

Abstract
Background. Psittacosis is a zoonosis caused by Chlamydia psittaci and is characterized by severe pneumonia and systemic infection. We sought to determine the basis of the 1000-fold difference in lethal dose of 2 C. psittaci 6BC strains in mice.
Methods. Genomes of the strains were sequenced. Mice were infected intraperitoneally and the growth kinetics, immune responses, and pathology were compared.
Results. The 2 strains differed by the presence of a 7.5-kb plasmid in the attenuated strain and 7 nonsynonomous single-nucleotide polymorphisms between the chromosomes, including a serine/threonine protein kinase gene pkn5. The plasmid was cured from the attenuated strain, but it remained nonlethal. Strains did not differ in growth kinetics in vitro or in vivo. Infection with the attenuated strain led to influx of activated macrophages with relatively minor organ damage. In contrast, the virulent strain caused an influx of nonactivated macrophages, neutrophils, and significant end organ damage. Mice infected with the virulent strain survived challenge when coinfected with either the plasmid-positive or plasmid-negative attenuated strain, indicating that an active process elicited by the attenuated strain reduces inflammation and disease.
Conclusions. C. psittaci modulates virulence by alteration of host immunity, which is conferred by small differences in the chromosome.
The genus Chlamydia is a ubiquitous obligate intracellular pathogen in which 3 species cause disease in humans. Chlamydia trachomatis is the most commonly reported bacterial cause of sexually transmitted infection [1] and the leading infectious cause of blindness [2]. Chlamydia pneumoniae causes atypical pneumonia and has been associated with coronary artery disease [3]. Chlamydia psittaci is a zoonotic pathogen that is distinct in disease spectrum and biology, especially by its propensity to cause severe systemic infections and sepsis [4]. C. psittaci is generally acquired from inhalation of aerosolized fecal material of infected birds and causes an acute disease called psittacosis. Clinical manifestations range from a flu-like illness, pneumonitis, to sepsis accompanied by hepatitis and myocarditis [5]. Although infections are relatively rare, highly virulent strains have caused fatal outbreaks as well as sporadic cases of zoonosis around the world [6–12].
Little is known regarding the virulence factors for chlamydiae due to lack of a tractable genetic system. Most inferences regarding disease-modifying virulence factors are derived from comparative genomic studies that correlate disease manifestation with specific genetic alterations [13]. Through such studies several candidates such as the tryptophan synthase, polymorphic membrane proteins, and plasmid-gene products have been implicated [14–18]. Recently, a Chlamydia-specific hypothetical protein CT135 has been identified as a single virulence factor that alters virulence in mouse models of genital tract infection [19]. The chlamydial secreted protease and other effector proteins have been shown to alter cellular functions and are candidate virulence factors. However, these factors have yet to be associated with actual disease severity [20, 21].
In our study, we investigated the differences in 2 stocks of C. psittaci 6BC that were maintained independently for about 30 years but were originally derived from a common source and, subsequently, passed numerous times in cell culture (see Materials and Methods). We found a 1000-fold difference in the lethal dose of these 2 stocks given to mice intraperitoneally. The purpose of our study was to identify genetic and pathogenic differences between these strains that account for the difference in virulence.
MATERIALS AND METHODS
Chlamydia psittaci Strains
The 2 C. psittaci 6BC strains used in this study were originally obtained in the mid-1970s from the laboratory of James Moulder, The University of Chicago. The strains were passaged multiple times and independently in cell cultures by Gerald Byrne and Thomas Hatch. The virulent Byrne strain, designated 6BC/B, and the attenuated Hatch strain, 6BC/H, were biologically cloned by limiting dilution. L cells were seeded to 48-well plates and grown to confluency in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum (FBS). Stocks of C. psittaci were diluted in sucrose-phosphate-glutamate buffer and L cells were inoculated at a rate of 1 inclusion-forming unit (IFU) per every 5 wells in medium containing 2 μg/mL cycloheximide, and plates were observed daily under light microscopy. On days 7–10, wells with obviously visible inclusions (3–5/plate) were collected by scraping and blindly passed to 75-cm2 flasks for propagation.
Chlamydia Infection
C. psittaci 6BC strains were propagated in L cells and titrated by standard methods [22]. Intraperitoneal infection with 104 IFU of C. psittaci 6BC was performed using the same stock source to minimize variations across experiments. Infected mice were monitored daily for weight changes and other signs of illness. At various times after infection, mice were euthanized and peritoneal lavage was collected and used for various assays as described (see below).
