

Abstract
Background
Clinically used general anesthetics, alone or in combination, are damaging to the developing mammalian brain. In addition to causing widespread apoptotic neurodegeneration in vulnerable brain regions, exposure to general anesthesia at the peak of synaptogenesis causes learning and memory deficiencies later in life. Our in-vivo rodent studies have suggested that activation of the intrinsic (mitochondria-dependent) apoptotic pathway is the earliest warning sign of neuronal damage, suggesting that a disturbance in mitochondrial integrity and function could be the earliest triggering events.
Methods
Since proper and timely mitochondrial morphogenesis is critical for brain development, we examined the long-term effects of a commonly used anesthesia combination (isoflurane, nitrous oxide, and midazolam) on the regional distribution, ultrastructural properties, and electron transport chain function of mitochondria, as well as synaptic neurotransmission, in the subiculum of rat pups.
Results
This anesthesia, administered at the peak of synaptogenesis, causes protracted injury to mitochondria, including significant enlargement of mitochondria (over 30%, p < 0.05), impairment of their structural integrity, about 28% increase in their complex IV activity (p < 0.05) and two-fold decrease in their regional distribution in presynaptic neuronal profiles (p < 0.05) where their presence is crucially important for the normal development and functioning of synapses. Consequently, we showed that impaired mitochondrial morphogenesis is accompanied by heightened autophagic activity, decrease in mitochondrial density (about 27%, p < 0.05) and long-lasting disturbances in inhibitory synaptic neurotransmission. The interrelation of these phenomena remains to be established.
Conclusion
Developing mitochondria are exquisitely vulnerable to general anesthesia and may be important early target of anesthesia-induced developmental neurodegeneration.
Introduction
Over the last decade, a myriad of studies have presented evidence that various mammalian species are susceptible to significant neurotoxicity when exposed to general anesthesia during early stages of their brain development. Anesthesia-induced neurotoxicity is described as apoptotic and its severity is age-dependent; i.e., the peak of susceptibility coincides with the peak of synaptogenesis.1
Since it has been suggested that general anesthetics used in clinical practice are not as innocuous as they were previously thought to be,2 recent research has been focused on deciphering the earliest cellular targets and mechanistic pathways of anesthesia-induced developmental neurotoxicity so that a method of effective and timely prevention can be devised. This is essential because the use of general anesthetics often cannot be avoided when a child’s life is in danger.
Based on presently available evidence, it appears that very early events in anesthesia-induced apoptotic neurodegeneration involve the activation of a mitochondria-dependent apoptotic cascade,1, 3 which leads to effector caspase activation and DNA fragmentation. Thus, mitochondria may be the initial and most vulnerable target of anesthesia-induced impairment of neuronal development. Since mitochondria are crucially important organelles for the formation, maintenance, and function of developing synapses, which can be permanently impaired by a single exposure to anesthesia,4,5 we did a series of experiments on the effects of general anesthesia on morphogenesis and regional distribution of mitochondria in the developing subiculum.
We found that early exposure to general anesthesia significantly modulates mitochondrial morphogenesis and the function of mitochondria electron transport chain enzyme activity. Regional distribution of mitochondria in presynaptic neuronal profiles is also significantly reduced, where their presence is crucially important for the normal development and function of synapses. We also found that impaired mitochondrial morphogenesis is accompanied by heightened autophagic activity, protracted neuropil destruction, and long-lasting disturbances in inhibitory synaptic neurotransmission.
Materials and Methods
Animals
We used Sprague-Dawley rat pups (Harlan Laboratories, Indianapolis, IN) at postnatal day (P) 7 for all experiments, since this is when they are most vulnerable to anesthesia-induced neuronal damage1. Experimental rat pups were exposed to 6 h of anesthesia; controls were exposed to 6 h of mock anesthesia (vehicle). After the administration of anesthesia, rats were allowed to recover and were reunited with their mothers. Each day, we weighed them and noted their general appearance. On P21, these rats were randomly divided into three groups, one for ultrastructural analysis of the subiculum, one for measuring electron transport chain enzyme activity, and the other for functional studies of synaptic activity (patch-clamp recordings of excitatory and inhibitory synaptic neurotransmission). Rats were used at P21 for ultrastructural and enzyme activity examination and at P21-P28 for electrophysiological studies, since synaptic maturation is generally completed at this age.
All experiments were approved by the Animal Use and Care Committee of the University of Virginia Health System, Charlottesville, Virginia, and were done in accordance with the Public Health Service’s Policy on Human Care and Use of Laboratory Animals. Efforts were made to minimize the number of animals used.
Anesthesia
Nitrous oxide and oxygen were delivered using a calibrated flowmeter. To administer a specific concentration of nitrous oxide/oxygen and isoflurane in a highly controlled environment, an anesthesia chamber was used.1,3,6 Isoflurane was administered using an agent-specific vaporizer that delivers a set percentage of anesthetic into the anesthesia chamber. Midazolam (Sigma-Aldrich Chemical, St. Louis, MO) was dissolved in 0.1 % dimethyl sulfoxide just before administration. For control animals, 0.1 % dimethyl sulfoxide was used alone. To administer a specific concentration of nitrous oxide/oxygen and isoflurane in a highly controlled environment, an anesthesia chamber was used 1,3,6. Rats were kept normothermic throughout the experiment, as previously described.7 For control experiments, air was substituted for the gas mixture. After initial equilibration of the nitrous oxide/oxygen/isoflurane or air atmosphere inside the chamber, the composition of the chamber gas was analyzed by infrared analyzer (Datex Ohmeda, Madison, WI) to establish the concentrations of nitrous oxide or nitrogen, isoflurane, carbon dioxide, and oxygen. We used our standard general anesthesia protocol, giving P7 rat pups a single injection of midazolam (9 mg/kg, intraperitoneally) followed by 6 h of nitrous oxide (75%), isoflurane (0.75%), and oxygen (approximately 24%). Several studies have shown that this protocol causes severe neurodegenerative damage to developing neurons1,3,5,6.
Histopathological studies
On P21, each pup was deeply anesthetized with phenobarbital (65 mg/kg, intraperitoneally) (University of Virginia Pharmacy, Charlottesville, Virginia). After cannulating the left ventricle, we clamped the descending aorta and did an initial flush with Tyrodes solution (30-40 ml) (Sigma-Aldrich Chemical). For morphometric analyses of the neuropil, this was followed by 10 min of continuous perfusion with freshly prepared paraformaldehyde (4%) and glutaraldehyde (0.5%).1,3,6,8 For morphometric analyses of pyramidal neurons, perfusion was done using paraformaldehyde (2%) and glutaraldehyde (2%). After the perfusion, we removed the rats’ brains and stored them in the same fixative overnight. Both control and experimental pups were perfused by an experienced experimenter on the same day, using the same solution to assure uniform tissue fixation. Any brains considered to have been inadequately perfused were not processed for electron microscopy analysis. Fixed brains were coronally sectioned (50-75 μm thick) with a DTK-1000 microslicer (Ted Pella, Tools for Science and Industry, Redding CA). The subiculum was localized as described in anatomical maps,9 fixed in 2% osmium tetroxide (Electron Microscopy Sciences, Hatfield, PA), stained with 4% uranyl acetate (Electron Microscopy Sciences), and embedded in aclar sheets using epon-araldite resins. The subiculum was then dissected from the aclar sheets and embedded in BEEM™ capsules (Electron Microscopy Sciences). To prepare capsules for microtome cutting (Sorvall MT-2 microtome, Ivan Sorvall, Norwalk, CT) the tips were manually trimmed so that ultrathin slices (silver interference color, 600-900 Å) could be cut using a diamond knife (Diatome, Hatfield, PA). Ultrathin sections were placed on grids and examined using a 1230 TEM electron microscope (Carl Zeiss, Oberkochen, Germany). Using a 16M-pixel digital camera (SIA-12C digital cameras, Scientific Instruments and Applications, Duluth, GA), we took 12 random, nonoverlapping electron micrographs (12,000X magnification) of each subicular layer (pyramidal, polymorphic, and molecular). Our electron micrographs depicted neuropils and large pyramidal neurons, depending on the type of analysis needed (see Results). The investigator analyzing electron micrographs was blinded to the experimental conditions.
