

Abstract
The unfolded protein response (UPR) detects the accumulation of unfolded proteins in the endoplasmic reticulum (ER) and adjusts the protein folding capacity to the needs of the cell. Under conditions of ER stress, the transmembrane protein Ire1 oligomerizes to activate its cytoplasmic kinase and RNase domains. It is unclear what feature of ER stress Ire1 detects. Here we found that the core ER-lumenal domain (cLD) of yeast Ire1 binds to unfolded proteins in vivo and to peptides primarily composed of basic and hydrophobic residues in vitro. Mutation of amino acid side chains exposed in a putative peptide-binding groove of Ire1 cLD impaired peptide binding. Peptide binding caused Ire1 cLD oligomerization in vitro, suggesting that direct binding to unfolded proteins activates the UPR.
Secretory and transmembrane proteins fold within the ER. Contingent on proper folding, they are either exported to the Golgi apparatus or degraded. An accumulation of unfolded proteins activates the unfolded protein response (UPR), a transcriptional program that restores ER protein folding homeostasis (1). The transmembrane protein Ire1 induces the UPR by oligomerizing. Oligomerization activates Ire1's cytoplasmic kinase and RNase domains and initiates a non-conventional mRNA splicing reaction of HAC1 mRNA. Spliced HAC1 mRNA is translated to produce the transcription factor Hac1, which upregulates protein folding machinery (2-4). The mechanism of Ire1 activation and signal transduction is conserved from yeast to mammals (5-9); however, it is unclear how Ire1 senses ER stress.
A direct binding model, in which Ire1 binding to unfolded proteins causes its oligomerization, is suggested by structural studies on the conserved core of yeast Ire1 ER-lumenal domain (cLD). The cLD structure contains two interfaces—Interface 1 creates a two-fold symmetric dimer containing a deep groove, while Interface 2 permits further oligomerization (Fig. S1) (10). Mutation of either interface diminishes oligomerization and decreases HAC1 mRNA splicing (11). The groove formed across Interface 1 has architectural similarity to the peptide-binding groove of MHCI, suggesting that Ire1 binds to unfolded proteins and that binding increases Ire1 oligomerization (10). Also, purified Ire1 cLD prevents the aggregation of unfolded proteins (12), suggesting that it binds exposed, aggregation-prone regions.
An alternative model poses that BiP, a predominant ER-resident Hsp70 chaperone that associates with inactive Ire1, detects ER stress by preferentially binding to accumulating unfolded proteins and releasing Ire1 for oligomerization (13, 14). In yeast, however, BiP release cannot be the primary activation step because mutations in Ire1 that impair BiP binding still properly respond to ER stress (10, 15, 16).
To test the hypothesis that unfolded proteins are Ire1-activating ligands, we determined if Ire1 interacts with a constitutively misfolded mutant (G255R) of carboxypeptidase Y (wt: CPY, mutant: CPY*) that is retained in the ER (17). Expression of CPY* induced the UPR in an Ire1-dependent manner, while expression of wild-type CPY induced the UPR to a lesser extent (Fig. 1A). To assess whether UPR induction correlated with binding to Ire1, we immunoprecipitated FLAG-tagged Ire1 from cells expressing HA-tagged CPY or CPY* at equivalent amounts. CPY*-HA co-immunoprecipitated with Ire1 in larger amounts than CPY-HA (Fig. 1B), mirroring the UPR-induction efficiency of each protein.
Fig. 1.
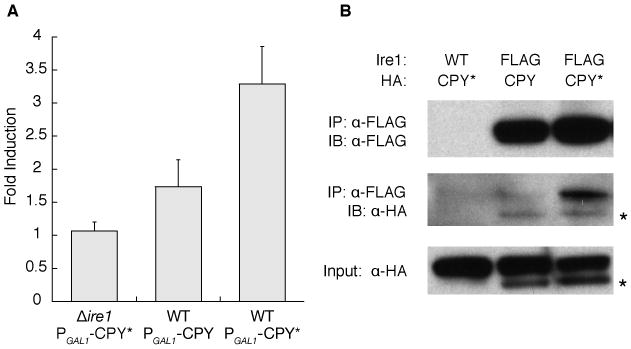
An unfolded protein co-immunoprecipitates with Ire1. A. The UPR activated by inducible CPY/CPY*-HA was measured with a 4×UPRE-GFP reporter. Fold induction is the ratio of median FITC intensity of cells in 2% galactose to cells in 2% raffinose after 4 hours. Error bars are SD; N≥4. B. FLAG-tagged Ire1 was immunoprecipitated from cells expressing CPY-HA or CPY*-HA (2 hours, 2% galactose); CPY/CPY*-HA was detected with anti-HA. The asterisk indicates unglycosylated CPY/CPY*.