Mice and Primary Cell Collection
Eight- to 9-week-old male mice were purchased from Jackson Laboratories. DBA2/J strains were used unless otherwise noted. C3H/HeJ mice mutated at Toll-like receptor (TLR) 4 and TLR2-/-B6.129-Tlr2tm1Kir/J mice were also purchased from Jackson Laboratories. Primary peritoneal macrophages were obtained by sterile elicitation by thioglycollate. Cells were plated to 48-well plates at 5 ×105 cells/mL in RPMI medium supplemented with 10% FBS and pretreated overnight with recombinant mouse interferon gamma (10 ng/mL) where indicated.
Macrophage Depletion
In vivo macrophage depletion was achieved by injection of clodronate liposomes [23, 24]. Clodronate or phosphate-buffered saline (PBS) was encapsulated in liposomes as described [25]. Clodronate was a gift of Roche Diagnostics GmbH. DBA2/J mice received 200 μL of clodronate liposome or PBS liposome control on day –1 (intravenously and intraperitoneally), day 1 (intraperitoneally), and day 3 (intraperitoneally) after infection. Mice were infected with plasmid-cured attenuated C. psittaci 6BC (104 IFU intraperitoneally). Depletion of macrophages in the peritoneal cavity was verified by flow cytometry analysis.
Genetic Analysis
Chlamydial DNA was obtained by using phenol extraction from density-gradient-purified elementary body preparations. Clones obtained from the original stocks, C. psittaci 6BC/B and C. psittaci 6BC/H, were used for genome sequencing. Plasmids were prepared by using Qiagen Miniprep kit. Polymerase chain reaction (PCR) was performed by using primers for plasmid-encoded plasmid open reading frame (ORF) 2 (pORF2) and pORF5, with ompA as control (ORF2 Fw 5- CGT ACA AAGTCT TCA GCT TAT AAC GCC C –3, ORF 2 Rev 5- CAC ACC GTT CTG TCT GAG AAG ACT –3, ORF5 Fw 5- CAA AGT CAA CAC CAA CAG CAG CCA –3, ORF5 Rev 5- ACA AGA GCG AGA ACG ACATTC CCT –3). Genome sequencing was performed by generating a DNA library using the Illumina preparation kit (PN 11251892), according to the manufacturer’s protocol. In brief, the genomic DNA is fragmented to <800 base pairs (bp) using a nebulization technique, The overhangs resulting from fragmentation is repaired into blunt ends, then an adenosine base is added to the 3' end, preparing the DNA fragments for ligation to the adapters. The adapter-modified DNA fragments are enriched by PCR. The library was analyzed by using the Illumina Genome Analyzer at the St Jude Children’s Hospital Hartwell Center. Readings were reconstructed using the original genome sequence, which was obtained by Dr G. Myers at the University of Maryland (genome accession: CP002586.1), then was annotated by using the Rapid Annotation using Subsystem Technology (RAST) server [26, 27]. Reads were aligned to the annotated sequence by using CLCbio Genomic Workbench. Average 230× coverage was achieved. Verification of single-nucleotide polymorphisms (SNPs) was performed by generating PCR fragments flanking the SNPs for pkn5, gatC, dhnA, pmp20G, and rsbU and then sequencing bidirectionally.
Expression of Recombinant Protein and Generation of Polyclonal Antibody
We generated recombinant C-terminal His-tagged Pgp3 protein by using pET23b+ vector and primers, as described by Storni et al [28]. We purified the His-tagged proteins under native conditions by using Ni-NTA superflow (Qiagen 30410) and Detoxigel (Thermo 20339) according to the manufacturers’ instructions to minimize the lipopolysaccharide (LPS) contamination. Polyclonal mouse antibody was raised in DBA2/J mice by intramuscular injection of 1μg of purified Pgp3 2 weeks apart and collection of serum after 6 weeks. In addition to our polyclonal mouse antibody, a rabbit anti-Pgp3 polyclonal antibody was generated (ProSci).
