Immunohistochemical analysis confirmed C9ORF72 expression in neurons of neuroanatomical regions affected in FTD and ALS with the staining pattern being consistent with predominantly cytoplasmic and synaptic localization. Quantitative mRNA expression analysis indicated that the GGGGCC repeat expansion abolished C9ORF72 transcript variant 1 expression from the mutant allele, leading to a significant overall reduction in C9ORF72 transcripts encoding C9ORF72 isoform a. Depending on the relative expression of the various transcripts, the loss of C9ORF72 transcript 1 may have a significant impact on selective tissues or cell types. Although preliminary analyses of C9ORF72 protein levels in cultured cells and whole brain tissue homogenate did not show an obvious change in the steady-state levels, we cannot exclude the possibility that reduced transcript levels of C9ORF72 affect protein translation under conditions of stress or may affect protein turnover and/or function. We also cannot guarantee the specificity of the commercial C9ORF72 antibodies used in this study since careful characterization of these antibodies has not yet been performed. In future experiments it will be crucial to generate more specific C9ORF72 antibodies and develop more quantitative approaches to measure C9ORF72 levels to further clarify the expression and localization of each of the C9ORF72 isoforms in different tissues and at various stages of disease progression. Although speculative at this time, it is possible that the expression pattern of C9ORF72 in individual patients may contribute to the variability in disease phenotype (FTD versus ALS) or course.
A common feature of non-coding repeat expansion disorders which has gained increased attention in recent years is the accumulation of RNA fragments composed of the repeated nucleotides as RNA foci in the nucleus and/or cytoplasm of affected cells (Todd and Paulson, 2010). In several disorders, the RNA foci have been shown to sequester RNA-binding proteins, leading to dysregulation of alternative mRNA splicing (Miller et al., 2000; Sofola et al., 2007; Timchenko et al., 1996; White et al., 2010). Using an oligonucleotide probe specific for the GGGGCC repeat we confirmed the presence of such nuclear RNA foci in post-mortem cerebral cortex and spinal cord tissue of C9ORF72 expanded repeat carriers. The GGGGCC sequence motif predicts the potential binding of several RNA-binding proteins, including the serine/arginine-rich splicing factor 1 (SRSF1) and the heterozygous nuclear ribonucleoprotein (hnRNP) A2/B1 (Cartegni et al., 2003; Smith et al., 2006; Sofola et al., 2007). Although future studies are needed to clarify whether these or other RNA-binding proteins play any role in disease pathogenesis, aberrant RNA splicing is a highly plausible mechanism in chromosome 9p-linked FTD/ALS given the accumulating evidence for RNA mis-processing in the pathogenesis of both ALS and FTD (Baumer et al., 2010). Dysregulation of hnRNP A2/B1 is a particularly interesting possibility since this protein is known to interact with the C/G-rich repeats that form RNA foci in another neurodegenerative condition (FXTAS) and because hnRNP A2/B1 has been shown to interact directly with TDP-43 (Buratti et al., 2005; Sofola et al., 2007). Identifying the aberrantly spliced RNA targets that are critical in disease may be the key to future therapeutic strategies.
The GGGGCC repeat length in healthy individuals ranged from 2-23 hexanucleotide units, whereas we estimated the repeat length to be 700-1600 units in FTD/ALS patients based on DNA from lymphoblast cell lines. Accurate sizing of the repeat is challenging, especially in DNA extracted from peripheral blood and brain tissue samples, where a smear of high molecular weight bands suggested somatic repeat instability (Figure S1). Notably, the large number of repeats observed in our patients is similar to other non-coding repeat expansion disorders where more than 1000 repeat copies are common (Liquori et al., 2001; Mahadevan et al., 1992; Moseley et al., 2006; Sato et al., 2009; Timchenko et al., 1996). However, the minimal repeat size needed to cause FTD/ALS remains to be determined and may be significantly smaller. Importantly, anticipation was not apparent in most of our families, although occasionally a significantly earlier onset was observed in the youngest generation. This could simply reflect heightened awareness by family members or caregivers; however, it remains possible that repeat length is correlated with the age of disease onset or clinical presentation. Future studies are needed to fully resolve this question.