To show that Ire1 can bind directly to CPY* and identify binding sites, we probed an array of peptide sequences derived from tiling along the sequence of CPY* with purified cLD tagged with GST (Fig. 2A). To compare GST-cLD binding between arrays and across CPY*'s sequence, we quantified the average signal intensity attributed to each amino acid in CPY* (Fig. 2B). GST-cLD bound to the signal peptide, propeptide, and C-terminal regions of CPY*. The GST tag did not contribute to binding because GST alone did not bind to the array. Notably, GST-cLD did not bind strongly to the region containing the G255R mutation, suggesting that this mutation exposed other sites through large-scale misfolding. A mutant of GST-cLD, W426A, which disrupts Interface 2 but preserves the putative peptide-binding groove (10, 11), bound to similar peptides on the array as GST-cLD (Fig. 2B). The lowered affinity of GST-cLDW426A to regions of weak binding by wild-type GST-cLD is likely due to reduced avidity in the absence of higher-order oligomers.
Fig 2.
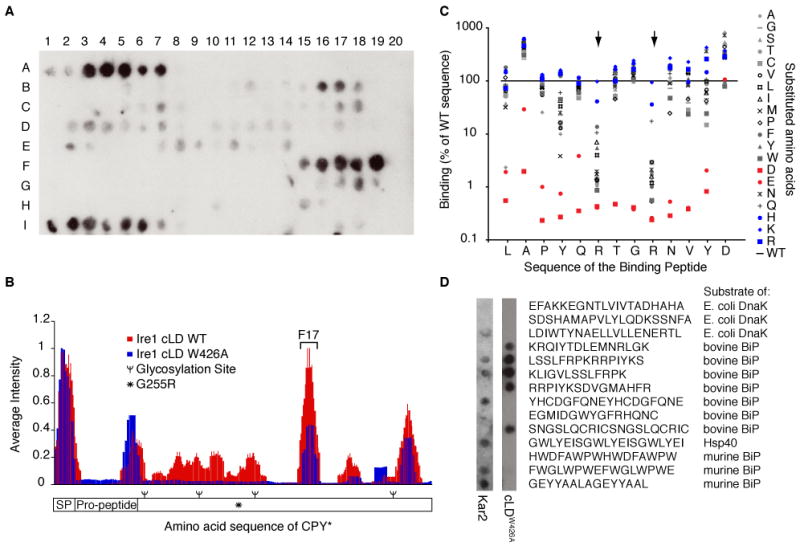
Ire1 binds specific regions in CPY* containing basic and hydrophobic residues. A. Binding of 500 nM GST-cLD to a peptide array tiling along the sequence of CPY* was detected with anti-GST. B. The contribution of each amino acid in CPY*'s sequence to Ire1-cLD binding (WT and W426A) was calculated by averaging the intensity of spots containing that amino acid (see Methods). Values were plotted along the sequence of CPY*. SP: signal peptide. C. An array composed of peptides with single amino acid substitutions in peptide F17 from the CPY* tiling array (AQLAPYQRTGRNVYD) was probed with GST-cLDW426A and GST-cLD (Fig. S3). The binding intensity of the mutated peptides was normalized to the intensity of the wild-type peptide. Large arrows highlight the basic residues in F17. Blue: introduced basic residues. Red: introduced acidic residues. D. A peptide array of chaperone substrates shows that GST-cLDW426A and Kar2 bind a different subset of peptides.