Morphometric analyses
To do morphometric analyses of mitochondria in the cytoplasmic soma of pyramidal neurons in their entirety which is not possible to perform with a single photo frame at such high magnification (12,000x) we took multiple sequential pictures and tiled them seamlessly together to make a mosaic of one whole cell body (n = 15 neurons in control group and anesthesia-treated group each). From these mosaic pictures, the cytoplasm area and mitochondrial area were measured using Image-Pro Plus 6.1 computer software (MediaCybernetics, Bethesda, MD). The number of animals necessary for complex and time-consuming ultrastructural histological studies was determined based on our previously published study.5
For morphometric analyses of mitochondria in presynaptic neuronal profiles, we first identified the synapses, using the following criteria: the presence of a postsynaptic density; the presence of more than one synaptic vesicle closely apposed to the presynaptic membrane; and the presence of a synaptic cleft delineated by parallel pre- and postsynaptic membranes.5,10 Both excitatory and inhibitory synapse-bearing presynaptic neuronal profiles were examined using the criteria specified by Crain et al.11 Once identified, the area of the presynaptic neuronal profile was measured and the presence or absence of mitochondria was noted. Using Image-Pro Plus 6.1 computer software, the area of mitochondria, where present, were measured so that the mitochondrial index could be calculated; the mitohondrial index is defined as the ratio between mitochondrial and presynaptic neuronal profile areas.
Spectrophotometric analyses of mitochondrial electron transport chain activity
After homogenizing and centrifuging subicular tissue, we measured mitochondrial electron transport chain (ETC) and citrate synthase activity using a Genesis 10-UV spectrophotometer (Thermo, Rochester, NY). The incubation temperatures for complexes I, II, and CS were 30°C and, for complex IV, 38°C. ETC complex activities were determined in supernatants as described by Perez-Carreras et al.12 and expressed as a percentage of the specific activity of citrate synthase to correct for the neuronal content of mitochondria. ETC assays were performed in triplicates.
Statistical analysis
Single comparisons among groups were made using unpaired two-tailed t-test. When ANOVAs with repeated measures were needed, the Bonferroni correction was used to help maintain prescribed alpha levels (e.g. 0.05). Using the standard version of GraphPad Prism 5.01 software (Media Cybernetics, Inc., Bethesda, MD) we considered p < 0.05 to be statistically significant. All the data are presented as mean ± standard error of mean (SEM).
Electrophysiology studies
All experiments were done on 300-μm-thick transverse rat brain slices from 21-28 day-old animals using procedures described previously.13 The subiculum was localized as described in anatomical maps.9
Rats were briefly anesthetized with isoflurane and decapitated. Their brains were rapidly removed and placed in chilled (4° C) cutting solution consisting, in mM, of 2 CaCl2, 260 sucrose, 26 NaHCO3, 10 glucose, 3 KCl, 1.25 NaH2PO4, and 2 MgCl2 equilibrated with a mixture of 95% oxygen and 5% carbon dioxide. A block of tissue containing the subiculum and hippocampus was glued to the chuck of a vibratome (World Precision Instruments, Sarasota, FL) and slices were cut in a transverse plane. The slices were incubated at 36° C in oxygenated saline for 1 h, then placed in a recording chamber that was perfused with extracellular saline at a rate of 1.5 ml/min. Incubating saline consisted, in mM, of 124 NaCl, 4 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 MgCl2, 10 glucose, and 2 CaCl2 equilibrated with a mixture of 95% oxygen and 5% carbon dioxide. Slices were maintained in the recording chamber at room temperature and remained viable for at least 1 h. Since the half-life of halogenated volatile anesthetics in nerve tissue after the induction of anesthesia is only about 10 min,14 it is unlikely that the isoflurane used to euthanize the rats could have interfered with the results of our experiments, which were performed at least 2 h later.
Recording procedures
Whole-cell recordings were obtained from pyramidal subiculum neurons visualized with an IR DIC camera (Hamamatsu C2400, Bridgewater, NJ) on a Zeiss 2 FS Axioscope (Carl Zeiss Jena, Thuringia, Germany) with a 40 X lens.
The standard extracellular saline for recording of evoked inhibitory postsynaptic currents (eIPSCs) and evoked excitatory postsynaptic currents (eEPSCs) consisted, in mM, of 2 CaCl2, 130 NaCl, 1 MgCl2, 10 glucose, 26 NaHCO3, 1.25 NaH2PO4, and 2 mM KCl. This solution was equilibrated with a mixture of 95% oxygen and 5% carbon dioxide for at least 30 min and had a resulting pH of about 7.4. For recording eIPSCs, we used an internal solution containing, in mM, 130 KCl, 4 NaCl, 0.5 CaCl2, 5 ethylene glycol tetraacetic acid (EGTA), 10 4-(2-hydroxyethil)-1-piperazineethanesulfonic acid (HEPES), 2 MgATP2, 0.5 Tris-GTP, and 5 lidocaine N-ethyl bromide. pH was adjusted with KOH to 7.25. For recordings of eEPSCS, this solution was modified by replacing KCl with equimolar K-gluconate. To eliminate glutamatergic excitatory currents, all recordings of eIPSCs were done in the presence of 5 μM NBQX (2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline-2,3-dione), 50 μM d-APV (2R)-amino-5-phosphonovaleric acid; AP5, (2R)-amino-5 phosphonopentanoate) in a bath solution. To eliminate inhibitory currents, all recordings of eEPSCs were done in the presence of 20 μM picrotoxin [a noncompetitive γ-amino butyric acidA antagonist] in the bath solution.
Synaptic stimulation of pyramidal subiculum neurons was achieved with a Constant Current Isolated Stimulator DS3 (Digitimer, Welwyn Garden City Hertfordshire, England). Electrical field stimulation was achieved by placing a stimulating electrode within the hippocampal CA1 soma layer with stimulation intervals of at least 20 s to allow recovery of synaptic responses. In all recordings, we first determined the current-output relationship, and then used current intensities in the stimulating electrode corresponding to the maximal amplitudes of eIPSCs and eEPSCs. Recordings were made with the standard whole-cell voltage clamp technique. Electrodes having final resistances of 2-4 MΩ were fabricated from thin-walled microcapillary glass. Membrane currents were recorded with an Axoclamp 200B amplifier (Molecular Devices, Foster City, CA). Voltage commands and digitization of membrane currents were done with Clampex 8.2 of the pClamp software package (Molecular Devices) running on an IBM-compatible computer (Dell, Inc., Round Rock, TX). Neurons were typically held at -70 mV.
Analysis of current
Current waveforms or extracted data were fitted using Clampfit 8.2 (Molecular Devices) and Origin 7.0 (OriginLab, Northhampton, MA). The decay of eIPSCs was estimated by a single-exponential term. All salts and chemicals were obtained from Sigma-Aldrich Chemical.
Results
I. General anesthesia disturbs the fine ultrastructural balance of developing neuronal mitochondria
Since mitochondrial ultrastructure dictates their function, 15 we studied the ultrastructural appearance of mitochondria with special emphasis on changes indicative of distorted mitochondrial integrity. Our work has been focused on the subiculum because this brain region is highly vulnerable to anesthesia-induced developmental neurodegeneration, as shown by substantial acute neuroapoptotic damage after the administration of anesthesia8,16 and chronic changes marked by substantial neuronal loss.16,17 The subiculum is part of the hippocampus proper and part of Papez’s circuit; it is intertwined with the hippocampal CA1 region, anterior thalamic nuclei, and both the entorhinal and cingulate cortices. Accordingly, the subiculum is important in cognitive development, especially the development of learning and memory.18
For ultrastuctural analysis of mitochondria, we examined the perikaryon of pyramidal subicular neurons (fig. 1A, B) and nerve terminals in subicular neuropil (fig. 1C, D) two weeks after exposure to anesthesia (at P21, see Materials and Methods). We noted that in control subiculi (fig. 1 A, C), the mitochondria appear normal with no evidence of swelling or injury; there was a typical homogeneous staining pattern of the matrix without excessive pallor. In contrast, in the experimental subiculum (fig. 1 B, D) many mitochondria displayed structural disorganization of cristae (see asterisks), including dilated intracristal spaces with vacuoles and overall swelling, which made the mitochondria appear substantially larger than normal. In addition, multiple mitochondrial profiles were suggestive of severe degenerative changes (fig. 1 D, black arrows).