In previous studies, we and others suggested that a single ~140kb ‘risk’ haplotype, broadly defined by SNP rs3849942 allele ‘A’, was shared by all affected family members of chromosome 9p-linked families and that this same haplotype was responsible for the ALS and FTLD-TDP GWAS hits at chromosome 9p (Mok et al., 2011). The presence of the ‘risk’ haplotype in all 75 unrelated expanded repeat carriers in our study further confirms the strong association of this haplotype with disease. While these findings are consistent with the previously proposed hypothesis of a single founder mutation, the identification of an expanded hexanucleotide repeat as the basis for disease in these patients now suggests the possibility that the abnormal repeat may occur on a predisposing haplotypic background that is prone to expansion. This alternative hypothesis is supported by our finding of significantly longer repeats on the ‘risk’ haplotype (defined by rs3849942 allele ‘A’) compared to the wild-type haplotype (defined as rs3849942 allele ‘G’) in the normal population. The somewhat unusual observation that the GGGGCC repeat was uninterrupted in control individuals carrying a range of normal allele sizes further supports this alternative hypothesis. De novo expansions of uninterrupted GGGGCC sequences at the long end of the normal spectrum could potentially explain the sporadic nature of the disease in a subset of our patients.
In summary, we identified a non-coding expanded GGGGCC hexanucleotide repeat in C9ORF72 as the cause of chromosome 9p-linked FTD/ALS and showed that this genetic defect is the most common cause of ALS and FTD identified to date. Our findings suggest multiple potential disease mechanisms associated with this repeat expansion, including a direct effect on C9ORF72 expression by affecting transcription (loss-of-function mechanism) and an RNA-mediated gain-of-function mechanism through the generation of toxic RNA foci. Future molecular studies are needed to explore how each mechanism contributes to neurodegeneration and pathological TDP-43 aggregation. Moreover, evaluation of larger numbers of patients with FTD and ALS associated with the expanded GGGGCC hexanucleotide repeat in C9ORF72 is warranted to further delineate the range of phenotypes and prevalence of these disorders, and to investigate the potential of the repeat for properties such as anticipation and spontaneous mutation. Finally, we suggest that in future publications this genetic defect be referred to as “c9FTD/ALS”.
While our manuscript was in preparation we learned of another group who independently identified repeat expansions in C9ORF72 as the cause of FTD and ALS linked to chromosome 9p (Renton et al. 2011).
EXPERIMENTAL PROCEDURES
Human samples
Four extensive FTD and ALS patient cohorts and one control cohort were included in this study. All individuals agreed to be in the study and biological samples were obtained after informed consent from subjects and/or their proxies. Demographic and clinical information for each cohort is summarized in Table S1. The proband of chromosome 9p-linked family VSM-20 is part of a series of 26 probands ascertained at UBC, Vancouver, Canada, characterized by a pathological diagnosis of FTLD with TDP-43 pathology (FTLD-TDP) and a positive family history of FTD and/or ALS (UBC FTLD-TDP cohort). Clinical and pathological evaluations of VSM-20 were conducted at UCSF, UBC and the Mayo Clinic (Boxer et al., 2011). A second cohort of 93 pathologically confirmed FTLD-TDP patients independent of family history was selected from the Mayo Clinic Florida (MCF) brain bank (MCF FTLD-TDP cohort) which focuses predominantly on dementia. The clinical FTD cohort (MC Clinical FTD cohort) represents a sequential series of patients seen by the Behavioral Neurology sections at MCF (n=197) and MCR (n=177), the majority of whom were participants in the Mayo Alzheimer’s Disease Research Center. Members of Family 118 were participants in the Mayo Alzheimer’s Disease Patient Registry. Clinical FTD patients underwent a full neurological evaluation and all who were testable had a neuropsychological evaluation. Structural neuroimaging was performed in all patients and functional imaging was performed in many patients. Patients with a clinical diagnosis of behavioral variant FTD (bvFTD), semantic dementia or progressive non-fluent aphasia based on Neary criteria (Neary et al., 1998) or patients with the combined phenotype of bvFTD and ALS were included in this study, while patients with a diagnosis of logopenic aphasia or corticobasal syndrome were excluded. In the MCF FTLD-TDP cohort and the MC Clinic FTD cohort, a positive family history was defined as a first or second degree relative with FTD and/or ALS or a first degree relative with memory problems, behavioral changes, parkinsonism, schizophrenia, or another suspected neurodegenerative disorder. It should be noted that information about family history was lacking in a significant proportion (23.7%) of the MCF FTLD-TDP cohort and these were included in the “sporadic” group. The MCF clinical ALS cohort represents a sequential series of 229 clinical ALS patients ascertained by the ALS Center at MCF. These patients underwent a full neurological evaluation including electromyography, clinical laboratory testing and imaging as appropriate to establish the clinical diagnosis of ALS. A positive family history in the MCF ALS series was defined as a first or second degree relative with ALS. The Control cohort (n=909) was comprised of DNA samples from 820 control individuals collected from the Department of Neurology and DNA extracted from 89 normal control brains from the MCF brain bank.
