

Abstract
Objective
Multinucleated cells are relatively resistant to classical apoptosis, and the factors initiating cell-death and damage in myositis are not well defined. We hypothesized that non-immune autophagic cell death may play a role in muscle fiber damage. Recent literature indicates that tumor necrosis factor-alpha-related apoptosis inducing ligand (TRAIL) may induce both NFκB (nuclear factor kappa-light chain enhancer of activated B cells) activation and autophagic cell death in other systems. Here, we have investigated its role in cell death and pathogenesis in vitro and in vivo using myositis (human and mouse) muscle tissues.
Methods
Gene expression profiling indicated that expression of TRAIL and several autophagy markers was specifically upregulated in myositis muscle tissue; these results were confirmed by immunohistochemistry and immunoblotting. We also analyzed TRAIL-induced cell death (apoptosis and autophagy) and NFκB activation in vitro in cultured cells.
Results
TRAIL was expressed predominantly in muscle fibers of myositis, but not in biopsies from normal or other dystrophic-diseased muscle. Autophagy markers were upregulated in human and mouse models of myositis. TRAIL expression was restricted to regenerating/atrophic areas of muscle fascicles, blood vessels, and infiltrating lymphocytes. TRAIL induced NFκB activation and IκB degradation in cultured cells that are resistant to TRAIL-induced apoptosis but undergo autophagic cell death.
Conclusion
Our data demonstrate that TRAIL is expressed in myositis muscle and may mediate both activation of NFκB and autophagic cell death in myositis. Thus, this non-immune pathway may be an attractive target for therapeutic intervention in myositis.
It is generally thought that the muscle dysfunction and weakness in idiopathic inflammatory myopathies (IIM) is the result of tissue damage caused by adaptive immune response to ubiquitously expressed autoantigens. There is growing evidence that non-immune mechanisms also equally contribute to muscle damage and dysfunction in myositis (1–3). The molecular mechanisms of muscle fiber death in IIM remain poorly understood. Cell death mechanisms can be broadly divided into two categories, Type I and Type II (4). Type I cell death (apoptosis) occurs in mononucleated cells and involves the condensation of cytoplasm and chromatin, controlled DNA and cellular fragmentation, and macrophage removal of cellular debris (4). Type II cell death (autophagy) has been demonstrated in multi-nucleated muscle fibers (5) and can affect cell damage through the formation of autophagic vesicles for removing cellular masses, damaged organelles, and/or proteins (6). Several cellular receptor/ligand complexes can induce cell death: Fas/Fas ligand (7), tumor necrosis factor-α (TNFα)/TNF receptor-1 or -2 (8), and TNFα-related apoptosis-inducing ligand (TRAIL)/death receptor-5 (DR5) (9). Downstream activation of these receptor/ligand complexes can activate caspase-dependent apoptosis (10). Normal human skeletal muscle cells are relatively resistant to classical apoptosis (11). Several independent studies have shown that classical apoptotic changes are absent from the skeletal muscle of myositis patients (11–13). Muscle fibers are known to atrophy and be replaced by fibrotic tissue and/or fat, but the exact underlying molecular pathways responsible for muscle fiber death in myositis remain obscure (11, 14).
TRAIL has been implicated in tissue remodeling and lumen formation (15), induces capase-3-independent autophagic cell death in other model systems (15), and is expressed in myositis muscle(16, 17). TRAIL is a type II transmembrane protein that is expressed on variety of cells (18) and can induce NFκB activation upon binding to its receptor (19). We propose that TRAIL-induced autophagy may be responsible for the skeletal muscle death that occurs in myositis and that the interaction between TRAIL and NFκB may modulate both the inflammation and cell death pathways of myositis. In the present study, the expression and role of TRAIL in muscle cell death have been investigated in humans with myositis, in MHC class I transgenic mouse model of the disease, and in cell culture. The results indicate that TRAIL mediates muscle fiber damage via autophagy in both humans and in a mouse model of myositis.
MATERIALS AND METHODS
Patient biopsies
Myositis muscle biopsy specimens from eight patients with myositis (four polymyositis [PM] and four dermatomyositis [DM]) and four healthy controls were used for these studies. Human samples were handled according to the National Institutes of Health and Johns Hopkins School of Medicine Institutional Review Board (IRB) guidelines. Patients with myositis met the Bohan and Peter criteria for probable or definite disease (20). Tissue was either: (1) formalin-fixed, processed in paraffin, and stained with hematoxylin and eosin, or (2) snap-frozen in isopentane chilled in liquid nitrogen, cut in 8-mm sections, and processed for immunohistochemical analysis. Histologic analysis showed a variable degree of inflammation between patients.
Mice
Transgenic mice conditionally expressing major histocompatibility complex class I (MHC) molecules, under the control of a muscle-specific creatine kinase promoter (HT double-transgenic), were generated as previously described (21). Unaffected, single-transgenic littermates (H or T) were used as controls. Gene induction and phenotypic assessments were performed as previously described (21). The double-transgenic animals (HT) showed several features of human myositis. Animals were cared for in accordance with our Institutional Animal Care and Use Committee (IACUC) guidelines, and all experimental protocols were IACUC-approved.
Immunohistochemical staining
Frozen muscle biopsy sections (5–8μm) were fixed in acetone, and endogenous peroxidase activity was blocked by applying hydrogen peroxide at room temperature for 10 min as described (11). Sections were incubated with the following primary antibodies: mouse anti-human developmental MHC (Novocastra, Newcastle-Upon-Tyne, UK), mouse anti-human TRAIL (1:20 dilution) (BD Pharmingen, San Diego, CA), and mouse anti-human DR5 (1:20) (R&D Systems) and developed as previously described (11). Negative controls were involved incubation with isotype-matched antibody or omitting the primary antibody.
Protein extraction and quantification
Tissue samples were homogenized as previously described (11). Western blotting using rabbit polyclonal anti-human Beclin (AbCam, Cambridge, MA) and rabbit polyclonal anti-human LC-3 (Novus Biologicals, Littleton, CO) was performed as described (11). Vinculin (mouse monoclonal anti-human, Sigma-Aldrich, St. Louis, MO) was used as a loading control. The autoradiograms were scanned using an Arcus II scanner (Agfa, Mortsel, Belgium), and volume analysis was carried out using Quantity One software (Bio-Rad Discovery Series; Bio-Rad, Richmond, CA).
Isolation of fixed single muscle fibers and immunofluorescence microscopy
Muscle fixation, isolation of single fibers, and immunostaining have been described in detail by Raben et al 2009 (22). In brief, the soleus muscles (slow-twitch type I) and the white portion of the gastrocnemius muscles (fast-twitch type II muscle) were removed from single- and double-transgenic mice. Single fibers were obtained by manual teasing under a light microscope and maintained in blocking solution in wells of a 24-well plate. The fibers were then permeabilized and incubated overnight at 4°C on a plate shaker with primary antibody (GM-130 (BD Transduction Laboratories, San Jose, CA), lysosome-associated membrane protein 1 (LAMP; BD Pharmingen, San Diego, CA); α-tubulin (Sigma-Aldrich, St. Louis, MO); LC-3 (microtubule-associated protein 1 light chain 3; a gift from Dr T. Ueno, Juntendo University School of Medicine, Japan). At least three animals from each genotype were used to obtain single muscle fibers for immunostaining. For each immunostaining and for confocal analysis, at least 20 fibers were isolated from each muscle type.