The regions of CPY* that GST-cLD (WT and W426A) recognized did not reveal a consensus sequence. However, comparison of the amino acid composition of binding peptides with that of CPY/CPY* showed a strong preference for arginine and an exclusion of acidic amino acids. Leucine and phenylalanine were also enriched in the binding peptides (Fig. S2). Further analysis by systematic mutation of a binding peptide (derived from spot F17 in Fig. 2A) confirmed the bias for basic residues (Fig. 2C). Mutation of either of the two arginines, and to a lesser extent the two tyrosines, in the binding peptide to non-basic amino acids reduced binding of GST-cLD (W426A and WT) (Fig. 2C and Fig. S3). Similarly, mutation of any amino acid to an acidic residue lowered the affinity of GST-cLDW426A for the peptide (Fig. 2C).
The Hsp70 family of chaperones, which includes BiP (Kar2 in yeast), also binds regions containing basic and hydrophobic residues (18-21). The overlap between Hsp70 and Ire1 substrates is not congruent, however, because Ire1 GST-cLDW426A did not bind measurably to several control peptides derived from known DnaK and BiP substrates (Fig. 2D). Furthermore, probing the same peptides with purified Kar2 showed that Kar2 and Ire1 bound an overlapping, but not identical, subset of peptides (Fig. 2D).
To determine the affinity of Ire1 cLD for peptides, we measured their binding to cLDW426A by fluorescence anisotropy. CPY*-derived peptide FAM-F17 bound with an affinity of K1/2 = 172 μM (Fig. 3A) and a Hill coefficient of 1.4. Notably, when a fragment of CPY containing this region was expressed within the ER, it induced the UPR and co-immunoprecipitated with Ire1 (Fig. S4). A signal peptide, ΔEspP-FAM, which contains hydrophobic and basic amino acids as preferred by Ire1 (22), bound to Ire1 cLDW426A with higher affinity (K1/2 = 0.75 μM, Hill coefficient=1.2). Although signal peptides are not normally displayed in the ER lumen, ΔEspP proved a useful tool to probe peptide/cLD interactions (Fig. 3A). Mutational analysis of ΔEspP on a peptide array confirmed that Ire1 cLDW426A bound ΔEspP with the same preference for basic and hydrophobic residues and intolerance of acidic residues (Fig. S5).
Fig 3.

A. Fluorescence anisotropy of ΔEspP-FAM (50 nM) and FAM-F17 (100 nM) binding to Ire1-cLDW426A. K1/2 for ΔEspP-FAM is 0.75±0.03 μM with a Hill coefficient of 1.2 and K1/2 for FAM-F17 is 172±4 μM with a Hill coefficient of 1.4. B. Induction of a HAC1 mRNA splicing reporter after a 3 hour incubation with 0 or 5 mM DTT in CRY1 Δire1∷KAN strains expressing no Ire1, WT Ire1, or Ire1-ΔMFY. N=5; error bars are SD; p value calculated by Student T-test between WT and ΔMFY. C. ΔMFY increases the K1/2 for Ire1-cLDW426A binding to ΔEspP-FAM to 42±2 μM, while the Hill coefficient decreases to 0.9. For A and C, N≥3; error bars are SD.
These K1/2's are composites of several equilibria: Ire1 cLD self-association and binding of Ire1 cLD monomer and dimer to peptide. The lack of significant Ire1 cLD monomer bound to peptide (Fig. 4B) and the Hill coefficient greater than 1 in the anisotropy assay suggest that Ire1 binds the peptides in a cooperative manner. By modeling Ire1 binding to peptide as a cooperative reaction in which there is little Ire1 monomer bound to peptide (Fig. 4B), we estimate the affinity of Ire1 dimer for ΔEspP-FAM is 0.068 μM (Fig. S6).
Fig 4.

The addition of peptide causes Ire1 oligomerization. A. C(s) analysis of velocity sedimentation of 7 μM Ire1-cLD WT (blue) and with 50 μM ΔEspP (red). Without peptide: RMSD: 0.0105, f/f0: 1.65. With peptide: RMSD: 0.0080, f/f0: 2.75. B. C(s) analysis of the velocity sedimentation of 9 μM Ire1-cLDW426A (blue) and with 20 μM of ΔEspP (red). Without peptide: RMSD=0.0104, f/f0: 1.46. With peptide: 0.0086, f/f0: 1.52. C. Comparison of the sedimentation equilibrium (20,000×g, 12 hrs) of Ire1-cLDW426A (blue) and with a 2:1 molar ratio of ΔEspP (red) with simulated Ire1 monomer (dotted) and dimer (solid). The full data set and fit to a monomer-dimer model is presented in Fig. S8.