Figure 1. Anesthesia causes long-lasting ultrastructural changes in mitochondria in subiculi of 21-day-old rats.
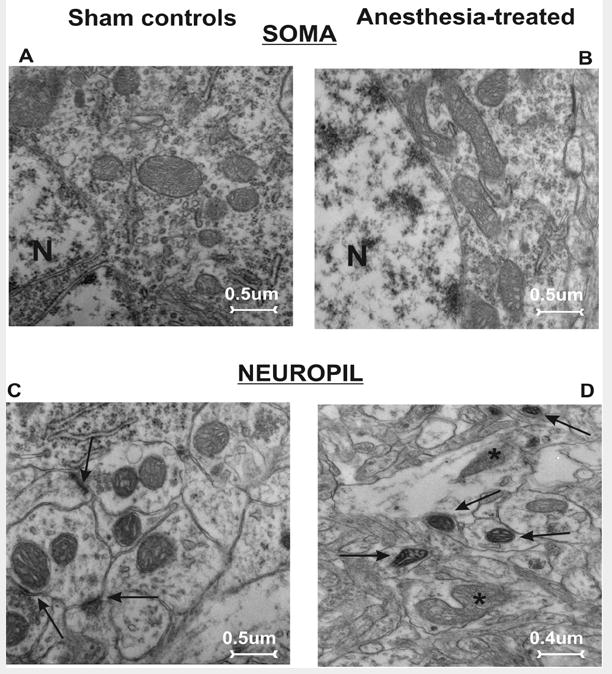
A, C) The pyramidal neuron (A) and neuropil (C; synaptic contacts are noted with arrows) in a subiculum from a control rat show abundant small mitochondria with no evidence of swelling or injury. B, D) Mitochondria in the perikarion of a pyramidal neuron (B) and nerve terminals in a neuropil (D) of subiculum from experimental rats display structural disorganization of cristae (asterisks), as well as dilated intracristal spaces with vacuoles and overall swelling. Note the presence of dark, condensed mitochondria in late stages of degeneration (arrows) (magnification 12,000×). N - nucleus.
Since degenerated organelles are removed by autophagy, we investigated whether the use of general anesthesia leads to heightened autophagic activity. We examined pyramidal neurons in control and experimental subiculi for ultrastructural signs of autophagosomes, lysosomes, and autophagic vacuoles. Using electron microscopy as the gold standard to examine the formation of autophagic profiles, we found that experimental neurons displayed many autophagic profiles (fig. 2A, B). Dispersed throughout the cytoplasm were many lysosomes (fig. 2A, double asterisks) and autophagic vacuoles. These single-membrane structures are formed by the fusion of lysosomes and autophagosomes to allow the digestion of biological “garbage”; they are also referred to as autolysosomes (fig. 2A, arrowheads). The morphological hallmark of autophagy is the formation of double-layered membrane structures called autophagosomes. Experimental neurons frequently contained multiple autophagosomes where parts of cannibalized mitochondria could be detected (fig. 2B, single asterisk). The presence of numerous autophagic profiles suggests that general anesthesia increases the autophagic load in immature neurons.
Figure 2. Anesthesia promotes autophagic activity, as shown in subicular pyramidal neurons of 21-day-old rats.
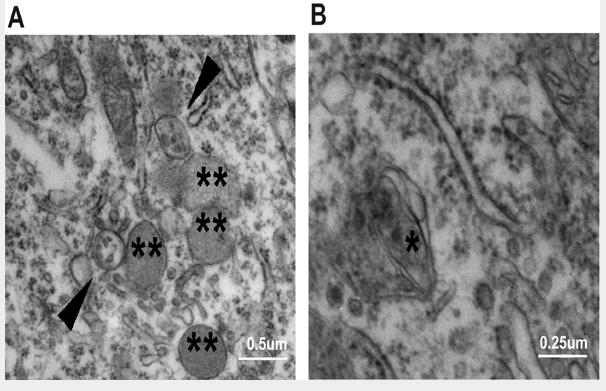
A) In experimental pyramidal neurons, numerous lysosomes (double asterisks) and autophagic vacuoles (arrowheads) were dispersed throughout the cytoplasm. B) Autophagosomes, double-layered membrane structures, were frequently noted in experimental neurons where parts of cannibalized mitochondria could be detected (single asterisk).
Since general anesthesia appears to cause mitochondrial enlargement (fig. 1 B, D), we did a detailed morphometric analysis of mitochondria in the somas of pyramidal subicular neurons. When measured as a percent of the cytoplasmic area of pyramidal neurons, we found that mitochondria in the experimental neurons occupied approximately twice as much area of the cytoplasmic soma area than did controls (22.5 ± 3.1% vs. 13.44 ± 1.2%, * p < 0.05) (fig. 3A, n = 15 neurons per group from 3 control and 3 experimental pups). This did not appear to be due to an increase in mitochondrial number, since mitochondrial density, presented as the number of mitochondria per unit area (μm2) of cytoplasmic soma, was significantly lower in experimental pyramidal neurons as compared to control pyramidal neurons (fig. 3B).
Figure 3. Morphometric analysis of mitochondria in the perikaryon of pyramidal subicular neurons of 21-day-old rats.
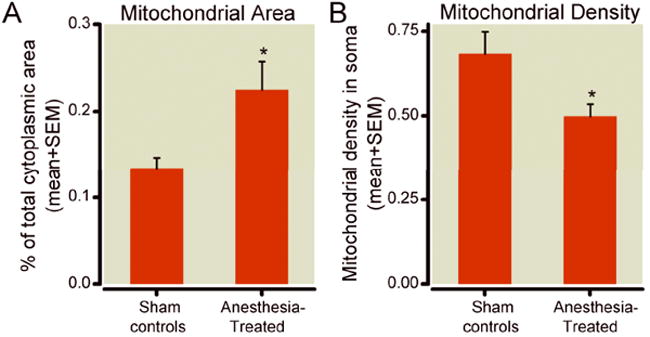
A) Mitochondria in the experimental neurons occupy significantly more cytoplasmic soma than do those in controls (22.5% vs. 13.44%,* p < 0.05) (n = 15 neurons per group from 3 animals each). B) In experimental animals, mitochondrial density, presented as the number of mitochondria per unit area (μm2) of cytoplasmic soma, is significantly lower in pyramidal neurons than that in controls (* p < 0.05, n = 15 neurons per group from 3 control and 3 experimental pups; control and experimental pups were litter-matched).
We confirmed that anesthesia causes mitochondrial enlargement when we categorized mitochondria as small (up to 0.05 μm2), medium (0.06 to 0.25 μm2), large (0.26 to 0.65 μm2), and extra-large (above 0.65 μm2) (fig. 4). Indeed, we found a complete reversal of the ratio between small and large mitochondria in control and experimental animals. For example, about 15% of the total mitochondria in control pyramidal neurons were small ones, whereas only 5% of mitochondria in experimental neurons were small (*** p < 0.001) (fig. 4A). Large mitochondria constituted only 5% of the total mitochondria in control animals, while more than 15% of the mitochondria in experimental pyramidal neurons were large (* p < 0.05) (fig. 4C). Interestingly, although extra-large mitochondria represent less than 1% of the total number of mitochondria, they appeared to be two-fold more prevalent in experimental pyramidal neurons (fig. 4D) than they were in control pyramidal neurons, suggesting that anesthesia causes a substantial imbalance in mitochondrial size, with a clear tendency toward mitochondrial enlargement (n = 15 neurons per group from 3 control and 3 experimental pups).