Gene expression profiling and analysis
Expression profiling was performed as previously described, using the Affymetrix mouse genome array 430 version 2.0 (23): Gastrocnemius and soleus muscles were removed from single- (n=4) and double-transgenic (n=4) mice, homogenized in Trizol reagent (Invitrogen, Carlsbad, CA) using a Polytron homogenizer (Brinkmann, Westbury, NY), and processed as previously described (23). Probe set analysis was done using both the dCHIP and MAS 5.0 expression algorithms. The signal intensity values (absolute analyses) of probe sets were then loaded into GeneSpring (Silicon Genetics, Redwood City, CA) for further analysis. Gene identification is listed in Supplemental Table 1.
Electron microscopy
Quadriceps from HT and control mice were removed and mid-belly portions were cubed to facilitate orientation during embedding and sectioning. Specimens were fixed in 3% glutaraldehyde in 0.1M phosphate buffer (pH 7.4) for 2h to 1 day, postfixed in 1% OsO4 in 0.1 M phosphate buffer (pH 7.4) overnight at 4°C, and rinsed in 10% saccharose 3 times (10 min each) before staining en bloc in 3% aqueous uranyl acetate for 1h at room temperature. Ultrathin sections were cut for staining with uranyl acetate and lead citrate. The sections were examined and photographed with an electron microscope at 75kV (H7000, Hitachi, Tokyo, Japan). These experiments were performed in Dr. Kunio Nagashima’s electron microscopy facility, SAIC-Frederick, Inc, Frederick, Maryland.
Immunoblotting and immunofluorescent staining of cultured cells
Cell culture and Western blotting were carried out as described previously (11). In brief, normal human skeletal muscle cells (SKMC Clonetics, San Diego, CA) were cultured in F10 growth medium (Ham’s F10 nutrient mixture [Life Technologies, Gaithersburg, MD] supplemented with 20% FBS [HyClone, Logan, UT], 2% chick embryo extract [Life Technologies, Grand Island, NY], 100 U/ml penicillin, and 100 μg/ml streptomycin). Human salivary gland (HSG)cells were passaged in RPMI containing 10% heat-inactivated calf serum according to standard tissue culture procedures. Cells were incubated for 24–48h with varying concentrations of recombinant TRAIL (rTRAIL) (1 to 20 ng/ml; R&D Systems, Minneapolis, MN) to induce cell death. Cells were lysed in buffer A (1% Nonidet P-40, 20 mM Tris [pH.7.4], 150 mM NaCl, 1 mM EDTA, and protease inhibitors), and 25 μg of each sample were separated on 10% SDS polyacrylamide gels and transferred to nitrocellulose membranes. Western blotting was performed using antibodies recognizing PARP (rabbit polyclonal anti-human, New England Biolabs, Beverly, MA), IkB (rabbit polyclonal anti-human, Cell Signaling Technology, Danvers, MA), and vinculin (as a loading control). SKMC were incubated with rTRAIL, and autophagic vesicles were identified by staining with monodansylcadaverine (MDC) (Sigma-Aldrich, St. Louis, MO).
RESULTS
Expression of TRAIL is specifically higher in skeletal muscle of myositis patients
TRAIL mRNA expression was determined in muscle biopsies from patients with various myopathies (Supplemental Figure 1). As compared to the expression in normal human muscle (NHM), the expression of TRAIL was not significantly higher in any of the 13 myopathic biopsies, with the exception of the myositis (JDM) patients’ muscle tissues. The relative expression of TRAIL in myositis muscle was significantly higher than that in NHM, indicating that elevated TRAIL expression is unique to myositis muscle tissue and may be involved in the disease pathogenesis pathway. Consistent with the higher expression of TRAIL in myositis muscle, TRAIL protein expression was higher in adult myositis (DM and PM) patient tissues than in muscle from normal or DMD biopsies (Figure 1). In normal and DMD tissues, there was no immunohistochemical staining for TRAIL, despite extensive degeneration of the muscle fibers in DMD muscle. DM myofibers showed extensive staining in the perifascicular area of the muscle and the infiltrating lymphocytes and blood vessels. PM muscle fibers showed sporadic staining in the muscle and in infiltrating cells surrounding the muscle fibers. To exclude the possibility that the TRAIL staining in the perifascicular region of DM muscle was actually non-specific staining related to atrophy and degeneration, we obtained serial sections for developmental myosin heavy chain (dMHC), a specific marker for regenerating muscle fibers. These results clearly indicated that TRAIL expression was restricted to the regenerating muscle fiber of myositis muscle(Figure 2A and B).
Figure 1.
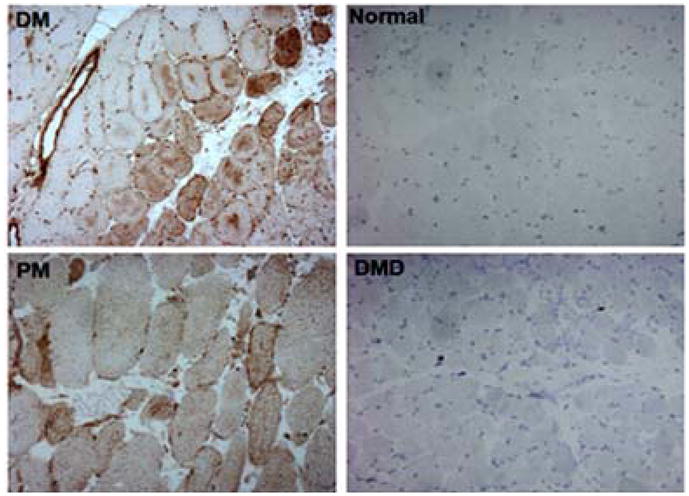
TRAIL expression is up-regulated in atrophic fibers of human myositis muscle. Tissue sections (n=4 per disease group) from myositis and muscular dystrophy muscle were stained for the presence of TRAIL. Intense intracellular TRAIL staining was seen in the perifascicular region of the DM muscle, whereas the PM muscle showed sporadic staining in some small degenerating fibers as well as interstitial cells. Normal and DMD tissues had no TRAIL staining on fibers. A representative example of the DM, normal, PM, and DMD samples is shown.
Figure 2.
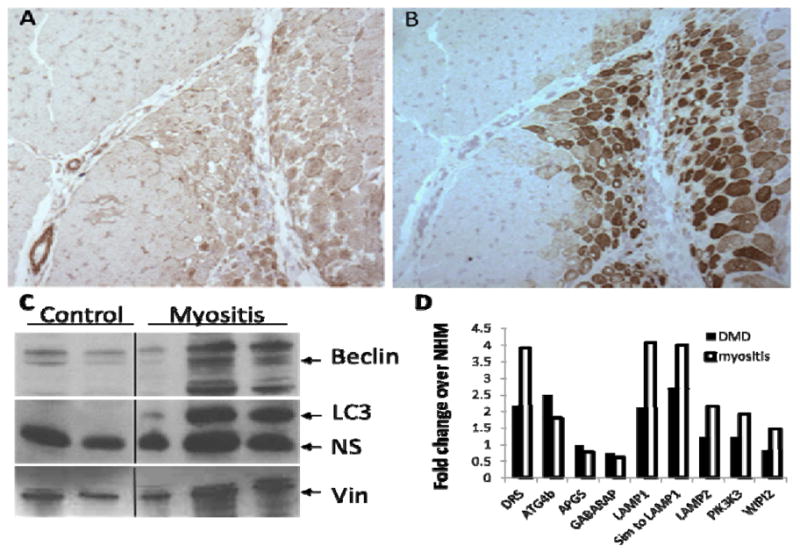
TRAIL expression is localized to perifascicular atrophic and regenerating areas of DM muscle. Serial sections from DM muscle biopsies were stained with antibodies against TRAIL. (A) (B) TRAIL is colocalized with developmental myosin heavy chain (dMHC) in perifascicular region. (C) Control and myositis human muscle homogenates were probed with antibodies against Beclin (top) or LC3 (middle) and Vinculin (bottom) expected sizes marked with arrows. (D) Differentially expressed autophagy pathway genes from myositis and DMD patients were compared to normal human muscle (NHM). All gene differences reported were statistically significant after multiple testing corrections. Data expressed as -fold change.