To determine if residues in the groove are important for peptide binding, we tested the mutant M229A-F265A-Y301A (ΔMFY), which changes three hydrophobic residues along the floor of the groove to alanines and reduces UPR signaling in vivo (10). We observed that cLD-ΔMFY bound ΔEspP-FAM with reduced affinity (K1/2=42 ± 2 μM) (Fig. 3D), corresponding with reduced UPR signaling (Fig. 3B) and impaired survival upon UPR-induction (Fig. S7). Thus, Ire1 binding with high affinity to ΔEspP-FAM depends on groove residues.
To interrogate whether cLD oligomerizes in response to peptide binding, as hypothesized by the direct binding model, we assessed the oligomeric state of cLD bound to ΔEspP. Velocity sedimentation of wild-type cLD showed a distribution of monomer, dimer, and tetramer species when assessed without peptide, and a shift towards heterogeneous larger oligomers in the presence of ΔEspP (Fig. 4A). To limit peptide-induced oligomerization to more discrete species, we repeated this experiment with Ire1 cLDW426A and observed only two predominant species of Ire1 cLDW426A corresponding to monomer and dimer (Fig. 4B). Addition of ΔEspP caused Ire1 cLDW426A to dimerize completely (Fig. 4B). In agreement, sedimentation equilibrium showed a shift from the simulated monomer towards the simulated dimer (Fig 4C). By fitting the data with a monomer-dimer model, we determined that the KD of Ire1 association without peptide was 8.2 μM (Fig. S8). The KD with peptide could not be determined because there was no remaining Ire1 monomer population; however, the average molecular weight of the species in solution shifted from 62 kDa to 88 kDa.
Taken together, Ire1-cLD binding to peptides causes it to oligomerize, as we predict occurs in cells when Ire1 binds to unfolded proteins. Furthermore, Ire1 co-immunoprecipitated with CPY*. This interaction was recently shown to be impaired by the ΔMFY mutation, but not the W426A mutation (23), consistent with our in vitro work. Because BiP dissociation from Ire1 during ER stress has been ruled out as the primary switch that governs the UPR, we suggest that BiP association fine-tunes Ire1 signaling (10, 15, 16), while unfolded proteins are activating ligands. The lack of congruity between Ire1 and BiP substrates suggests that Ire1 does not always compete with BiP for binding sites and is not dependent on BiP saturation.
Ire1's preference for basic and hydrophobic residues and intolerance of acidic residues was maintained despite the wide range in affinity between the peptides analyzed here. In vivo, where the substrates are unfolded proteins rather than short peptides, this sequence selectivity must be accompanied by selecting for stretches of misfolded proteins that are sufficiently exposed to sample the depth of the groove. Importantly, acidic features prominent in many ER resident proteins, e.g., ER-retention signals, would render them invisible to Ire1 even if unstructured and exposed.
By sequence, Ire1-cLD is conserved from yeast to mammalian Ire1α/Ire1β, and including the additional metazoan ER-stress sensor PERK. This evolutionary conservation and the central importance of unfolded protein recognition by all of these sensors strongly suggest that these molecules deploy similar mechanisms. However, unlike yeast Ire1, the crystal structure of human Ire1α displays a groove too narrow for peptide binding (24). These two static structures can be reconciled if the structure of hIre1 represents a “closed” conformation formed in the absence of bound peptide. In this conformation, Interface 2 cannot form, and Ire1 cannot oligomerize. Peptide binding would induce a conformational switch towards an “open” conformation, represented by the yeast Ire1 structure, establishing Interface 2 oligomerization and leading to Ire1 activation. In this way, the conformational change is induced by peptide binding, forming the core module of the signaling machine that senses ER stress. The unique peptide-binding groove and oligomerization interfaces are attractive candidates for targeted drug design that could help alleviate the mis-regulation of hIre1 and PERK apparent in disease.
Supplementary Material
Acknowledgments
We thank: J. Weissman, G. Narlikar, and D. Mullins for insightful comments; S. Neher and A. Korennykh for their invaluable guidance; the Walter Lab, especially H. Li, E. van Anken, D. Pincus, C. Gallagher, and M. Diaz for their assistance and scientific discussions. Purified Kar2 was a generous gift from J. Brodsky. We are grateful to the MIT Biopolymers Institute for synthesizing the peptide arrays. B.G. was supported by an NSF predoctoral fellowship. P.W. is an Investigator of the Howard Hughes Medical Institute.