Figure 4. Mitochondrial size classification in the perikaryon of pyramidal subicular neurons of 21-day-old rats.
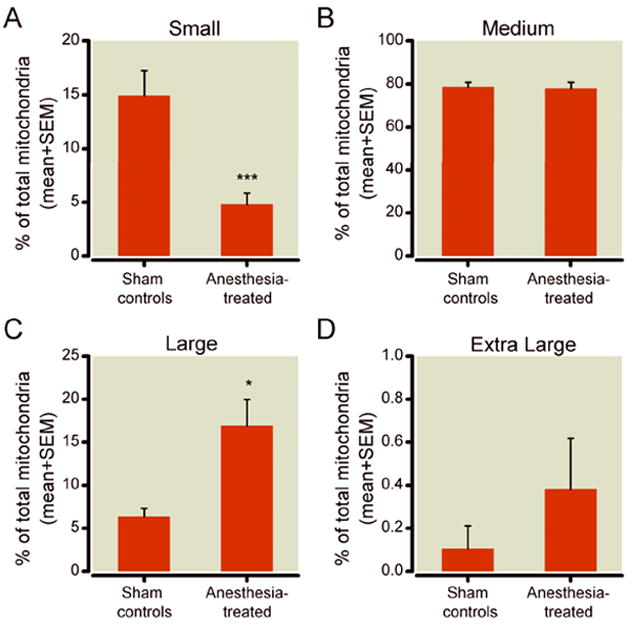
A) A small fraction of mitochondria (up to 0.05 μm2) constitutes about 15% of total mitochondria in control pyramidal neurons, but only 5% in the experimental pyramidal neurons (*** p < 0.001). B) A medium-sized fraction of mitochondria (0.06 to 0.25 μm2) represents the largest population of mitochondria in subicular pyramidal neurons. This fraction remains unchanged after anesthesia treatment. C) A large fraction of mitochondria (0.26 to 0.65 μm2) constitutes only about 5% of total mitochondria in control pyramidal neurons, but 15% of those in experimental pyramidal neurons (* p < 0.05). D) An extra-large fraction of mitochondria (more than 0.65 μm2) represents less than 1% of the total number of mitochondria in experimental pyramidal neurons and shows over two-fold higher prevalence in these neurons (fig. 3D) although it did not reach statistical significance (n = 15 neurons per group from 3 control and 3 experimental pups; control and experimental pups were litter-matched).
II. General anesthesia disturbs regional distribution of developing neuronal mitochondria
The proper development and function of synapses depends on mitochondrial support, since synaptogenesis has high metabolic requirements.19-22 Therefore, regional distribution of mitochondria in presynaptic nerve terminals and their strategic placement in the vicinity of developing synapses is crucially important. Having demonstrated that general anesthesia causes substantial enlargement of mitochondria in neuronal somas, as shown in figures 3 and 4, we then examined whether similar morphometric changes could be detected in presynaptic neuronal terminals. To compare regional distribution of mitochondria in presynaptic neuronal profiles in experimental and control subiculi, we quantified the number of presynaptic neuronal profiles, out of all those present in any given electron microscopy photo frame of subicular neuropils (at 12,000x magnification), that contained mitochondrial profiles. We expressed the findings as a percentage of presynaptic profiles containing mitochondria. We found a significantly higher (* p < 0.05) percentage of mitochondria-containing presynaptic profiles in control as compared to experimental subiculi (fig. 5) (n = 28 photo frames/group from 4 control and 4 experimental pups from two different litters).
Figure 5. Anesthesia decreases mitochondrial density in presynaptic neuronal terminal subicular neuropils of 21-day-old rats.
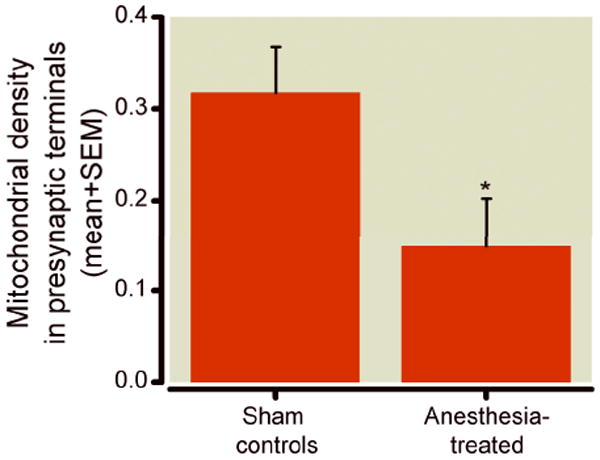
Compared to controls, fewer presynaptic neuronal terminals in experimental subicular neuropils contain mitochondrial profiles. When the findings are presented as a percentage of presynaptic profiles containing mitochondria, a significantly (about two-fold) higher percentage of mitochondria-containing presynaptic profiles occurs in control subiculi than in experimental subiculi (* p < 0.05) (n = 28 photo frames/group from 4 control and 4 experimental pups from two different litters; control and experimental pups were litter-matched).
To further assess our ultrastructural observation suggesting substantial swelling of mitochondrial profiles in presynaptic neuron terminals (fig. 1D), we did detailed morphometric analysis of the area of mitochondrial profiles (in μm2). We found that experimental mitochondrial profiles were, on average, 38% larger than control mitochondrial profiles (* p < 0.05) (fig. 6A) (n = 30 photo frames obtained from 5 control pups; n = 45 photo frames obtained from 5 experimental pups; control and experimental pups were litter-matched; total of three different litters were used). When the areas of mitochondria-containing presynaptic nerve terminals were measured, we found no difference between control and experimental animals (fig. 6B) (n = 30 photo frames obtained from 5 control pups; n = 45 photo frames obtained from 5 experimental pups; control and experimental pups were litter-matched; total of three different litters were used). Consequently, when we calculated the mitochondrial index, which is the ratio between mitochondrial area and mitochondria-containing presynaptic area, we found that this index was significantly higher in experimental subicular neuropils (* p < 0.05) indicating that terminally distributed mitochondria display morphometric changes that are similar to those in mitochondria located in the soma (fig. 6C).
Figure 6. Morphometric analysis of mitochondria in presynaptic neuronal terminals in subicular neuropils of 21-day-old rats.
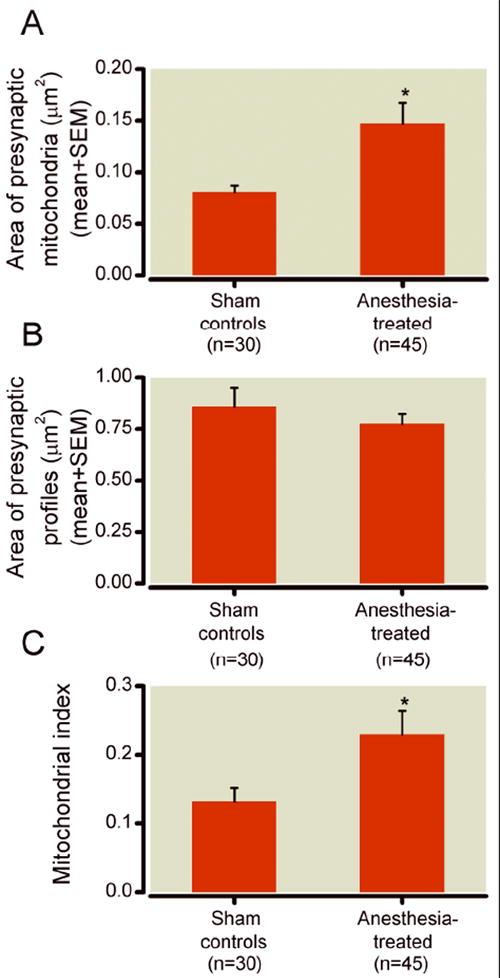
A) Morphometric analysis of the area of mitochondrial profiles shows that experimental mitochondrial profiles were approximately 30% larger than that of controls (* p < 0.05)) (n = 30 photo frames obtained from 5 control pups; n = 45 photo frames obtained from 5 experimental pups; control and experimental pups were litter-matched; total of three different litters were used). B) There is no difference between control and experimental subiculi with regard to the areas of mitochondria-containing presynaptic nerve terminals (n = 30 photo frames obtained from 5 control pups; n = 45 photo frames obtained from 5 experimental pups; control and experimental pups were litter-matched; total of three different litters were used). C) The calculated mitochondrial index (ratio between mitochondrial area and mitochondria-containing presynaptic area) was significantly higher in experimental subicular neuropils (* p < 0.05) than in control neuropils.