Plasmid Curing
We cured the plasmid from the attenuated C. psittaci 6BC/H strain by using the DNA gyrase inhibitor novobiocin (Sigma), as reported in literature [29]. L cells were infected with the attenuated, plasmid-containing C. psittaci in the presence of novobiocin (125 μg/mL), and chlamydiae were harvested 48 hours after infection. We assessed the efficiency of the novobiocin in curing the plasmid by taking a portion of the treated sample and infecting a monolayer of L cells and examining inclusions by dual immunofluoresence staining with antibodies against chlamydial LPS and the plasmid encoded protein Pgp3. The curing efficiency was low (<5%), and chlamydiae were passaged in the presence of novobiocin treatment 4 times to enrich for the plasmid-negative population. Clonal strains were obtained by limiting dilutions and then screened for the presence or absence of the plasmid by PCR for plasmid ORF2.
Flow Cytometry
Murine peritoneal exudates were blocked with Fc block (BD 553141) and incubated with fluorochrome-conjugated antibodies (Abs). Stained cells were analyzed on a BD LSR II by using FACSDiva software. The following Abs were used: myeloid, CD11b-PECy7 (BD552850); macrophage, F4/80-APC (MF48005); neutrophil, Ly6G-PE (BD clone 1A8 551461); and major histocompatibility complex (MHC) class II, IA/AE-Alexa488 (BD 557000).
Cytokine Analysis
Peritoneal lavage supernatant fluids were stored at −80°C until assessment. Tumor necrosis factor alpha (TNF-α; Biosource) and chemokine (C-X-C motif) ligand 1 (CXCL1) (Biosource CMC 1063) were measured by enzyme-linked immunosorbent assay with the Cytoset kit, according to the manufacturer’s protocol.
Immunofluorescence
HeLa cells were grown on glass coverslips and then infected. Host cells were fixed in methanol and stained or formalin fixed and permealized by saponin for intracellular staining, then incubated with primary antibody against Pgp3 and goat antimouse secondary antibody labeled with FITC. Rabbit polyclonal anti-Chlamydia LPS antibody (Fitzgerald 20-CR19) was used with antirabbit secondary antibody labeled with Texas red.
Histology
Various organs were obtained from mice euthanized 7 days after infection and fixed in 10% formalin. Specimens were sent to RADIL (Research Animal Diagnostic Laboratory) for histological diagnosis and digital photography.
RESULTS
Two Related Strains of C. psittaci 6BC Differ in Lethal Dose by 1000-Fold
Intraperitoneal infection with C. psittaci 6BC has been shown to cause a predictable disease course in susceptible strains of mice such as DBA2/J with a lethal dose as low as 10 infectious particles [30]. C. psittaci disseminates to multiple organs and causes peritonitis, pericarditis, and myocarditis characterized by significant neutrophil influx, weight loss, and death approximately 1 week after infection (Figure 1) [30]. We were surprised to find that 2 stocks of C. psittaci 6BC, which were maintained independently for over 30 years, varied considerably in their ability to kill mice by the intraperitoneal route. We cloned the 2 stocks by limiting dilution and found a >1000-fold difference in lethal dose (LD100) between the more virulent strain (6BC/B) and the attenuated strain (6BC/H) (Figure 1). The original attenuated uncloned C. psittaci 6BC/H stock had a lower LD100 than the cloned strain, indicating that the uncloned stock contained a heterogeneous population of virulent and attenuated organisms (Figure 1).
Figure 1.

A–C, Pathology in mice infected with Chlamydia psittaci 6BC/B. DBA2/J mice were infected intraperitoneally (104 inclusion-forming units [IFU]) and euthanized on day 7 after infection for histological analysis. (A) Lung: infiltration of inflammatory cells in the interstitial tissue and serosa. B, Heart: inflammatory cells in the pericardium. C, Liver: thick layer of inflammatory cells on surface and microabscess in the parenchyma. D, Lethal dose determination of C. psittaci 6BC strains. Original and clonal stocks of virulent C. psittaci 6BC/B and attenuated C. psittaci 6BC/H were prepared at inocula of 102–105 IFU and injected intraperitoneally. Percentage of mice (n = 5) that survived challenge is shown.
Virulent and Attenuated Strains Have Similar Growth Kinetics
We investigated the in vitro growth of the 2 cloned strains in fibroblasts and peritoneal macrophages but did not find significant differences (Figure 2). Furthermore, the in vivo growth of both strains was also nearly identical in DBA2/J mice infected intraperitoneally (Figure 2). Both strains disseminated systemically to the liver, lungs, and spleen, although the pathogen load in the liver tended to be 3–10-fold higher for the virulent strain (Figure 2).