Characterization of hexanucleotide repeat insertion in C9ORF72 genomic region
The GGGGCC hexanucleotide repeat in C9ORF72 was PCR amplified in family VSM-20 and in all patient and control cohorts using the genotyping primers listed in Table S2 using one fluorescently labeled primer followed by fragment length analysis on an automated ABI3730 DNA-analyzer (Applied Biosystems). The PCR reaction was carried out in a mixture containing 1M betaine solution, 5% dimethylsulfoxide and 7-deaza-2-deoxy GTP in substitution for dGTP. Allele identification and scoring was performed using GeneMapper v4.0 software (Applied Biosystems). To determine the number of GGGGCC units and internal composition of the repeat, 48 individuals homozygous for different fragment lengths were sequenced using the PCR primers.
Repeat-primed PCR analysis
To provide a qualitative assessment of the presence of an expanded (GGGGCC)n hexanucleotide repeat in C9ORF72, we performed a repeat-primed PCR reaction in the presence of 1M betaine, 5% dimethyl sulfoxide and complete substitution of 7-deaza-2-deoxy GTP for dGTP using a previously optimized and described cycling program (Hantash et al., 2010). Primer sequences are provided in Table S2. PCR products were analyzed on an ABI3730 DNA Analyzer and visualized using GeneMapper software.
Probe labeling, agarose gel electrophoresis, southern transfer, hybridization and detection
A 241bp digoxigenin (DIG)-labeled probe was generated using primers listed in Table S2 from 10ng gDNA by PCR reaction using PCR DIG Probe Synthesis Kit Expand High fidelity mix enzyme and incorporating 0.35mM DIG-11-dUTP: 0.65mM dTTP (1:6) in the dNTP labeling mix as recommended in the DIG System User’s Guide (Roche Applied Science). A total of 2μl of PCR labeled probe per ml of hybridization solution was used as recommended in the DIG System User’s Guide. A total of 5-10μg of gDNA was digested with XbaI at 37 °C overnight and electrophoresed in 0.8% agarose gels in 1X TBE. DNA was transferred to positively charged nylon membrane (Roche Applied Science) by capillary blotting and crosslinked by UV irradiation. Following prehybridization in 20 ml DIG EasyHyb solution at 47°C for 3h, hybridization was carried out at 47°C overnight in a shaking water bath. The membranes were then washed two times in 2X standard sodium citrate (SSC), 0.1% sodium dodecyl sulfate (SDS) at room temperature for 5 min each and twice in 0.1x SSC, 0.1% SDS at 68°C for 15 min each. Detection of the hybridized probe DNA was carried out as described in the User’s Guide. CDP-star chemiluminescent substrate was used and signals were visualized on X-ray film after 5 to 15h.
SNP genotyping
SNP rs3844942 was genotyped using a custom-designed Taqman SNP genotyping assay on the 7900HT Fast Real Time PCR system. Primers are included in Table S2. Genotype calls were made using the SDS v2.2 software (Applied Biosystems, Foster City, CA).