III. General anesthesia acutely disturbs functional balance in developing neuronal mitochondria
Since we have previously reported that general anesthesia causes acute disturbances of cytochrome c homeostasis within the first 4 h of anesthesia exposure,1 we question whether anesthesia modulates the function of ETC complexes, particularly complex IV (cytochrome c oxidoreductase). Complex IV is of interest for three reasons: it depends on the availability of cytochrome c, since it transfers electrons from cytochrome c to oxygen, thus controlling the final steps of electron transport and adenosine triphosphate (ATP) synthesis; it is encoded in part by mitochondrial deoxyribonucleic acid, which makes it particularly vulnerable to mitochondrial dysfunction; modulation of complex IV activity has been shown to cause elevated free oxygen radical production23 thus making neurons vulnerable to excessive lipid peroxidation and protein oxidation. We measured complex IV activity in mitochondrial homogenate prepared from fresh subicular tissue of rat pups on P8, 24 h after anesthesia treatment. Since citrate synthase activity is directly proportional to mitochondrial content, the activity of complex IV was expressed as a ratio (per the activity of citrate synthase). As shown in figure 7A, there was a significant increase in complex IV activity 24 h after anesthesia treatment (* p < 0.05) (n = 8 pups in control group; n = 6 pups in experimental group). When we measured the activity of complexes I and II/III, we found no change in experimental groups as compared to controls (fig. 7B, C, respectively) (n = 5 pups in control group; n = 5 pups in experimental group for complex I activity; n=3 in control group; n = 4 pups in experimental group for complex II/III activity).
Figure 7.
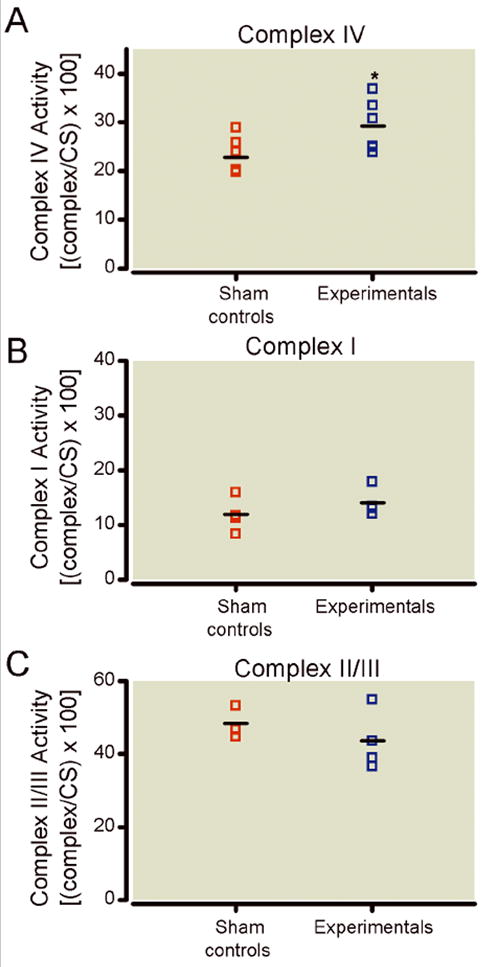
Anesthesia differentially modulates the activity of mitochondrial respiratory chain proteins. A) Compared to control subiculi, the activity of complex IV in experimental subiculi is significantly increased at 24 h after anesthesia (* p < 0.05) (n = 8 pups in control group; n = 6 pups in experimental group). B) The activity of complex I was unchanged in the anesthesia-treated group as compared to sham controls (n = 5 pups per group). C) The activity of complex II was unchanged in the anesthesia-treated group as compared to sham controls (n = 3 pups in control group; n = 4 pups in experimental group). The activity of complexes I, II, and IV were expressed as ratios [per the activity of citrate synthase] since citrate synthase activity is directly proportional to mitochondrial content.
IV. General anesthesia impairs developmental synaptic transmission
Given the fact that general anesthesia impairs synapse formation during early brain development,4,5 as well as our findings suggesting that general anesthetics impair mitochondrial morphogenesis and decrease the numbers of mitochondria in presynaptic neuronal profiles, we ask whether the diminished presence of mitochondria in anesthesia-treated subiculum has any bearing on the functional integrity of its synapses. To address this question, we examined inhibitory (eIPSC) and excitatory (eEPSC) synaptic transmission by recording from the pyramidal layer of control and anesthesia-treated rat subicular slices (see Methods). The traces in figure 8A show representative eIPSCs from the control (red line) and experimental group (blue line), both of which received anesthesia at P7. Paired stimulation of afferent fibers resulted in pair-pulse depression, a highly characteristic finding for subicular neurons (fig. 8A). This test is done by analyzing changes in the ratio of eIPSCs elicited by two identical presynaptic stimuli delivered in rapid succession. This paired-pulse depression of test eIPSCs (P2) relative to conditioning eIPSCs (P1) is thought to be due to depletion of a fraction of readily available synaptic sites.
Figure 8. Alterations occurred in inhibitory synaptic transmission in pyramidal cells of subiculi after exposure to anesthesia early in life.
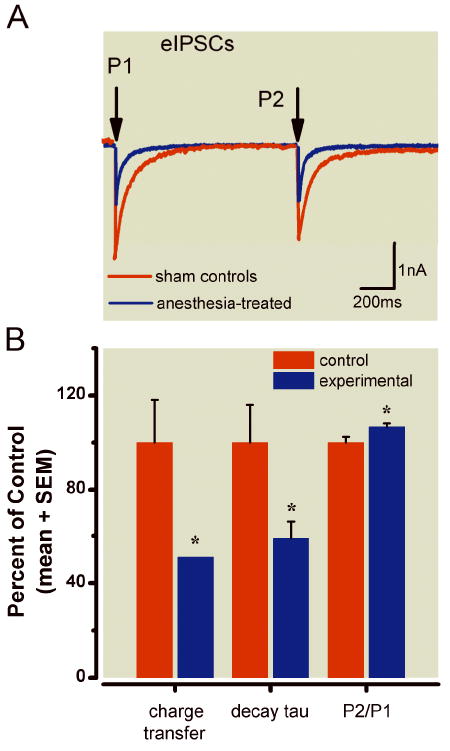
A) Representative eIPSCs obtained using a paired-pulse protocol to record from two pyramidal cells in the subiculi of rats in the control (red trace) and experimental groups (blue trace). Note that the experimental group had decreased current amplitude and faster decay. Arrows indicate the time of paired-pulse stimulus application (interval 1.1 s). Stimulus transients have been removed for clarity of the current traces. B) Histograms showing average data from control cells (n = 14) and experimental cells (n = 12). Black solid bars indicate control cells; gray bars represent experimental cells; vertical lines indicate the SEM of multiple determinations. All data are normalized to 100% of average responses in the control group. Left panel shows a decrease in net charge transfer of eIPSCs from 100 ± 18% to 51 ± 9% (p < 0.05) in the experimental group; middle panels show a decrease in decay of tau from 100 ± 16% to 59 ± 7%; right panels show a small but significant increase in P2/P1 from 100 ± 2% to 107 ± 1% (p < 0.05).
In comparison to the control group, the experimental group had ~ 49% decrease in net charge transfer of eIPSCs measured as the area under the curve (p < 0.05); a decreased decay time constant (tau) from 58 ± 9 ms (n = 14) to 34 ± 4ms (n = 12, p < 0.05); and a significantly altered paired-pulse ratio P2/P1 (amplitude of eIPSC-2: amplitude of eIPSC-1) from 0.81 ± 0.02 (n = 14) to 0.87 ± 0.01 (n = 12, p < 0.05). The data are summarized in the histogram in figure 8B.