Markers of autophagy are upregulated in human myositis muscle biopsies and the MHC class I mouse model of myositis (HT)
Since classical apoptosis is not observed in muscle fibers from myositis patients (11–13, 24–26), we analyzed normal and myositis muscle biopsies for the presence of specific autophagic markers, Beclin and LC3 (Figure 2C). Both proteins were significantly higher in myositis patients than in controls. In addition, gene expression profiling revealed that autophagy-related Atg4b and LAMP mRNA were up-regulated in muscle biopsies from myositis patients, indicating an activation of the autophagic pathway (Figure 2D). Likewise, the expression of DR5, the receptor for TRAIL, was several-fold higher in myositis and DMD muscle biopsies than in NHM (Figure 2D).
Since the HT mouse model of myositis recapitulates several features of human myositis, we performed similar analyses in these mice. HT muscles displayed autophagy-related characteristics similar to those of human myositis muscle. Protein and mRNA from the muscle biopsies were probed for the autophagic markers Beclin and LC3; both were seen to be up-regulated in fast-twitch myositis muscle (Figure 3D). As was seen for the human biopsies (Figure 2C), mouse HT muscle tissues had a higher expression of Beclin and LC3 proteins than did the control mouse muscle, as measured by immunoblot assay (Figure 3A). These data indicate that the autophagic cell death pathway is active in both human patients and HT mice.
Figure 3.

Autophagic markers, Beclin and LC3, are up-regulated in HT myositis mice. Western blotting (A); Autophagic pathway genes expression at protein (A) and mRNA level (B) (n=3 mice/group/muscle type). (C) Electron microscopy (quadriceps) Black arrows and open arrowhead indicate double-membrane autophagic vacuoles and multilamellar structures, respectively. Open arrows and black arrowhead indicate cytoplasmic granules and small dense bodies, respectively. Sizing bar, 1 μm. (D) Activated LC3-Gastroc 1 (control) and Gastroc -2 (HT); LC3 (green) and GM130 (red–Golgi marker) Gastroc 3 and soleus-4. GM130 and LAMP (green–lysosomal marker) co-localized in HT gastrocnemius muscle (5). Control and HT gastrocnemius muscles with LAMP (green) and α-tubulin (red) (6–7).
Because Type II (fast-twitch) skeletal muscle is more drastically affected in human myositis, we used gene array analysis of HT and control mouse muscle to determine whether there is differential expression of autophagy-related genes in type I (slow-twitch) and type II (fast-twitch) muscle. Gene expression profiling analysis of gastrocnemius (Type II) and soleus (Type I) muscle revealed that several autophagy-related genes had a several-fold higher level of expression in HT muscle (slow- and fast-twitch) than in the muscles of control mice: Atg4b, Atg5, Atg7, Gabarap, Gabarapl1, Map1lc3a, Pik3c3, Pik3r4, Rab24, Ulk2, and Wipi (Figure 3B and Supplemental Table 1). Some of the autophagy-related genes showed preferential up-regulation in gastrocnemius muscle, suggesting that Type II muscle may be more susceptible than Type I muscle to autophagic cell death.
Autophagic markers are predominantly present on Type II muscle of HT mice
Muscle (gastrocnemius and/or soleus) tissue was collected from control and HT mice. Gastrocnemius muscles were stained for LC3, to determine the localization of LC3 and characterize autophagosome formation. Control mouse muscle fibers showed minimal LC3 staining along the length of the tissue (Figure 3D1), whereas the muscle of HT mice displayed a punctate, striated expression pattern for LC3 (Figure 3D2).
Since the slow and fast muscle fibers are differentially affected in the HT mice, we performed a comparative analysis of LC3 expression by immunofluorescent staining in gastrocnemius and soleus muscle to determine the approximate subcellular localization and muscle type specificity of LC3 in the HT muscle. LC3 co-localized with GM 130 (a cis-Golgi matrix marker) in the gastrocnemius and soleus muscles; when the images were merged (Figure 3D3 and 3D4), we saw overlapping expression of the intensely stained LC3 (green, presence of autophagy) and GM 130 (red, Golgi localization) in the gastrocnemius, whereas the soleus displayed minimal or no expression of LC3 or co-localization with GM 130. These data suggest that autophagic vacuoles are highly abundant in fast fibers but absent from the slow muscle of the HT mouse.
The subcellular localization of LC3 and GM 130 was further investigated by examining its potential co-localization with LAMP, in order to further define the autophagic processes that occur in HT mice. LAMP is required for the fusion of autophagosomes and lysosomes during autophagic progression. In myositis muscle, LAMP (green) co-localized with GM 130 (red) (Figure 3D5). Gastrocnemius muscles from control and HT mice were stained for the presence of LAMP and α-tubulin. In control mice, some LAMP expression was detected; however it did not overlap substantially with α-tubulin expression (Figure 3D6). In HT mice, LAMP expression was higher and was co-localized with tube-like staining of α-tubulin, as seen in the merged panel (Figure 3D7). Also, the muscle α-tubulin expression patterns differed between control and HT mice (red staining in Figure 3D6 vs. 3D7). HT gastronemius muscle clearly showed a disruption and rearrangement of the microtubules (α-tubulin staining) into longitudinal tunnels that co-localized with LAMP staining (Figure 3D7). These findings demonstrate a co-localization of LAMP, α-tubulin, the Golgi marker GM 130, and LC3 (Figure 3D) in the HT muscle (individual, non-merged images for each are available in Supplemental Figure 2), suggesting activation of the autophagic pathway in the type II muscle of HT mice.
Autophagic vesicles are present in electron micrographs of HT muscle
Autophagy in HT myositis muscle was further validated by electron microscopy (EM) of the quadriceps muscle (Figure 3C). Ultrathin sections from HT muscles were negatively stained with uranyl acetate and lead citrate to visualize the features of classic autophagic vesicles by electron microscopy. Several previously described features of autophagy (27, 28) were noted in the quadriceps muscle: Typical double-membrane autophagic vacuoles could be identified in muscle (black arrows). Multilamellar structures (open arrowhead) with heterogenous cytoplasmic contents (open arrows) were also present. The small densely stained bodies could represent glycogen granules or lysosomal fragments in the autophagic areas (black arrowhead). Thus, these data clearly point to a role for autophagic cell death in myositis skeletal muscle.