C9ORF72 quantitative real-time PCR
Total RNA was extracted from lymphoblast cell lines and brain tissue samples with the RNAeasy Plus Mini Kit (Qiagen) and reverse transcribed to cDNA using Oligo dT primers and the SuperScript III Kit (Invitrogen). RNA integrity was checked on an Agilent 2100 Bioanalyzer. Following standard protocols, real-time PCR was performed with inventoried TaqMan gene expression assays for GAPDH (Hs00266705) and C9ORF72 (Hs00945132) and one custom-designed assay specific to the C9ORF72 variant 1 transcript (Table S3) (Applied Biosystems) and analyzed on an ABI Prism 7900 system (Applied Biosystems). All samples were run in triplicate. Relative Quantification was determined using the ΔΔCt method after normalization to GAPDH. For the custom designed C9ORF72 variant 1 Taqman assay, probe efficiency was determined by generation of a standard curve (slope:−3.31459, r2: 0.999145).
C9ORF72 gDNA and cDNA sequencing
To determine the genotype for rs10757668 in gDNA, C9ORF72 exon 2 was amplified using flanking primers c9orf72-2aF and c9orf72-2aR (Table S3). PCR products were purified using AMPure (Agencourt Biosciences) then sequenced in both directions with the same primers using the Big Dye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems). Sequencing reactions were purified using CleanSEQ (Agencourt Biosciences) and analyzed on an ABI3730 Genetic Analyzer (Applied Biosystems). Sequence data was analyzed with Sequencher 4.5 software (Gene Codes). For cDNA sequencing, total RNA was isolated from frontal cortex tissue using the RNAeasy Plus Mini Kit (Qiagen). Reverse transcription reactions were performed using SuperScript III Kit (Invitrogen). RT-PCR was performed using primers specific for each of the three C9ORF72 mRNA transcripts; V1: cDNA-V1-1F with cDNA-2F, V2: cDNA-V2-1F with cDNA-2F, V3: cDNA-V3-1F with cDNA-2F (Table S2). PCR products were sequenced as described, and sequence data from each of the three transcripts were visualized for the genotype status of rs10757668.
C9orf72 Westernblot analysis
Human-derived lymphoblast cells and frontal cortex tissue were homogenized in radioimmunoprecipitation assay (RIPA) buffer and protein content was measured by the BCA assay (Pierce). Twenty and fifty micrograms of protein were loaded for the lymphoblast and brain tissue lysates, respectively, and run on 10% SDS gels. Proteins were transferred onto Immobilon membranes (Invitrogen) and probed with antibodies against C9ORF72 (Santa Cruz 1:5000 for lymphoblast cell lines and GeneTex 1:2000 for frontal cortex brain samples). The epitopes used to raise these antibodies are amino acids 1-158 (GeneTex) and 165-215 (Santa Cruz), and the antibodies are therefore predicted to recognize C9ORF72 isoforms a and b. A GAPDH antibody (Meridian Life Sciences 1:500,000) was used as an internal control to verify equal protein loading between samples.
RNA-FISH
For in situ hybridization two 2′-O-methyl RNA 5′oligos labeled with Cy3 were ordered from IDT (Coralville, IA): (GGCCCC)4 predicted to hybridize to the expanded GGGGCC repeat identified in this study and (CAGG)6 predicted to hybridize only to CCTG repeats observed in DM2 and included in this experiment as a negative control. Slides were pre-treated following the in situ hybridization protocol from AbCam with minor modifications. Lyophilized probe was re-constituted to 100ng/μl in nuclease free water. Probe working solutions of 5ng/μl were used for paraffin specimens, and diluted in LSI/WCP Hybridization Buffer (Abbott Molecular). Following overnight hybridization, slides were washed 3 times in 1X PBS at 37°C for 5 min each. DAPI counterstain (VectaShield®) was applied to each specimen and coverslipped. For each patient, 100 cells were scored for the presence of nuclear RNA foci per tissue section.