Figure 2.

Growth characteristics of virulent and attenuated Chlamydia psittaci strains. A, Primary fibroblasts were grown from the peritoneal membrane of DBA2/J mice in 24-well plates and infected with C. psittaci in triplicates with or without interferon gamma (IFN-γ) (10 ng/mL). Infected cells were collected 48 hours after infection and titrated along with the initial inoculum. The burst size (output/input ratio) is compared on a log scale. B, Primary peritoneal macrophages were plated at 5 × 105 cells per well, incubated with or without IFN-γ (10 ng/mL) overnight, then infected at a multiplicity of infection of 1 for 48 hours in triplicates. The burst size is compared on a log scale. C, DBA2/J mice were infected intraperitoneally and euthanized on days 2, 5, and 7 and peritoneal lavage was obtained. The number of C. psittaci in samples was calculated by inclusion-forming unit (IFU) assay. No significant difference in load was observed (n = 3). D, Mice were infected intraperitoneally and killed on day 7, and the number of chlamydiae (IFU) in the peritoneal lavage and the liver was determined. Data are shown as a ratio of chlamydial load in the liver vs the amount in the peritoneal lavage. The ratio was greater for the virulent strain by 2–3-fold but was not statistically significant (n = 3: P = .20).
Virulent and Attenuated Strains Induce Qualitative Differences in Inflammation
Gross pathology in mice infected intraperitoneally with the virulent strain was characterized by a purulent infiltrate and pale appearance of the liver, whereas the organs of mice infected with the attenuated strains appeared relatively normal (Figure 3). The liver from mice infected with attenuated strains had numerous microgranulomas scattered throughout the hepatic parenchyma, whereas liver tissue of virulent strain–infected mice showed numerous microgranulomas but also microabscesses throughout the parenchyma of liver with granular degeneration of hepatocytes. Characterization of the cellular infiltrates by flow cytometry analysis showed a significant difference in the number of neutrophils recruited to the peritoneal cavity (Figure 3) by mice with virulent strains: 4.25 ± 2.0 × 105 vs attenuated strains: 6.31 ± 2.1 × 104; P < .04). There was also a modest but significant difference in the quantity of neutrophil-recruiting chemokines (Figure 3). Histological findings and morphological differences in mononuclear cells from the peritoneal cavity (Figure 4) led us to investigate the macrophage activation status, as measured by the ratio of F4/80 positive cells expressing MHC class II molecules over time (Figure 4). We found that macrophages from mice infected with the virulent strain became partially activated initially, but failed to fully activate and may revert to a nonactivated status, whereas the majority of the macrophages in mice infected with the attenuated strains gradually became activated over time. Given this apparent importance of macrophages in pathogenesis, we depleted macrophages and infected mice with the attenuated strain. This led to significant weight loss and the mice became moribund (Figure 4). We also observed a significant increase in neutrophil influx and a modest increase in pathogen load (8.2 ± 7.7 × 105 IFU vs 3.4 ± 2.2 × 106 IFU; P = .03), substantiating a role for activated macrophages in disease attenuation.
Figure 3.