Significant variation occurs in the muscle fibers of HT mice
To assess the consequences of autophagic cell death in myositis muscle, we examined muscle samples from control and HT mice (three control and three HT mice) after hematoxylin and eosin (H&E) staining. HT muscle showed endomysial infiltration and fibrosis, centralized nuclei, and variation in fiber size (Figure 4A and 4B). Images were randomly selected from non-overlapping sections of muscle (2–5 images per sample), and individual muscle fibers were measured using ImageJ software (National Institutes of Health, Bethesda, MD) to determine: (1) the number of cells per panel (Figure 4C1), (2) the cross-sectional area of the individual fibers (Figure 4C2), and (3) Feret’s minimal diameter of the fibers (Figure 4C3). There were no statistically significant differences in the average number of cells per panel or the average cell area between the control and HT samples. As illustrated in Figures 4A and 4B, HT samples showed a broad variability in the number of cells per panel, ranging from <50 cells to >170 cells per non-overlapping image, whereas the number of cells per panel in the normal images did not exceed 110 (Figure 4C1). Likewise, in affected mice, the individual cellular areas ranged from <2000 nm2 to >4000 nm2, whereas the normal muscle cellular area peaked at 3001–3500 nm2 (Figure 4C2), suggesting that the muscle fibers in affected tissue are much smaller than in healthy control tissue. Consistent with the cell images seen in the representative H&E samples, the average Feret’s minimal diameter for the HT samples was statistically significantly lower than for normal myofibers (Figure 4C3), indicating that some form of cell death had occurred in these muscle fibers.
Figure 4.
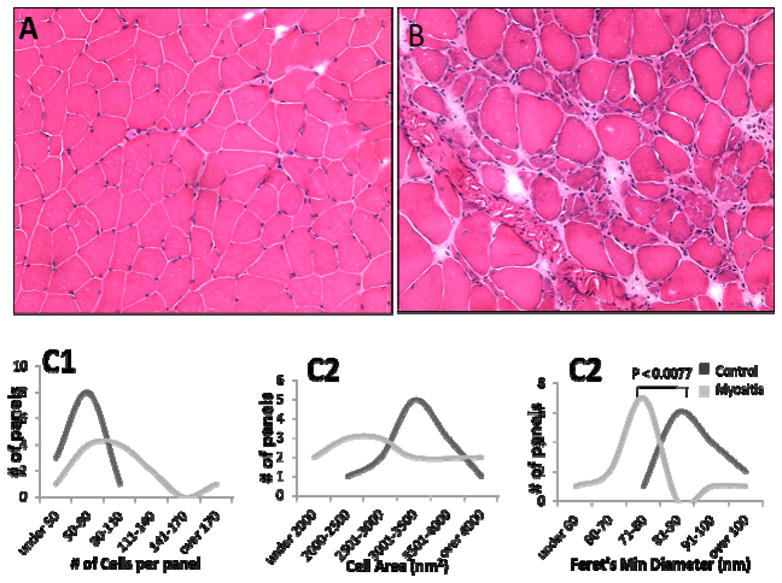
H&E staining of muscle of HT myositis mice: Representative sections of non-diseased mouse control (A) and HT mouse (B) muscle sections are shown (3 mice/genotype). Five panels per stained section were used to measure Cell count per panel (C1), cross-sectional area (C2) and Feret’s minimal diameter (C3). Cellular measurements were determined using ImageJ software downloaded from the NIH (http://rsbweb.nih.gov/ij/).
Addition of exogenous recombinant TRAIL (rTRAIL) induces NFkB activation and autophagy but not caspase-3-dependent apoptosis in muscle cells
Incubation of cells with human rTRAIL efficiently induced NFκB activation, as shown by the nuclear translocation of NFkB p65 and IκB degradation. This activation was efficiently blocked by IKK inhibitor in a dose-dependent manner (Supplementary Figure 3 A & B). Poly (ADP-ribose) polymerase (PARP) cleavage is a classical marker of caspase-3 activation and cell death in multiple cell types. Intact PARP is approximately 116 kDa, whereas activated, cleaved PARP is approximately 85 kDa (Figure 5A). rTRAIL-induced PARP cleavage was monitored in HSG and SKMC. In the absence of rTRAIL, PARP remained intact in both HSG and SKMC cells (Lanes 1 and 5). With increased addition of rTRAIL, PARP cleavage was increased in HSG, but not in SKMC, suggesting that the caspase-3 apoptotic pathway was not activated in SKMC. The effect of rTRAIL on induction of autophagy was then investigated in SKMC. Monodansylcadaverin (MDC) is a lysosomotrophic agent that marks autophagic vesicles in cells. SKMC clearly showed more autophagic vesicles upon rTRAIL treatment in these cells, suggesting that TRAIL-induced autophagy, not apoptosis, occurs in skeletal muscle cells (Figure 5B).
Figure 5.
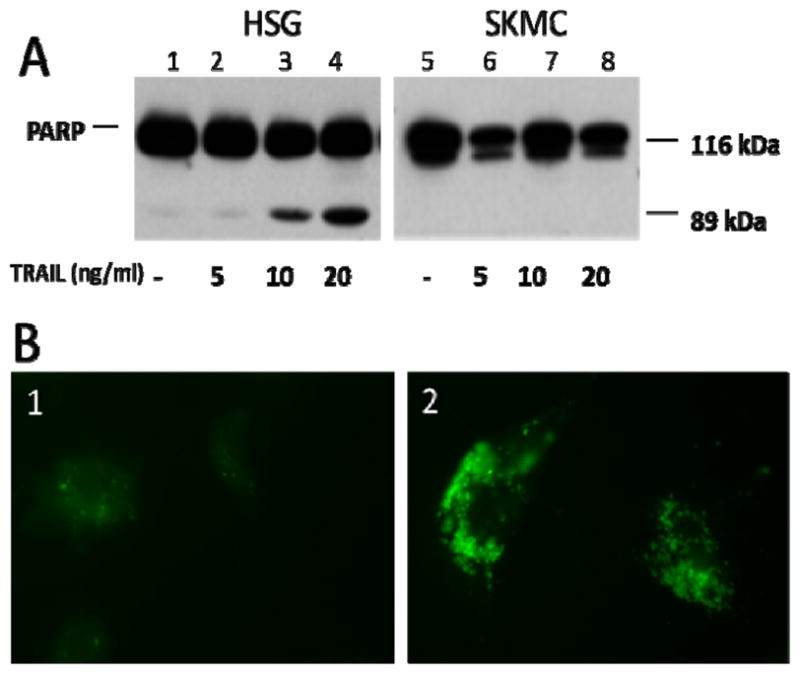
TRAIL induces autophagy in skeletal muscle cells, but caspase-3-dependent apoptosis is absent. (A) Human salivary gland cells (HSG) and human skeletal muscle cells (SKMC) were incubated with rTRAIL protein (0, 5, 10, and 20 ng/mL). HSG cells had higher levels of cleaved PARP (Lanes 1–4), whereas SKMC cells were resistant to PARP cleavage even at the highest concentration of TRAIL (Lanes 5–8). (B)Autophagy was monitored in SKMC after rTRAIL addition. B1/2, monodansyl cadaverine (MDC), a marker of autophagic vacuoles. Addition of rTRAIL (B2) to SKMC resulted in the formation of many vesicles and a larger cell size than seen in SKMC exposed to media only (B1). Representative images are shown.