It is generally accepted that presynaptic depressants that change the probability of transmitter release from presynaptic terminals cause a smaller fraction of the readily releasable pool of vesicles to undergo exocytosis and therefore decrease pair-pulse depression. 24 This means that in the presence of presynaptic modulators, the ratio P2:P1 becomes larger. On the other hand, if a modulator acts on postsynaptic sites, pair-pulse depression should remain unchanged while current amplitude and/or decay time might be changed. Changes in decay time constant, as well as decreased current amplitudes and alterations in the P2:P1 ratio strongly suggest that both postsynaptic and presynaptic mechanisms contribute to the decreased synaptic strength of inhibitory transmission in the experimental group. In contrast, we found no significant difference in the synaptic strength (net charge transfer) of eEPSCs between the control (n = 20 cells) and experimental groups (n = 10 cells, data not shown).
Discussion
General anesthesia administered to 7-day-old rat pups causes long-lasting alterations in mitochondrial morphogenesis and regional distribution, heightened autophagocytic activity and ongoing neuropil destruction, as well as significant disturbances in synaptic neurotransmission in the subiculum.
Mitochondrial regeneration in neurons depends on balancing two opposing processes, mitochondrial fusion and fission.25 Deranged fusion leads to mitochondrial fragmentation; deranged fission leads to mitochondrial enlargement. Since our ultrastructural analyses indicate that anesthesia causes enlargement of mitochondria, general anesthetics may modulate the fine equilibrium between fusion and fission which can lead to disturbances in mitochondrial functioning. This may not be well tolerated by immature and functionally busy mammalian neurons, which are in need of adequate metabolic support. Indeed, impairment of mitochondrial morphogenesis may, at least in part, be the cause of reported anesthesia-induced developmental neurodegeneration,1,6,8,26-28, especially since an imbalance between fission and fusion appears to have a causal role in initiating several neurodegenerative diseases.29,30 Interestingly, large (and “giant”) mitochondria are often described in aging neurons.31 Although it is tempting to draw a parallel between certain elements of neurodegeneration unique to aging and anesthesia-induced neurodegeneration unique to developmental brain it remains to be determined whether they share similar cellular pathways. In particular, it remains to be determined how anesthesia affects developmental fusion and fission using an easy-to-manipulate in-vitro system while focusing on various GTPase proteins (e.g., Drp1, fis 1, OPA 1, mitofusin 1 and 2) that are crucial for proper pathway activation.32-34
It is possible that fusion and fission are modulated by anesthesia and thus are the main causes of mitochondrial enlargement. However, our ultrastructural observations indicate that mitochondria are swollen and plagued by deranged, fragmented cristae and inner membranes suggesting that the impairment of mitochondrial membrane integrity may be the main cause of their “leakiness,” allowing the indiscriminate entry of colloids and water. In support of this notion is our previous finding that anesthetics causes significant downregulation of bcl-xL proteins, which are important in maintaining mitochondrial membrane integrity. This downregulation leads to cytochrome c leakage suggestive of increased mitochondrial permeability.1
Mitochondria have been classified by size in chronic neurodegenerative diseases such as Parkinson’s and Alzheimer’s, in which large mitochondria predominate, while the population of medium-sized mitochondria remains unchanged.35 We observed a similar tendency toward mitochondrial enlargement with a seemingly stable population of medium-sized mitochondria. Although medium-sized mitochondria may show a lower propensity for swelling than small ones, a more likely explanation is that the observed phenomenon is due to the shift in mitochondrial size distribution toward medium and large size category caused by the swelling of small and medium-sized mitochondria, respectively rather than a focal increase in mitochondria of any one size. Therefore, a detailed ultrastructural analysis of mitochondria should always accompany the size analysis.
Mitochondria are generated in the soma and move within the cytoplasm to distribute within cells.36 Since neurons have multiple compartments (e.g., dendrites, axons, and synapses) that are located far from the cell body, they depend heavily on proper mitochondrial distribution.19 As the main regulators of ATP production, mitochondria are frequently found in the vicinity of active growth cones of developing neurons19 and in terminals with active synapses.20,21 We report that significantly fewer mitochondria are located in presynaptic neuronal profiles in anesthesia-treated subiculi than are in controls. Also, the mitochondrial profiles in presynaptic neuronal profiles are significantly larger than those in controls, suggesting that anesthesia-induced morphological changes shift the regional distribution of mitochondria away from very distant, thin, and highly arborized dendritic branches at a time when their presence is crucial for normal synapse formation and development. It remains to be determined whether anesthetics impair proper mitochondrial trafficking which may explain anesthesia-induced impairment of the morphogenesis and plasticity of dendritic spines and synapses.4, 5
We report a decrease in mitochondrial density in both neuronal soma and presynaptic terminals which may suggest mitochondrial “dropout” due to mitochondrial degeneration and removal via autophagy. However, a decrease in mitochondrial density could be relative, caused by the fact that large mitochondria represent a bigger fraction of cytoplasmic and presynaptic terminal areas due to mitochondrial swelling or, perhaps, improper fission/fusion.
We show that the activity of complex IV is significantly up-regulated while the activity of complexes I and II/III remains unchanged. It is possible that an increase in complex IV activity provides anesthesia-treated neurons with increased ATP levels,37 which would result in a higher energy state. Although it would be tempting to consider higher ATP levels to be beneficial for developing neurons, there is a drawback to the increased neuronal energy level based on an isolated increase in complex IV activity. Instead of leading to decreased oxidative stress by decreasing ROS production, as previously thought,38 a recent report suggests that acute elevation of complex IV activity is associated with increased ROS production.23 In other words, a higher complex IV electron transfer rate onto oxygen, in view of intact activity of complexes I, II, and III, results in a higher degree of oxidation of the ubiquinone pool. This allows complexes II and III to transfer electrons to oxygen, causing an increase in ROS production. If anesthesia leads to elevated ROS production, protein oxidation, and lipid peroxidation, this may, at least in part, explain the ongoing neuropil destruction and prominent neurite degradation. 5
Here we focus on subiculum. However, the effects of anesthesia on complex IV activity could be brain- region specific. For example, in a neurotoxic model of Parkinson’s disease it has been shown that striatal, but not cortical neurons demonstrate elevated complex IV activity, resulting in ROS upregulation and neuronal cell death.37 Hence, it remains to be determined how anesthesia affects the activity of complex IV and other ETC proteins in other brain regions that are vulnerable to anesthesia-induced developmental neurodegeneration.