References and Notes
- 1.Ron D, Walter P. Nat Rev Mol Cell Biol. 2007;8:519. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 2.Cox JS, Walter P. Cell. 1996;87:391. doi: 10.1016/s0092-8674(00)81360-4. [DOI] [PubMed] [Google Scholar]
- 3.Sidrauski C, Walter P. Cell. 1997;90:1031. doi: 10.1016/s0092-8674(00)80369-4. [DOI] [PubMed] [Google Scholar]
- 4.Kawahara T, Yanagi H, Yura T, Mori K. Mol Biol Cell. 1997;8:1845. doi: 10.1091/mbc.8.10.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang XZ, et al. EMBO J. 1998;17:5708. doi: 10.1093/emboj/17.19.5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shen X, et al. Cell. 2001;107:893. doi: 10.1016/s0092-8674(01)00612-2. [DOI] [PubMed] [Google Scholar]
- 7.Calfon M, et al. Nature. 2002;415:92. doi: 10.1038/415092a. [DOI] [PubMed] [Google Scholar]
- 8.Li H, Korennykh AV, Behrman SL, Walter P. Proc Natl Acad Sci USA. 2010;107:16113. doi: 10.1073/pnas.1010580107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tirasophon W, Welihinda AA, Kaufman RJ. Genes Dev. 1998;12:1812. doi: 10.1101/gad.12.12.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Credle JJ, Finer-Moore JS, Papa FR, Stroud RM, Walter P. Proc Natl Acad Sci USA. 2005;102:18773. doi: 10.1073/pnas.0509487102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aragón T, et al. Messenger RNA targeting to endoplasmic reticulum stress signalling sites. Nature. 2009;457:736–740. doi: 10.1038/nature07641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kimata Y, et al. J Cell Biol. 2007;179:75. doi: 10.1083/jcb.200704166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bertolotti A, Zhang Y, Hendershot LM, Harding HP, Ron D. Nat Cell Biol. 2000;2:326. doi: 10.1038/35014014. [DOI] [PubMed] [Google Scholar]
- 14.Okamura K, Kimata Y, Higashio H, Tsuru A, Kohno K. Biochem Biophys Res Commun. 2000;279:445. doi: 10.1006/bbrc.2000.3987. [DOI] [PubMed] [Google Scholar]
- 15.Pincus D, et al. PLoS Biol. 2010;8:e1000415. doi: 10.1371/journal.pbio.1000415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kimata Y, Oikawa D, Shimizu Y, Ishiwata-Kimata Y, Kohno K. J Cell Biol. 2004;167:445. doi: 10.1083/jcb.200405153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Finger A, Knop M, Wolf DH. Eur J Biochem. 1993;218:565. doi: 10.1111/j.1432-1033.1993.tb18410.x. [DOI] [PubMed] [Google Scholar]
- 18.Rüdiger S, Germeroth L, Schneider-Mergener J, Bukau B. EMBO J. 1997;16:1501. doi: 10.1093/emboj/16.7.1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Crouy-Chanel A, Kohiyama M, Richarme G. J Biol Chem. 1996;271:15486. doi: 10.1074/jbc.271.26.15486. [DOI] [PubMed] [Google Scholar]
- 20.Flynn GC, Chappell TG, Rothman JE. Science. 1989;245:385. doi: 10.1126/science.2756425. [DOI] [PubMed] [Google Scholar]
- 21.Blond-Elguindi S, et al. Cell. 1993;75:717. doi: 10.1016/0092-8674(93)90492-9. [DOI] [PubMed] [Google Scholar]
- 22.Peterson JH, Woolhead CA, Bernstein HD. J Biol Chem. 2003;278:46155. doi: 10.1074/jbc.M309082200. [DOI] [PubMed] [Google Scholar]
- 23.Ishiwata-Kimata Y, et al. Mol Biol Cell. 2011 doi: 10.1091/mbc.E11-04-0295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou J, et al. Proc Natl Acad Sci USA. 2006;103:14343. doi: 10.1073/pnas.0606480103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.