Discussion
In this study, we have addressed the role of TRAIL in mediating muscle fiber damage in myositis. We have shown that TRAIL is expressed in PM and DM biopsies but not in normal control muscle or in other myopathic conditions such as Duchenne muscular dystrophy. We have also shown that TRAIL is expressed in perifasicular atrophic areas of DM muscle where there is muscle atrophy/degeneration coupled with muscle regeneration, as indicated by the positivity for dMHC. We have further shown that the TRAIL receptor DR5 is coordinately expressed together with several genes that are more prominently expressed in myositis skeletal muscle than in normal muscle and also confirmed the expression of two of autophagic genes (Beclin and LC-3) at the protein level in myositis but not control biopsies. These observations have been further confirmed in the MHC class I transgenic mouse model of myositis, in which we found atrophic muscle fibers, together with up-regulation of the autophagic markers Beclin and LC-3 and the presence of typical autophagic vesicles by electron microscopy. In the mouse model, autophagy appeared to be more prominent in the fast-twitch Type II skeletal muscle than in the slow-twitch Type I muscle. Finally, using in vitro cell culture experiments, we demonstrated that TRAIL induces autophagy and NFkB activation but not apoptosis in skeletal muscle cells, suggesting a potential role for TRAIL in mediating muscle fiber death(via autophagy)in myositis.
TRAIL expression has been previously demonstrated in biopsies from juvenile myositis patients, and TRAIL is known to be involved in autophagic tissue remodeling (15, 16). By analyzing publicly available microarray data, we found that TRAIL was also specifically up-regulated in juvenile myositis muscle but not in other myopathic patients’ muscle biopsies. Gene expression was analyzed in mRNA samples from muscle biopsies of various myopathic patients and normal human controls, and despite the fact that several of the myopathies share characteristic features (muscle inflammation and fibrosis), only muscle from juvenile DM patients had distinctly higher TRAIL expression (Figure 1 and Supplemental Figure 1). This finding suggested that the muscle fiber damage that is seen in myositis patients may occur through a TRAIL-mediated mechanism, dissimilar to that seen in other myopathies.
Atrophy and degeneration/regeneration are among the histological features of several myopathies, including myositis. The atrophy is uniquely localized to perifascicular area in DM. Perifascicular regions of myositis muscle express higher amounts of autoantigens and MHC class I; these sites are highly vulnerable to cytotoxic T-cell-mediated damage, resulting in muscle fiber degeneration and atrophy (29, 30). These perifascicular regions stain positively for dMHC, indicating active degeneration and regeneration cycles.
TRAIL/DR5 activation can activate apoptotic cell death (Type I cell death) through the Fas-associated protein with death domain (FADD)/caspase pathway (31, 32). TRAIL/DR5 activation can also stimulate a non-apoptotic autophagic cell death (Type II cell death) pathway through NFκB and tissue remodeling (15, 33, 34). Previous studies in human muscle biopsies have shown that DR5, DR4, and Fas are expressed in normal muscle tissue and are up-regulated in DM muscle biopsies (16). Our data from the current study have shown that in myositis muscle, TRAIL and DR5 are both upregulated, as are the genes in the autophagic pathway, not only in humans but also in the mouse myositis model. The molecular pathways that connect TRAIL-induced autophagy to inflammation (NFkB activation) are not yet fully understood. However, there is some evidence that TRAIL and TRAIL receptor-induced NFkB activation is mediated by TRAF2-NIK-I-kappaB kinase alpha/beta signaling and that autophagy is mediated by JNK activation via TRAF2-MEKK1-MKK4, suggesting that TRAIL receptors induce apoptosis, inflammation, and autophagy through distinct signaling pathways (35, 36). Activation of NFkB in skeletal muscle has a number of consequences, including the production of pro-inflammatory cytokines, initiation of ER stress (ER overload and Unfolded protein response), and suppression of new muscle formation via myoD inhibition. Our data have not demonstrated that autophagy causes inflammation, but there is emerging evidence in the literature to suggest that autophagy controls the cellular inflammatory response via IFN-gamma-induced signaling (Jak2-STAT1 activation)(37).
Autophagy can be activated in response to the accumulation of misfolded or improperly glycosylated proteins, which can trigger an endoplasmic reticulum (ER) stress response and activate autophagy (38, 39). In response to autophagic activation, a cascade of protein mediators is mobilized to form the autophagosome, which fuses with the lysosome to cause the concerted destruction of cellular organelles and macromolecules by lysosomal hydrolytic enzymes (39). Several of the genes identified in this paper have a well-defined role in this process. Apg5 and Apg12 are localized to autophagosome membranes (40) and recruit LC3 (also known as Atg8) to the autophagosome, whereas LAMP1 and LAMP2 are the major glycoproteins localized between the lysosomal and plasma membranes (41).
TRAIL-mediated cell death occurring via the autophagic pathway has previously been shown to be protective, particularly in apoptosis-defective tumor cells (33) and in tissue morphogenesis and remodeling, in which TRAIL has been shown to play a critical role in cellular activation (15). In myositis, TRAIL could initiate autophagic pathways not only to mediate cell death but also to facilitate the fusion of regenerating muscle fibers. The identification of high levels of TRAIL expression in regenerating areas in DM muscle supports such a role in myositis. Consistent with a role for TRAIL in caspase-independent cell death is the up-regulation of markers of autophagy in both myositis patients and HT mouse muscle, but not in control muscle. Beclin and LC3 are specific markers of autophagic cell death (33, 36, 42). Beclin is targeted to the trans-Golgi network and induces the phosphorylation of Bcl-2 and p53 in autophagic cell death through a JNK-mediated pathway (34, 36, 43), whereas the activation and lipidation of LC3 (LC3II) are markers for autophagosome formation (44). Staining of muscle fibers revealed that LC3 was prominently colocalized with the Golgi marker GM 130 in the fast-twitch HT muscle (Figure 3D2/3) but absent from the type I and control mouse muscle (Figure 3D1/4). This differential expression of LC3 between muscle types is interesting, but it is currently unclear whether this difference reflects differential distribution of the ER-Golgi network in type I and type II muscle fibers or differential sensitivities to TRAIL-mediated damage (45). The restriction of LC3 expression to fast-twitch muscles could be partially explained by previous studies that have suggested that cell damage is more prominent in Type II than in Type I muscles in myositis (3).
Variation in fiber size is one of the typical features of human myositis skeletal muscle. Our analysis of muscle fiber size, cell area, and Feret’s minimum diameter clearly indicated that myositis skeletal muscle also shows significant variation in fiber size, indicating an active degeneration and regeneration process. These data indicate that autophagic death is active and may contribute to the atrophy and variation in fiber size in the skeletal muscle of myositis as a non-immune form of muscle damage.
From an analysis of muscle tissues (human or mouse), it is difficult judge whether TRAIL has a causative role in inducing inflammation and autophagy in myositis; therefore, we also performed in vitro cell culture experiments, which suggested that TRAIL induces both inflammation (NFkB activation) and autophagy in cells. We speculate that TRAIL-mediated activation of NFκB could lead to an ER overload response/ER stress, which could in turn result in enhanced muscle inflammation and propagation of muscle disease. Our current and previously published data indicate potential for a significant non-immune component (autophagy and ER stress) in the pathogenesis of inflammatory myopathies (1, 2, 46). Future genetic experiments using TRAIL knockout mice or pharmacological interventions (anti-TRAIL or -DR5) antibodies should clarify the role of TRAIL in muscle fiber damage in myositis.