We question whether our patch-clamp results showing that a single exposure to anesthesia leads to lasting depression of inhibitory transmission in subicular neurons could be due to impaired regional distribution of mitochondria. This is based on the fact that defective synaptic transmission is associated with the loss of mitochondria from axon terminals.22 Interestingly, although inhibitory neurotransmission was impaired, excitatory neurotransmission was spared. Our earlier study using hippocampal slices of rats exposed to general anesthesia at age P7 demonstrated that excitatory synaptic transmission was not affected.8 This is intriguing considering that our morphometric studies of the subiculum show nonselective synapse loss5 and nonselective changes in mitochondrial regional distribution/ morphometry when excitatory and inhibitory synapses were examined (data not shown). The obvious reason for this selective functional synaptic plasticity is not known. However, it is of interest that functioning mitochondria and their ATP production are essential for maintaining normal synaptic physiology.34,39,40
Acute application of isoflurane or midazolam potentiates the inhibitory drive by heightening inhibitory synaptic activity mediated by gamma amino-butyric acidA receptors.41 In contrast, nitrous oxide silences excitatory (N-methyl-D-aspartate-mediated) synaptic transmission.42 Thus, it is possible that, as a consequence of an inadequate metabolic ratio of supply to demand, anesthesia-induced degenerative changes in mitochondria and a potential decrease in ATP production preferentially impair highly activated inhibitory synaptic function. ROS signaling may be important in modulating synaptic transmission. For example, acute applications of hydrogen peroxide, the common donor of ROS, preferentially reduced inhibitory over excitatory synaptic transmission in thalamocortical,43 hippocampal, 44,45 cortical, and striatal slices45 by both pre- and post-synaptic mechanisms. Since the strength of eIPSCs but not eEPSCs in subiculi is greatly diminished in slices of rats exposed to clinical anesthesia early in life, it is tempting to speculate that this can be at least in part a result of the production of ROS in response to anesthetic-induced mitochondrial dysfunction. However, the precise mechanism for the selective homeostatic changes in neuronal function under extensive γ amino-butyric acidA stimulation associated with various physiological and pathological conditions remains to be examined.46
Because damaged mitochondria could become an uncontrollable source of ROS and therefore would have to be degraded to ensure neuronal survival, it came as no surprise that anesthesia created a substantial amount of biological “garbage” and heightened autophagy. Autophagy is initiated by the formation of autophagosomes, double-membrane-bound cellular structures that enter lysosomes, acidic vacuolar compartments containing various lytic enzymes that have pH optima in the acidic range.47-49 Lysosomes may slowly leak enzymes, which, in turn, can induce apoptosis via activation of a variety of pro-caspases. Our observation of a substantial number of autophagic bodies, in addition to impaired mitochondrial morphogenesis, neuropil damage, and synapse loss5 raises the important possibility that anesthesia kills developing neurons simply by overwhelming natural autophagy with a massive production of defective mitochondria. Further studies will be necessary to test this possibility.
Although a reliable model for studying developmental neurodegeneration our anesthesia protocol is based on the use of anesthetics in combination. As such it prevents us from deciphering the relative contribution of each agent. Further studies of individual anesthetics will help us decipher their relative importance in inducing mitochondrial morphological impairments and dysfunction.
We show that general anesthesia causes significant impairment in mitochondrial morphogenesis and function in developing rat brain thus suggesting that mitochondria may be the most vulnerable initial target of anesthesia-induced developmental neurotoxicity.
Acknowledgments
The authors thank Jan A. Redick, (Laboratory Director) from the Advanced Microscopy Facility at the University of Virginia, Charlottesville, Virgnia; Alev Erisir, M.D., Ph.D., (Associate Professor) from Department of Psychology at the University of Virginia, Charlottesville, Virginia; the Advanced Microscopy Facility at the University of Virginia, Charlottesville, Virginia, for technical assistance with electron microscopy and data analyses.
Funding Statement: This study was supported by the grants from the National Institute of Health/ National Institute on Child and Human Development (Bethesda, Maryland) (NIH/NICHD) HD44517 (to Vesna Jevtovic-Todorovic) and HD44517-S1 (to Vesna Jevtovic-Todorovic); Harold Carron endowment (to Vesna Jevtovic-Todorovic) and the grant from the National Institute of Health/National Institute on General Medical Sciences (Bethesda, Maryland) (NIH/NIGMS) GM070726 (to Slobodan M. Todorovic).
References
- 1.Yon J-H, Daniel-Johnson J, Carter LB, Jevtovic-Todorovic V. Anesthesia induces neuronal cell death in the developing rat brain via the intrinsic and extrinsic apoptotic pathways. Neuroscience. 2005;35:815–27. doi: 10.1016/j.neuroscience.2005.03.064. [DOI] [PubMed] [Google Scholar]
- 2.Wilder RT, Flick RP, Sprung J, Katusic SK, Barbaresi WJ, Mickelson C, Gleich SJ, Schroeder DR, Weaver AL, Warner DO. Early exposure to anesthesia and learning disabilities in a population-based birth cohort. Anesthesiology. 2009;110:796–804. doi: 10.1097/01.anes.0000344728.34332.5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yon J-H, Carter LB, Reiter RJ, Jevtovic-Todorovic V. Melatonin reduces the severity of anesthesia-induced apoptotic neurodegeneration in the developing rat brain. Neurobiol Dis. 2006;21:522–30. doi: 10.1016/j.nbd.2005.08.011. [DOI] [PubMed] [Google Scholar]
- 4.Head BP, Patel HH, Niesman IR, Drummond JC, Roth DM, Patel PM. Inhibition of p75 neurotrophin receptor attenuates isoflurane-mediated neuronal apoptosis in the neonatal central nervous system. Anesthesiology. 2009;110:813–25. doi: 10.1097/ALN.0b013e31819b602b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lunardi N, Ori C, Erisir A, Jevtovic-Todorovic V. General anesthesia causes long-lasting disturbances in the ultrastructural properties of developing synapses in young rats. Neurotox Res. 2010;17:179–88. doi: 10.1007/s12640-009-9088-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu LX, Yon J-H, Carter LB, Jevtovic-Todorovic V. General anesthesia activates BDNF-dependent neuroapoptosis in the developing rat brain. Apoptosis. 2006;11:1603–15. doi: 10.1007/s10495-006-8762-3. [DOI] [PubMed] [Google Scholar]
- 7.Jevtovic-Todorovic V, Benshoff N, Olney JW. Ketamine potentiates cerebrocortical damage induced by the common anaesthetic agent nitrous oxide in adult rats. Br J Pharmacol. 2000;130:1692–8. doi: 10.1038/sj.bjp.0703479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jevtovic-Todorovic V, Hartman RE, Izumi Y, Benshoff ND, Dikranian K, Zorumski CF, Olney JW, Wozniak DF. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. J Neurosci. 2003;23:876–82. doi: 10.1523/JNEUROSCI.23-03-00876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Sydney: Academic; 1986. [DOI] [PubMed] [Google Scholar]
- 10.Erisir A, Harris JL. Decline of the critical period of visual plasticity is concurrent with the reduction of NR2B subunit of the synaptic NMDA receptor in layer 4”. J Neurosci. 2003;23:5208–18. doi: 10.1523/JNEUROSCI.23-12-05208.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crain B, Cotman C, Taylor D, Lynch G. A quantitative electron microscopic study of synaptogenesis in the dentate gyrus of the rat. Brain Res. 1973;63:195–204. doi: 10.1016/0006-8993(73)90088-7. [DOI] [PubMed] [Google Scholar]
- 12.Pérez-Carreras M, Del Hoyo P, Martín MA, Rubio JC, Martín A, Castellano G, Colina F, Arenas J, Solis-Herruzo JA. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology. 2003;38:999–1007. doi: 10.1053/jhep.2003.50398. [DOI] [PubMed] [Google Scholar]