Immunohistochemistry
Immunohistochemistry for C9ORF72 was performed on sections of post-mortem brain and spinal cord tissue from patients with FTLD-TDP pathology known to carry the GGGGCC repeat expansion (N=4), patients with FTLD-TDP without the repeat expansion (N=4), ALS without the repeat expansion (N=4), other molecular subtypes of FTLD (N=4), Alzheimer’s disease (N=2) and neurologically normal controls (N=4). Immunohistochemistry was performed on 3μm thick sections of formalin fixed, paraffin embedded post mortem brain and spinal cord tissue using the Ventana BenchMark® XT automated staining system (Ventana, Tuscon, AZ) with anti-C9ORF72 primary antibody (Sigma-Aldrich, anti-C9orf72, generated using amino acid 110-199 as epitope; 1:50 overnight incubation following microwave antigen retrieval) and developed with aminoethylcarbizole (AEC).
Supplementary Material
HIGHLIGHTS.
Non-coding repeat expansion in C9ORF72 causes FTD and ALS linked to chromosome 9p.
C9ORF72 repeat expansion forms nuclear RNA foci in brain and spinal cord.
Repeat expansion results in loss of one alternatively spliced C9ORF72 transcript.
Repeat expansion in C9ORF72 is a major cause of both FTD and ALS.
ACKNOWLEDGEMENTS
We are grateful to all patients, family members and caregivers who participated in this study. The expert technical assistance of Pamela Desaro, Amelia Johnston and Thomas Kryston in the collection of DNA and post mortem tissue in the Mayo Clinic Florida ALS Center and of Margaret Luk in performing immunohistochemistry at UBC is also acknowledged. We also thank Richard Crook, Jennifer Gass and Ashley Cannon for technical assistance with the genetic and expression analyses. This research was funded as part of the Mayo Clinic ADRC grant from the National Institute on Aging (P50 AG016574), and members of one family were participants in the Mayo Clinic Alzheimer’s Disease Patient Registry (ADPR) from the National Institute on Aging (U01 AG006786). Research was further funded by the ALS Association (RR), Mayo Foundation and MCF ALS Center donor funds (KB). RR is also funded by NIH grants R01 NS065782, R01 AG026251. Some TDP-43 analysis was funded by NIH grant R01 AG037491 (KAJ). ZKW is partially supported by the NIH/NINDS 1RC2NS070276, NS057567, P50NS072187, Mayo Clinic Florida (MCF) Research Committee CR program (MCF #90052030), Dystonia Medical Research Foundation, and the gift from Carl Edward Bolch, Jr., and Susan Bass Bolch (MCF #90052031/PAU #90052).The UBC studies were funded by the Canadian Institutes of Health Research (CIHR) Operating Grants #179009 and #74580 and by the Pacific Alzheimer’s Research Foundation (PARF) Center Grant C06-01. G-YRH is supported by a Clinical Genetics Investigatorship award from the CIHR. ALB is funded by R01AG038791, R01AG031278, the John Douglas French Foundation; the Hellman Family Foundation and the Tau Research Consortium. BLM is funded by P50AG023501, P01AG019724, the Larry Hillblom Foundation and the State of CA and P50 AG1657303 to BLM and WWS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
SUPPLEMENTAL INFORMATION Supplemental Information includes 2 figures and 3 tables.