Supplementary Material
Supplemental Figure 1: TRAIL mRNA expression is up-regulated in juvenile dermatomyositis muscle. Expression of TRAIL mRNA in muscle biopsies was analyzed. As compared to control muscles, there were no major differences in the expression of TRAIL in any of the muscle biopsies, except for juvenile dermatomyositis muscle; the expression of TRAIL was statistically higher than for all other muscle biopsy samples. ALS, amyotrophic lateral sclerosis; AQM, cute quadriplegic myopathy; BMD, Becker muscular dystrophy; Lamin, laminin 2α deficiency/limb girdle muscular dystrophy (LGMD); Cal-3, calpain deficiency/LGMD Type 2a; DMD, Duchenne muscular dystrophy; Dys, dysferlin deficiency/LGMD Type 2b; Emerin, Emery-Dreifuss muscular dystrophy; FKRP, fukutin-related protein muscular dystrophy; FSHD, fascioscapulohumeral muscular dystrophy; HSP, Hereditary Spastic paraplegia (HSP); JDM, juvenile dermatomyositis; control, normal human muscle. Numbers in parenthesis indicate the number of muscle biopsies (47).
Supplemental Figure 2: Muscle biopsies from control and HT mice were immunofluorescently stained with antibodies recognizing markers of autophagy (LC3), Golgi (GM 130), autophagosome: lyososome fusion (LAMP), and microtubules (α-tubulin). (A) Gastrocnemius (Type II fast-twitch) and soleus (Type I slow-twitch) muscle from HT mice were stained to determine the differential expression of LC3 (green) between muscle types and to demonstrate co-localization with the Golgi with GM 130 (red) staining. LC3 and GM 130 were expressed and co-localized in the gastrocnemius, but virtually absent in the soleus. (B) The Golgi marker GM130, which co-localized with the autophagic LC3 marker, was then immunostained with LAMP, which is a marker for the fusion of autophagosomes and lysosomes during the autophagic process. GM130 (green) and LAMP (red) were co-localized in the striated ribbons of the HT gastrocnemius. (C) Control and HT gastrocnemius muscle biopsies were stained for LAMP (green) and α-tubulin (red) to illustrate microtubule structural integrity and colocalization of LAMP with α-tubulin in sections of the HT gastrocnemius. The structural integrity of the myositis muscle had been compromised, as indicated by the α-tubulin staining.
Supplementary Figure 3: TRAIL induces NFkB nuclear translocation and I-kB alpha degradation. Cells were cultured either in 6-cm dishes or dishes containing coverslips for 2 days at 37°C. Sub-confluent cells were washed with medium and incubated with different concentrations of the IKK inhibitor (0, 25, 2.5 and 25 uM) for 60 min before the addition of recombinant human TRAIL (10 ng/ml). Cells were collected at 15, 30, and 60 min after treatment and analyzed by indirect immunofluorescent staining and western blotting. (A). Western blotting: Protein concentration was determined with a colorimetric assay (Bio-Rad). Proteins were separated by SDS-page, transferred to a nylon membrane, and probed with rabbit anti-I-kB alpha antibody and detected with an anti-rabbit serum coupled to horseradish peroxidase (Santa Cruz Biotechnology), then developed by using an enhanced chemiluminescence system(Pierce). Anti-vinculin antibodies were used to assess protein loading. (B) Immunofluorescent staining: Cells were washed with PBS and fixed with 5% paraformaldehyde in PBS at 37°C for 15 min. After being blocked, cells were incubated with anti-NFkB antibody (Santa Cruz Biotechnology) in PBS plus 1% BSA for 2 h. The coverslips were developed with goat anti-rabbit IgG labeled with Texas red (Molecular Probes) and counterstained with DAPI to stain nuclei. Coverslips were mounted in ProLong antifade (Molecular Probes) and analyzedin a fluorescent microscope (Zeiss).
Supplementary Figure 4: DR5 is expressed in human myositis muscle. Tissue sections (n=4 per disease group) from polymyositis patients were stained for the presence of DR5 by immunohistochemical staining. Diffuse DR5 staining was seen in the myositis muscle (A), which showed no staining when the primary antibody was replaced by an isotype control (B). A representative example of the polymyositis muscle is shown.
Acknowledgments
The authors wish to acknowledge Megan Mistak for her valuable technical skills and contributions to this manuscript and Dr. Deborah McClellan for editorial assistance. Dr. Nagaraju is supported by the National Institutes of Health (RO1-AR050478 and 5U54HD053177). Y-W Chen is partially supported by NIH/NIAMS 1 R01 AR052027-01A2, NIH/NICHD 1 U54 HD053177-01A1, and NIH/NICHD 1R24HD050846-01. Dr. Lundberg is supported by The Myositis Association and Association Francaise contre les Myopathies.
Abbreviations used in this manuscript
- CHX
cyclohexamide
- DM
dermatomyositis
- DMD
Duchenne muscular dystrophy
- dMHC
developmental myosin heavy chain
- DR#
death receptor-#
- EDL
extensor digitorum longus
- HSG
human salivary gland
- HT
myositis (in the double-transgenic myositis model)
- IIM
idiopathic inflammatory myopathies
- Iκβ
inhibitor of kappaB-α
- IKKI
Iκβ kinase inhibitor
- LAMP
lysosome-associated membrane protein
- NFκB
nuclear factor kappa-light chain enhancer of activated B cells
- NHM
normal human muscle
- PM
polymyositis
- PARP
poly(ADP-ribose) polymerase
- SKMC
human skeletal muscle cells
- TNFα
tumor necrosis factor-α
- TRAIL
tumor necrosis factor-α-related apoptosis inducing ligand
References
- 1.Nagaraju K, Casciola-Rosen L, Lundberg I, Rawat R, Cutting S, Thapliyal R, et al. Activation of the endoplasmic reticulum stress response in autoimmune myositis: potential role in muscle fiber damage and dysfunction. Arthritis Rheum. 2005;52(6):1824–35. doi: 10.1002/art.21103. [DOI] [PubMed] [Google Scholar]
- 2.Henriques-Pons A, Nagaraju K. Nonimmune mechanisms of muscle damage in myositis: role of the endoplasmic reticulum stress response and autophagy in the disease pathogenesis. Curr Opin Rheumatol. 2009;21(6):581–7. doi: 10.1097/BOR.0b013e3283319265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dastmalchi M, Alexanderson H, Loell I, Stahlberg M, Borg K, Lundberg IE, et al. Effect of physical training on the proportion of slow-twitch type I muscle fibers, a novel nonimmune-mediated mechanism for muscle impairment in polymyositis or dermatomyositis. Arthritis Rheum. 2007;57(7):1303–10. doi: 10.1002/art.22996. [DOI] [PubMed] [Google Scholar]
- 4.Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16(6):663–9. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 5.Fukuda T, Ewan L, Bauer M, Mattaliano RJ, Zaal K, Ralston E, et al. Dysfunction of endocytic and autophagic pathways in a lysosomal storage disease. Ann Neurol. 2006;59(4):700–8. doi: 10.1002/ana.20807. [DOI] [PubMed] [Google Scholar]
- 6.Bursch W, Karwan A, Mayer M, Dornetshuber J, Frohwein U, Schulte-Hermann R, et al. Cell death and autophagy: cytokines, drugs, and nutritional factors. Toxicology. 2008;254(3):147–57. doi: 10.1016/j.tox.2008.07.048. [DOI] [PubMed] [Google Scholar]