- 13.Joksovic PM, Weiergraber M, Lee WY, Struck H, Schneider T, Todorovic SM. Isoflurane-sensitive presynaptic R-type calcium channels contribute to inhibitory synaptic transmission in the rat thalamus. J Neurosci. 2009;29:1434–45. doi: 10.1523/JNEUROSCI.5574-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stevens WC, Kingston HGG. Inhalation anesthesia. In: Barash PG, Lippincott JB, editors. Clinical Anesthesia. 2. Philadelphia: 1992. pp. 429–65. [Google Scholar]
- 15.Mannella CA. The relevance of mitochondrial membrane topology to mitochondrial function. Biochim Biophys Acta. 2006;1762:140–7. doi: 10.1016/j.bbadis.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 16.Rizzi S, Carter LB, Ori C, Jevtovic-Todorovic V. Clinical anesthesia causes permanent damage to the fetal guinea pig brain. Brain Pathology. 2008;18:198–210. doi: 10.1111/j.1750-3639.2007.00116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nikizad H, Yon J-H, Carter LB, Jevtovic-Todorovic V. Early exposure to general anesthesia causes significant neuronal deletion in the developing rat brain. Ann NY Acad Sci. 2007;1122:69–82. doi: 10.1196/annals.1403.005. [DOI] [PubMed] [Google Scholar]
- 18.McNaughton N. The role of the subiculum within the behavioural inhibition system. Behav Brain Res. 2006;174:232–50. doi: 10.1016/j.bbr.2006.05.037. [DOI] [PubMed] [Google Scholar]
- 19.Morris RL, Hollenbeck PJ. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J Cell Sci. 1993;104:917–27. doi: 10.1242/jcs.104.3.917. [DOI] [PubMed] [Google Scholar]
- 20.Rowland KC, Irby NK, Spirou GA. Specialized synapse-associated structures within the calyx of Held. J Neurosci. 2000;20:9135–44. doi: 10.1523/JNEUROSCI.20-24-09135.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shepherd GM, Harris KM. Three-dimensional structure and composition of CA3→CA1 axons in rat hippocampal slices: Implications for presynaptic connectivity and compartmentalization. J Neurosci. 1998;18:8300–10. doi: 10.1523/JNEUROSCI.18-20-08300.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stowers RS, Megeath LJ, Gorska-Andrzejak J, Meinertz IA, Schwarz TL. Axonal transport of mitochondria to synapses depends on Milton, a novel Drosophila protein. Neuron. 2002;36:1063–77. doi: 10.1016/s0896-6273(02)01094-2. [DOI] [PubMed] [Google Scholar]
- 23.Dröse S, Brandt U. The mechanism of mitochondrial superoxide production by cytochrome bc1 complex. J Biol Chem. 2008;283:21649–54. doi: 10.1074/jbc.M803236200. [DOI] [PubMed] [Google Scholar]
- 24.Zucker RS, Regehr WG. Short-term synaptic plasticity. Ann Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]
- 25.Chan DC. Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol. 2006;22:79–99. doi: 10.1146/annurev.cellbio.22.010305.104638. [DOI] [PubMed] [Google Scholar]
- 26.Ikonomidou C, Bosch F, Miksa M, Bittigav P, Vockler J, Dikranian K, Tenkova TI, Stefovska V, Turski L, Olney JW. Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science. 1999;283:70–4. doi: 10.1126/science.283.5398.70. [DOI] [PubMed] [Google Scholar]
- 27.Ikonomidou C, Bittigau P, Ishimaru MJ, Wozniak DF, Kock C, Genz K, Price MT, Stefovska V, Horster F, Tenkova T, Dikranian K, Olney JW. Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome. Science. 2000;287:1056–60. doi: 10.1126/science.287.5455.1056. [DOI] [PubMed] [Google Scholar]
- 28.Slikker W, Jr, Zou X, Hotchkiss CE, Divine RL, Sadovova N, Twaddle NC, Doerge DR, Scallet AC, Patterson TA, Hanig JP, Paule MG, Wang C. Ketamine-induced neuronal cell death in the perinatal rhesus monkey. Toxicol Sci. 2007;98:145–58. doi: 10.1093/toxsci/kfm084. [DOI] [PubMed] [Google Scholar]
- 29.Bossy-Wetzel E, Barsoum MJ, Godzik A, Schwarzenbacher R, Lipton SA. Mitochondrial fission in apoptosis, neurodegeneration and aging. Curr Opin Cell Biol. 2003;15:706–16. doi: 10.1016/j.ceb.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 30.Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci. 2009;29:9090–103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Navarro A, Boveris A. Brain mitochondrial dysfunction in aging, neurodegeneration, and Parkinson’s disease. Front Aging Neurosci. 2010;2:pii:34. doi: 10.3389/fnagi.2010.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smirnova E, Griparic L, Shurland DL, van der Bliek AM. Dynamin-related protein Drp1 is required for mitochondrial division in mammalian cells. Mol Biol Cell. 2001;12:2245–56. doi: 10.1091/mbc.12.8.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zorzano A, Liesa M, Sebastiáan D, Segalés J, Palacín M. Mitochondrial fusion proteins: Dual regulators of morphology and metabolism. Semin Cell Dev Biol. 2010;21:566–74. doi: 10.1016/j.semcdb.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 34.Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–87. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 35.Trimmer PA, Swerdlow RH, Parks JK, Keeney P, Bennett JP, Jr, Miller SW, Davis RE, Parker WD., Jr Abnormal mitochondrial morphology in sporadic Parkinson’s and Alzheimer’s disease cybrid cell lines. Exp Neurol. 2000;162:37–50. doi: 10.1006/exnr.2000.7333. [DOI] [PubMed] [Google Scholar]
- 36.Yaffe MP. The machinery of mitochondrial inheritance and behavior. Science. 1999;283:1493–7. doi: 10.1126/science.283.5407.1493. [DOI] [PubMed] [Google Scholar]
- 37.Singh S, Misiak M, Beyer C, Arnold S. Brain region specificity of 3-nitropropionic acid-induced vulnerability of neurons involves cytochrome c oxidase. Neurochem Int. 2010;57:297–305. doi: 10.1016/j.neuint.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 38.Cadenas E, Boveris A, Ragan CI, Stoppani AO. Production of superoxide radicals and hydrogen peroxide by NADH-ubiquinone reductase and ubiquinol-cytochrome c reductase from beef-heart mitochondria. Arch Biochem Biophys. 1977;180:248–57. doi: 10.1016/0003-9861(77)90035-2. [DOI] [PubMed] [Google Scholar]
- 39.Inquimbert P, Rodeau JL, Schlichter R. Regional differences in the decay kinetics of GABA(A) receptor-mediated miniature IPSCs in the dorsal horn of the rat spinal cord are determined by mitochondrial transport of cholesterol. J Neurosci. 2008;28:3427–37. doi: 10.1523/JNEUROSCI.5076-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jonas E. Regulation of synaptic transmission by mitochondrial ion channels. J Bioenerg Biomembr. 2004;36:357–61. doi: 10.1023/B:JOBB.0000041768.11006.90. [DOI] [PubMed] [Google Scholar]
- 41.Franks NP. General anaesthesia: From molecular targets to neuronal pathways of sleep and arousal. Nat Rev Neurosci. 2008;9:370–86. doi: 10.1038/nrn2372. [DOI] [PubMed] [Google Scholar]
- 42.Mennerick S, Jevtovic-Todorovic V, Todorovic SM, Shen W, Olney JW, Zorumski CF. Effect of nitrous oxide on excitatory and inhibitory synaptic transmission in hippocampal cultures. J Neurosci. 1998;18:9716–26. doi: 10.1523/JNEUROSCI.18-23-09716.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Frantseva MV, Perez Velazquez JL, Carlen PL. Changes in membrane and synaptic properties of thalamocortical circuitry caused by hydrogen peroxide. J Neurophysiol. 1998;80:1317–26. doi: 10.1152/jn.1998.80.3.1317. [DOI] [PubMed] [Google Scholar]
- 44.Müller M, Fontana A, Zbinden G, Gähwiler BH. Effects of interferons and hydrogen peroxide on CA3 pyramidal cells in rat hippocampal slice cultures. Brain Res. 1993;619:157–62. doi: 10.1016/0006-8993(93)91607-t. [DOI] [PubMed] [Google Scholar]
- 45.Sah R, Galeffi F, Ahrens R, Jordan G, Schwartz-Bloom RD. Modulation of the GABA(A)-gated chloride channel by reactive oxygen species. J Neurochem. 2002;80:383–91. doi: 10.1046/j.0022-3042.2001.00706.x. [DOI] [PubMed] [Google Scholar]
- 46.Mody I. Aspects of the homeostatic plasticity of GABAA receptor-mediated inhibition. J Physiol. 2005;562:37–46. doi: 10.1113/jphysiol.2004.077362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Brunk UT, Terman A. The mitochondrial-lysosomal axis theory of aging: Accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur J Biochem. 2002;269:1996–2002. doi: 10.1046/j.1432-1033.2002.02869.x. [DOI] [PubMed] [Google Scholar]
- 48.Terman A, Gustafsson B, Brunk UT. Autophagy, organelles and ageing. J Pathol. 2007;211:134–43. doi: 10.1002/path.2094. [DOI] [PubMed] [Google Scholar]
- 49.Levine B, Yuan J. Autophagy in cell death: An innocent convict? J Clin Invest. 2005;115:2679–88. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]