REFERENCES
- Ahmed Z, Sheng H, Xu YF, Lin WL, Innes AE, Gass J, Yu X, Wuertzer CA, Hou H, Chiba S, et al. Accelerated lipofuscinosis and ubiquitination in granulin knockout mice suggest a role for progranulin in successful aging. Am J Pathol. 2010;177:311–324. doi: 10.2353/ajpath.2010.090915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- Baumer D, Ansorge O, Almeida M, Talbot K. The role of RNA processing in the pathogenesis of motor neuron degeneration. Expert Rev Mol Med. 2010;12:e21. doi: 10.1017/S1462399410001523. [DOI] [PubMed] [Google Scholar]
- Boxer AL, Mackenzie IR, Boeve BF, Baker M, Seeley WW, Crook R, Feldman H, Hsiung GY, Rutherford N, Laluz V, et al. Clinical, neuroimaging and neuropathological features of a new chromosome 9p-linked FTD-ALS family. J Neurol Neurosurg Psychiatry. 2011;82:196–203. doi: 10.1136/jnnp.2009.204081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, Hunter K, Stanton VP, Thirion JP, Hudson T, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;68:799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- Buratti E, Brindisi A, Giombi M, Tisminetzky S, Ayala YM, Baralle FE. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J Biol Chem. 2005;280:37572–37584. doi: 10.1074/jbc.M505557200. [DOI] [PubMed] [Google Scholar]
- Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–3571. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- Daughters RS, Tuttle DL, Gao W, Ikeda Y, Moseley ML, Ebner TJ, Swanson MS, Ranum LP. RNA gain-of-function in spinocerebellar ataxia type 8. PLoS Genet. 2009;5:e1000600. doi: 10.1371/journal.pgen.1000600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway JN, Nelson DL. Evidence for RNA-mediated toxicity in the fragile X-associated tremor/ataxia syndrome. Future Neurol. 2009;4:785. doi: 10.2217/fnl.09.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijselinck I, Engelborghs S, Maes G, Cuijt I, Peeters K, Mattheijssens M, Joris G, Cras P, Martin JJ, De Deyn PP, et al. Identification of 2 Loci at chromosomes 9 and 14 in a multiplex family with frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Arch Neurol. 2010;67:606–616. doi: 10.1001/archneurol.2010.82. [DOI] [PubMed] [Google Scholar]
- Giordana MT, Ferrero P, Grifoni S, Pellerino A, Naldi A, Montuschi A. Dementia and cognitive impairment in amyotrophic lateral sclerosis: a review. Neurol Sci. 2011;32:9–16. doi: 10.1007/s10072-010-0439-6. [DOI] [PubMed] [Google Scholar]
- Graff-Radford NR, Woodruff BK. Frontotemporal dementia. Semin Neurol. 2007;27:48–57. doi: 10.1055/s-2006-956755. [DOI] [PubMed] [Google Scholar]
- Gros-Louis F, Gaspar C, Rouleau GA. Genetics of familial and sporadic amyotrophic lateral sclerosis. Biochim Biophys Acta. 2006;1762:956–972. doi: 10.1016/j.bbadis.2006.01.004. [DOI] [PubMed] [Google Scholar]
- Guo A, Tapia L, Bamji SX, Cynader MS, Jia W. Progranulin deficiency leads to enhanced cell vulnerability and TDP-43 translocation in primary neuronal cultures. Brain Res. 2010;1366:1–8. doi: 10.1016/j.brainres.2010.09.099. [DOI] [PubMed] [Google Scholar]
- Hantash FM, Goos DG, Tsao D, Quan F, Buller-Burckle A, Peng M, Jarvis M, Sun W, Strom CM. Qualitative assessment of FMR1 (CGG)n triplet repeat status in normal, intermediate, premutation, full mutation, and mosaic carriers in both sexes: implications for fragile X syndrome carrier and newborn screening. Genet Med. 2010;12:162–173. doi: 10.1097/GIM.0b013e3181d0d40e. [DOI] [PubMed] [Google Scholar]
- Kabashi E, Valdmanis PN, Dion P, Spiegelman D, McConkey BJ, Vande Velde C, Bouchard JP, Lacomblez L, Pochigaeva K, Salachas F, et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat Genet. 2008;40:572–574. doi: 10.1038/ng.132. [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Abe K, Matsuura T, Ikeda Y, Hitomi T, Akechi Y, Habu T, Liu W, Okuda H, Koizumi A. Expansion of Intronic GGCCTG Hexanucleotide Repeat in NOP56 Causes SCA36, a Type of Spinocerebellar Ataxia Accompanied by Motor Neuron Involvement. Am J Hum Genet. 2011;89:121–130. doi: 10.1016/j.ajhg.2011.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski TJ, Jr., Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C, Davis A, Gilchrist J, Kasarskis EJ, Munsat T, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–1208. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- Laaksovirta H, Peuralinna T, Schymick JC, Scholz SW, Lai SL, Myllykangas L, Sulkava R, Jansson L, Hernandez DG, Gibbs JR, et al. Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: a genome-wide association study. Lancet Neurol. 2010;9:978–985. doi: 10.1016/S1474-4422(10)70184-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Hum Mol Genet. 2010;19:R46–64. doi: 10.1093/hmg/ddq137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Ber I, Camuzat A, Berger E, Hannequin D, Laquerriere A, Golfier V, Seilhean D, Viennet G, Couratier P, Verpillat P, et al. Chromosome 9p-linked families with frontotemporal dementia associated with motor neuron disease. Neurology. 2009;72:1669–1676. doi: 10.1212/WNL.0b013e3181a55f1c. [DOI] [PubMed] [Google Scholar]
- Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- Lomen-Hoerth C, Anderson T, Miller B. The overlap of amyotrophic lateral sclerosis and frontotemporal dementia. Neurology. 2002;59:1077–1079. doi: 10.1212/wnl.59.7.1077. [DOI] [PubMed] [Google Scholar]
- Lomen-Hoerth C, Murphy J, Langmore S, Kramer JH, Olney RK, Miller B. Are amyotrophic lateral sclerosis patients cognitively normal? Neurology. 2003;60:1094–1097. doi: 10.1212/01.wnl.0000055861.95202.8d. [DOI] [PubMed] [Google Scholar]
- Luty AA, Kwok JB, Thompson EM, Blumbergs P, Brooks WS, Loy CT, Dobson-Stone C, Panegyres PK, Hecker J, Nicholson GA, et al. Pedigree with frontotemporal lobar degeneration--motor neuron disease and Tar DNA binding protein-43 positive neuropathology: genetic linkage to chromosome 9. BMC Neurol. 2008;8:32. doi: 10.1186/1471-2377-8-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, Kwong LK, Forman MS, Ravits J, Stewart H, et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61:427–434. doi: 10.1002/ana.21147. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J, Kovacs GG, Ghetti B, Halliday G, Holm IE, et al. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathol. 2009;117:15–18. doi: 10.1007/s00401-008-0460-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, Neville C, Narang M, Barcelo J, O’Hoy K, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 1992;255:1253–1255. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- Miller JW, Urbinati CR, Teng-Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, Swanson MS. Recruitment of human muscleblind proteins to (CUG)(n) expansions associated with myotonic dystrophy. EMBO J. 2000;19:4439–4448. doi: 10.1093/emboj/19.17.4439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok K, Traynor B, Schymick J, Tienari P, Laaksovirta H, Peuralinna T, Myllykangas L, Chio A, Shatunov A, Boeve B, et al. The chromosome 9 ALS and FTD locus is probably derived from a single founder. Neurobiol Aging. 2011 doi: 10.1016/j.neurobiolaging.2011.08.005. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita M, Al-Chalabi A, Andersen PM, Hosler B, Sapp P, Englund E, Mitchell JE, Habgood JJ, de Belleroche J, Xi J, et al. A locus on chromosome 9p confers susceptibility to ALS and frontotemporal dementia. Neurology. 2006;66:839–844. doi: 10.1212/01.wnl.0000200048.53766.b4. [DOI] [PubMed] [Google Scholar]
- Moseley ML, Zu T, Ikeda Y, Gao W, Mosemiller AK, Daughters RS, Chen G, Weatherspoon MR, Clark HB, Ebner TJ, et al. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet. 2006;38:758–769. doi: 10.1038/ng1827. [DOI] [PubMed] [Google Scholar]
- Neary D, Snowden JS, Gustafson L, Passant U, Stuss D, Black S, Freedman M, Kertesz A, Robert PH, Albert M, et al. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Pearson JP, Williams NM, Majounie E, Waite A, Stott J, Newsway V, Murray A, Hernandez D, Guerreiro R, Singleton AB, et al. Familial frontotemporal dementia with amyotrophic lateral sclerosis and a shared haplotype on chromosome 9p. J Neurol. 2011;258:647–655. doi: 10.1007/s00415-010-5815-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phukan J, Pender NP, Hardiman O. Cognitive impairment in amyotrophic lateral sclerosis. Lancet Neurol. 2007;6:994–1003. doi: 10.1016/S1474-4422(07)70265-X. [DOI] [PubMed] [Google Scholar]
- Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng HX, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- Sato N, Amino T, Kobayashi K, Asakawa S, Ishiguro T, Tsunemi T, Takahashi M, Matsuura T, Flanigan KM, Iwasaki S, et al. Spinocerebellar ataxia type 31 is associated with “inserted” penta-nucleotide repeats containing (TGGAA)n. Am J Hum Genet. 2009;85:544–557. doi: 10.1016/j.ajhg.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatunov A, Mok K, Newhouse S, Weale ME, Smith B, Vance C, Johnson L, Veldink JH, van Es MA, van den Berg LH, et al. Chromosome 9p21 in sporadic amyotrophic lateral sclerosis in the UK and seven other countries: a genome-wide association study. Lancet Neurol. 2010;9:986–994. doi: 10.1016/S1474-4422(10)70197-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PJ, Zhang C, Wang J, Chew SL, Zhang MQ, Krainer AR. An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum Mol Genet. 2006;15:2490–2508. doi: 10.1093/hmg/ddl171. [DOI] [PubMed] [Google Scholar]
- Sofola OA, Jin P, Qin Y, Duan R, Liu H, de Haro M, Nelson DL, Botas J. RNA-binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat-induced neurodegeneration in a Drosophila model of FXTAS. Neuron. 2007;55:565–571. doi: 10.1016/j.neuron.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, Ackerley S, Durnall JC, Williams KL, Buratti E, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672. doi: 10.1126/science.1154584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassone F, Iwahashi C, Hagerman PJ. FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS) RNA Biol. 2004;1:103–105. doi: 10.4161/rna.1.2.1035. [DOI] [PubMed] [Google Scholar]
- Timchenko LT, Miller JW, Timchenko NA, DeVore DR, Datar KV, Lin L, Roberts R, Caskey CT, Swanson MS. Identification of a (CUG)n triplet repeat RNA-binding protein and its expression in myotonic dystrophy. Nucleic Acids Res. 1996;24:4407–4414. doi: 10.1093/nar/24.22.4407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd PK, Paulson HL. RNA-mediated neurodegeneration in repeat expansion disorders. Ann Neurol. 2010;67:291–300. doi: 10.1002/ana.21948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdmanis PN, Dupre N, Bouchard JP, Camu W, Salachas F, Meininger V, Strong M, Rouleau GA. Three families with amyotrophic lateral sclerosis and frontotemporal dementia with evidence of linkage to chromosome 9p. Arch Neurol. 2007;64:240–245. doi: 10.1001/archneur.64.2.240. [DOI] [PubMed] [Google Scholar]
- Van Deerlin VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR, Dickson DW, Rademakers R, Boeve BF, Grossman M, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010;42:234–239. doi: 10.1038/ng.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Es MA, Veldink JH, Saris CG, Blauw HM, van Vught PW, Birve A, Lemmens R, Schelhaas HJ, Groen EJ, Huisman MH, et al. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet. 2009;41:1083–1087. doi: 10.1038/ng.442. [DOI] [PubMed] [Google Scholar]
- Vance C, Al-Chalabi A, Ruddy D, Smith BN, Hu X, Sreedharan J, Siddique T, Schelhaas HJ, Kusters B, Troost D, et al. Familial amyotrophic lateral sclerosis with frontotemporal dementia is linked to a locus on chromosome 9p13.2-21.3. Brain. 2006;129:868–876. doi: 10.1093/brain/awl030. [DOI] [PubMed] [Google Scholar]
- Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J, Hu X, Smith B, Ruddy D, Wright P, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MC, Gao R, Xu W, Mandal SM, Lim JG, Hazra TK, Wakamiya M, Edwards SF, Raskin S, Teive HA, et al. Inactivation of hnRNP K by expanded intronic AUUCU repeat induces apoptosis via translocation of PKCdelta to mitochondria in spinocerebellar ataxia 10. PLoS Genet. 2010;6:e1000984. doi: 10.1371/journal.pgen.1000984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin F, Banerjee R, Thomas B, Zhou P, Qian L, Jia T, Ma X, Ma Y, Iadecola C, Beal MF, et al. Exaggerated inflammation, impaired host defense, and neuropathology in progranulin-deficient mice. J Exp Med. 2010;207:117–128. doi: 10.1084/jem.20091568. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.