- 7.Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science. 1995;270(5239):1189–92. doi: 10.1126/science.270.5239.1189. [DOI] [PubMed] [Google Scholar]
- 8.Tartaglia LA, Rothe M, Hu YF, Goeddel DV. Tumor necrosis factor’s cytotoxic activity is signaled by the p55 TNF receptor. Cell. 1993;73(2):213–6. doi: 10.1016/0092-8674(93)90222-c. [DOI] [PubMed] [Google Scholar]
- 9.Pan G, Ni J, Wei YF, Yu G, Gentz R, Dixit VM. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science. 1997;277(5327):815–8. doi: 10.1126/science.277.5327.815. [DOI] [PubMed] [Google Scholar]
- 10.Los M, Gibson SB. Apoptotic pathways as targets for novel therapies in cancer & other diseases. New York: Springer; 2005. [Google Scholar]
- 11.Nagaraju K, Casciola-Rosen L, Rosen A, Thompson C, Loeffler L, Parker T, et al. The inhibition of apoptosis in myositis and in normal muscle cells. J Immunol. 2000;164(10):5459–65. doi: 10.4049/jimmunol.164.10.5459. [DOI] [PubMed] [Google Scholar]
- 12.Schneider C, Gold R, Dalakas MC, Schmied M, Lassmann H, Toyka KV, et al. MHC class I-mediated cytotoxicity does not induce apoptosis in muscle fibers nor in inflammatory T cells: studies in patients with polymyositis, dermatomyositis, and inclusion body myositis. J Neuropathol Exp Neurol. 1996;55(12):1205–9. doi: 10.1097/00005072-199612000-00003. [DOI] [PubMed] [Google Scholar]
- 13.Tews DS, Goebel HH. Cell death and oxidative damage in inflammatory myopathies. Clin Immunol Immunopathol. 1998;87(3):240–7. doi: 10.1006/clin.1998.4527. [DOI] [PubMed] [Google Scholar]
- 14.Li M, Dalakas MC. Expression of human IAP-like protein in skeletal muscle: a possible explanation for the rare incidence of muscle fiber apoptosis in T-cell mediated inflammatory myopathies. J Neuroimmunol. 2000;106(1–2):1–5. doi: 10.1016/s0165-5728(99)00162-9. [DOI] [PubMed] [Google Scholar]
- 15.Mills KR, Reginato M, Debnath J, Queenan B, Brugge JS. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is required for induction of autophagy during lumen formation in vitro. Proc Natl Acad Sci U S A. 2004;101(10):3438–43. doi: 10.1073/pnas.0400443101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao Y, Fedczyna TO, McVicker V, Caliendo J, Li H, Pachman LM. Apoptosis in the skeletal muscle of untreated children with juvenile dermatomyositis: impact of duration of untreated disease. Clin Immunol. 2007;125(2):165–72. doi: 10.1016/j.clim.2007.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tezak Z, Hoffman EP, Lutz JL, Fedczyna TO, Stephan D, Bremer EG, et al. Gene expression profiling in DQA1*0501+ children with untreated dermatomyositis: a novel model of pathogenesis. J Immunol. 2002;168(8):4154–63. doi: 10.4049/jimmunol.168.8.4154. [DOI] [PubMed] [Google Scholar]
- 18.Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, et al. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3(6):673–82. doi: 10.1016/1074-7613(95)90057-8. [DOI] [PubMed] [Google Scholar]
- 19.Kim YS, Schwabe RF, Qian T, Lemasters JJ, Brenner DA. TRAIL-mediated apoptosis requires NF-kappaB inhibition and the mitochondrial permeability transition in human hepatoma cells. Hepatology. 2002;36(6):1498–508. doi: 10.1053/jhep.2002.36942. [DOI] [PubMed] [Google Scholar]
- 20.Bohan A, Peter JB. Polymyositis and dermatomyositis (first of two parts) N Engl J Med. 1975;292(7):344–7. doi: 10.1056/NEJM197502132920706. [DOI] [PubMed] [Google Scholar]
- 21.Nagaraju K, Raben N, Loeffler L, Parker T, Rochon PJ, Lee E, et al. Conditional up-regulation of MHC class I in skeletal muscle leads to self-sustaining autoimmune myositis and myositis-specific autoantibodies. Proc Natl Acad Sci U S A. 2000;97(16):9209–14. doi: 10.1073/pnas.97.16.9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raben N, Shea L, Hill V, Plotz P. Monitoring autophagy in lysosomal storage disorders. Methods Enzymol. 2009;453:417–49. doi: 10.1016/S0076-6879(08)04021-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lamason R, Zhao P, Rawat R, Davis A, Hall JC, Chae JJ, et al. Sexual dimorphism in immune response genes as a function of puberty. BMC Immunol. 2006;7:2. doi: 10.1186/1471-2172-7-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Behrens L, Bender A, Johnson MA, Hohlfeld R. Cytotoxic mechanisms in inflammatory myopathies. Co-expression of Fas and protective Bcl-2 in muscle fibres and inflammatory cells. Brain. 1997;120 (Pt 6):929–38. doi: 10.1093/brain/120.6.929. [DOI] [PubMed] [Google Scholar]
- 25.Inukai A, Kobayashi Y, Ito K, Doyu M, Takano A, Honda H, et al. Expression of Fas antigen is not associated with apoptosis in human myopathies. Muscle Nerve. 1997;20(6):702–9. doi: 10.1002/(sici)1097-4598(199706)20:6<702::aid-mus7>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 26.Olive M, Martinez-Matos JA, Montero J, Ferrer I. Apoptosis is not the mechanism of cell death of muscle fibers in human muscular dystrophies and inflammatory myopathies. Muscle Nerve. 1997;20(10):1328–30. doi: 10.1002/(sici)1097-4598(199710)20:10<1328::aid-mus20>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 27.Engel A, Franzini-Armstrong C. Myology: basic and clinical. 3. New York: McGraw-Hill, Medical Pub. Division; 2004. [Google Scholar]
- 28.Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol. 2005;64(2):113–22. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 29.Nevo Y, Pestronk A. Neonatal perifascicular myopathy. Pediatr Neurol. 1996;15(2):150–2. doi: 10.1016/0887-8994(96)00127-0. [DOI] [PubMed] [Google Scholar]
- 30.Casciola-Rosen L, Nagaraju K, Plotz P, Wang K, Levine S, Gabrielson E, et al. Enhanced autoantigen expression in regenerating muscle cells in idiopathic inflammatory myopathy. J Exp Med. 2005;201(4):591–601. doi: 10.1084/jem.20041367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ichikawa K, Liu W, Fleck M, Zhang H, Zhao L, Ohtsuka T, et al. TRAIL-R2 (DR5) mediates apoptosis of synovial fibroblasts in rheumatoid arthritis. J Immunol. 2003;171(2):1061–9. doi: 10.4049/jimmunol.171.2.1061. [DOI] [PubMed] [Google Scholar]
- 32.Schneider P, Thome M, Burns K, Bodmer JL, Hofmann K, Kataoka T, et al. TRAIL receptors 1 (DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate NF-kappaB. Immunity. 1997;7(6):831–6. doi: 10.1016/s1074-7613(00)80401-x. [DOI] [PubMed] [Google Scholar]
- 33.Han J, Hou W, Goldstein LA, Lu C, Stolz DB, Yin XM, et al. Involvement of protective autophagy in TRAIL resistance of apoptosis-defective tumor cells. J Biol Chem. 2008;283(28):19665–77. doi: 10.1074/jbc.M710169200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Park KJ, Lee SH, Kim TI, Lee HW, Lee CH, Kim EH, et al. A human scFv antibody against TRAIL receptor 2 induces autophagic cell death in both TRAIL-sensitive and TRAIL-resistant cancer cells. Cancer Res. 2007;67(15):7327–34. doi: 10.1158/0008-5472.CAN-06-4766. [DOI] [PubMed] [Google Scholar]
- 35.Hu WH, Johnson H, Shu HB. Tumor necrosis factor-related apoptosis-inducing ligand receptors signal NF-kappaB and JNK activation and apoptosis through distinct pathways. J Biol Chem. 1999;274(43):30603–10. doi: 10.1074/jbc.274.43.30603. [DOI] [PubMed] [Google Scholar]
- 36.Park KJ, Lee SH, Lee CH, Jang JY, Chung J, Kwon MH, et al. Upregulation of Beclin-1 expression and phosphorylation of Bcl-2 and p53 are involved in the JNK-mediated autophagic cell death. Biochem Biophys Res Commun. 2009;382(4):726–9. doi: 10.1016/j.bbrc.2009.03.095. [DOI] [PubMed] [Google Scholar]
- 37.Chang YP, Tsai CC, Huang WC, Wang CY, Chen CL, Lin YS, et al. Autophagy facilitates IFN-gamma-induced Jak2-STAT1 activation and cellular inflammation. J Biol Chem. 285(37):28715–22. doi: 10.1074/jbc.M110.133355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26(24):9220–31. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem. 2006;281(40):30299–304. doi: 10.1074/jbc.M607007200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, et al. Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001;152(4):657–68. doi: 10.1083/jcb.152.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eskelinen EL. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Aspects Med. 2006;27(5–6):495–502. doi: 10.1016/j.mam.2006.08.005. [DOI] [PubMed] [Google Scholar]
- 42.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, et al. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19(21):5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mijaljica D, Prescott M, Devenish RJ. Endoplasmic reticulum and Golgi complex: Contributions to, and turnover by, autophagy. Traffic. 2006;7(12):1590–5. doi: 10.1111/j.1600-0854.2006.00495.x. [DOI] [PubMed] [Google Scholar]
- 44.Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004;36(12):2503–18. doi: 10.1016/j.biocel.2004.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ralston E, Lu Z, Ploug T. The organization of the Golgi complex and microtubules in skeletal muscle is fiber type-dependent. J Neurosci. 1999;19(24):10694–705. doi: 10.1523/JNEUROSCI.19-24-10694.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li CK, Knopp P, Moncrieffe H, Singh B, Shah S, Nagaraju K, et al. Overexpression of MHC Class I Heavy Chain Protein in Young Skeletal Muscle Leads to Severe Myositis. Implications for Juvenile Myositis. Am J Pathol. 2009 doi: 10.2353/ajpath.2009.090196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bakay M, Wang Z, Melcon G, Schiltz L, Xuan J, Zhao P, et al. Nuclear envelope dystrophies show a transcriptional fingerprint suggesting disruption of Rb-MyoD pathways in muscle regeneration. Brain. 2006;129(Pt 4):996–1013. doi: 10.1093/brain/awl023. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: TRAIL mRNA expression is up-regulated in juvenile dermatomyositis muscle. Expression of TRAIL mRNA in muscle biopsies was analyzed. As compared to control muscles, there were no major differences in the expression of TRAIL in any of the muscle biopsies, except for juvenile dermatomyositis muscle; the expression of TRAIL was statistically higher than for all other muscle biopsy samples. ALS, amyotrophic lateral sclerosis; AQM, cute quadriplegic myopathy; BMD, Becker muscular dystrophy; Lamin, laminin 2α deficiency/limb girdle muscular dystrophy (LGMD); Cal-3, calpain deficiency/LGMD Type 2a; DMD, Duchenne muscular dystrophy; Dys, dysferlin deficiency/LGMD Type 2b; Emerin, Emery-Dreifuss muscular dystrophy; FKRP, fukutin-related protein muscular dystrophy; FSHD, fascioscapulohumeral muscular dystrophy; HSP, Hereditary Spastic paraplegia (HSP); JDM, juvenile dermatomyositis; control, normal human muscle. Numbers in parenthesis indicate the number of muscle biopsies (47).
Supplemental Figure 2: Muscle biopsies from control and HT mice were immunofluorescently stained with antibodies recognizing markers of autophagy (LC3), Golgi (GM 130), autophagosome: lyososome fusion (LAMP), and microtubules (α-tubulin). (A) Gastrocnemius (Type II fast-twitch) and soleus (Type I slow-twitch) muscle from HT mice were stained to determine the differential expression of LC3 (green) between muscle types and to demonstrate co-localization with the Golgi with GM 130 (red) staining. LC3 and GM 130 were expressed and co-localized in the gastrocnemius, but virtually absent in the soleus. (B) The Golgi marker GM130, which co-localized with the autophagic LC3 marker, was then immunostained with LAMP, which is a marker for the fusion of autophagosomes and lysosomes during the autophagic process. GM130 (green) and LAMP (red) were co-localized in the striated ribbons of the HT gastrocnemius. (C) Control and HT gastrocnemius muscle biopsies were stained for LAMP (green) and α-tubulin (red) to illustrate microtubule structural integrity and colocalization of LAMP with α-tubulin in sections of the HT gastrocnemius. The structural integrity of the myositis muscle had been compromised, as indicated by the α-tubulin staining.
Supplementary Figure 3: TRAIL induces NFkB nuclear translocation and I-kB alpha degradation. Cells were cultured either in 6-cm dishes or dishes containing coverslips for 2 days at 37°C. Sub-confluent cells were washed with medium and incubated with different concentrations of the IKK inhibitor (0, 25, 2.5 and 25 uM) for 60 min before the addition of recombinant human TRAIL (10 ng/ml). Cells were collected at 15, 30, and 60 min after treatment and analyzed by indirect immunofluorescent staining and western blotting. (A). Western blotting: Protein concentration was determined with a colorimetric assay (Bio-Rad). Proteins were separated by SDS-page, transferred to a nylon membrane, and probed with rabbit anti-I-kB alpha antibody and detected with an anti-rabbit serum coupled to horseradish peroxidase (Santa Cruz Biotechnology), then developed by using an enhanced chemiluminescence system(Pierce). Anti-vinculin antibodies were used to assess protein loading. (B) Immunofluorescent staining: Cells were washed with PBS and fixed with 5% paraformaldehyde in PBS at 37°C for 15 min. After being blocked, cells were incubated with anti-NFkB antibody (Santa Cruz Biotechnology) in PBS plus 1% BSA for 2 h. The coverslips were developed with goat anti-rabbit IgG labeled with Texas red (Molecular Probes) and counterstained with DAPI to stain nuclei. Coverslips were mounted in ProLong antifade (Molecular Probes) and analyzedin a fluorescent microscope (Zeiss).
Supplementary Figure 4: DR5 is expressed in human myositis muscle. Tissue sections (n=4 per disease group) from polymyositis patients were stained for the presence of DR5 by immunohistochemical staining. Diffuse DR5 staining was seen in the myositis muscle (A), which showed no staining when the primary antibody was replaced by an isotype control (B). A representative example of the polymyositis muscle is shown.