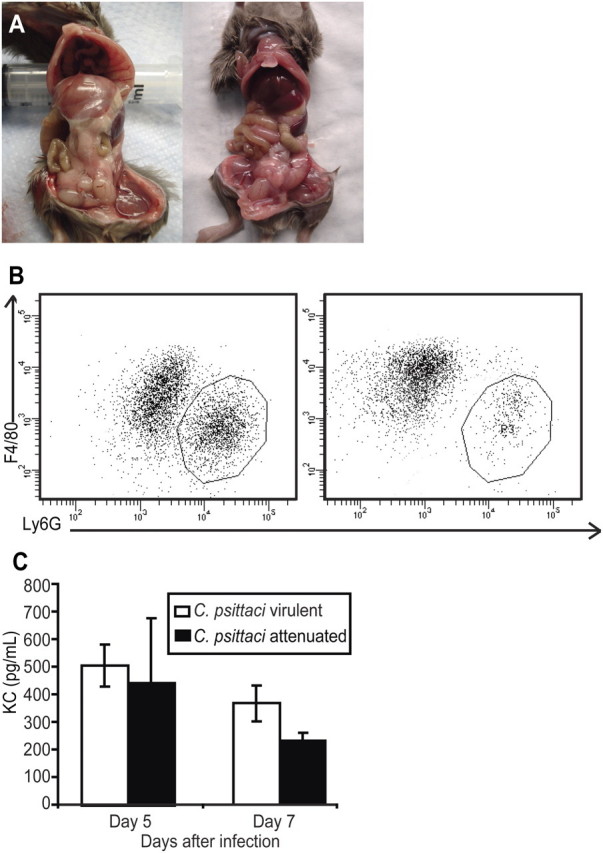
A, Gross pathology of mice infected intraperitoneally. Purulent material was detected in the peritoneal cavity of mice infected for 7 days with the virulent strain (left) and the attenuated strain (right). B, Flow cytometry analysis of the peritoneal lavage: virulent strain (left) and attenuated strain (right). The population was gated on CD11b-expressing myeloid cells, then differentiated by the neutrophil marker (Ly6G) and the macrophage marker (F4/80). The circled area signifies the neutrophil population. Overall, there were 5–10-fold more neutrophils in the peritoneal cavity of mice infected with the virulent strain. C, Enzyme-linked immunosorbent assay of the neutrophil-recruiting chemokine (C-X-C motif) ligand 1 (CXCL1). Cells were collected from the peritoneal lavage fluid of mice infected with the virulent or attenuated strains on days 5 and 7. There was a statistically significant difference on day 7 (P = .012).
Figure 4.
A, Morphology of cells in the peritoneal lavage fluid. Cells were collected from mice at 6 days after infection and plated in a 24-well culture dish, then washed and fixed after a few hours. Cells from mice infected with the attenuated strain had an epitheloid morphology (left), whereas cells from mice infected with the virulent strain had a monocytic appearance (right). Macrophage (mΦ) activation status was monitored over time by flow cytometry using the F4/80 macrophage marker and I-A/I-E class II marker. B, Representative flow cytometry plots of lavage from mice infected with virulent or attenuated strains. The activated macrophage population appears in the right upper quadrant. C, Rate of F4/80-positive macrophages expressing class II markers plotted over time. Each data point represents average of 3 mice. Macrophages gradually became activated when infected with the attenuated strain in contrast to a leveling off and apparent loss of macrophage activation in mice infected with the virulent strain. D, Weight loss in macrophage-depleted mice infected with the attenuated stains. Mice were treated with clodronate or phosphate-buffered saline (PBS) control on days –1, 1, and 3 after infection with the attenuated strain. Macrophage-depleted mice lost significant weight and became moribund by day 4.
Genomic Characterization
We sought to identify candidate virulence factors by comparative genomics. The C. psittaci genome of both the attenuated and virulent strains consisted of 1 171 660 bp encoding 1029 ORFs. We found that the attenuated strain possessed a 7.5-kb plasmid, which is relatively well conserved across various Chlamydia species but was absent in the virulent strain (Figure 5). We found that the plasmid-positive attenuated C. psittaci strain expressed the plasmid-encoded peptide Pgp3 mid to late in the developmental cycle with a punctate staining pattern on immunofluoresence assay, suggesting that only a small subpopulation of dividing chlamydiae expressed this protein (Figure 5). At very late times after infection, inclusions became uniformly stained, suggesting that Pgp3 may have been secreted from chlamydial cell bodies within the inclusion (not shown). At no time was Pgp3 detected above background levels outside of the inclusion. Inclusions from the plasmid-negative virulent strain were negative for Pgp3 (not shown). Apart from the plasmid, the 1.1-Mb chromosome was nearly identical for both strains except for 11 SNPs that included 1-bp differences in 7 chromosomal genes, resulting in 1 amino acid alteration in each of these proteins (Table 1). None of the amino acid changes encoded alterations in putative functional domains for annotated gene products. Full sequence information is available in GenBank (genome accession: CP002586.1).
Figure 5.
A, Plasmid content of virulent and attenuated clones. PCR was performed on total DNA preparations from four virulent (V) and attenuated (A) clones. Top: amplification of chromosomally encoded ompA (control); bottom: PCR for plasmid ORF2. All attenuated strains had evidence of the plasmid, whereas the virulent clones were plasmid-free. B, Location of Pgp3 protein in a HeLa cell infected with a Chlamydia psittaci plasmid-positive clone for 26 hours. Staining was performed with anti-RpoD (Sigma66) antibody and Texas red secondary antibody, anti-Pgp3 antibody and FITC secondary antibody, and Hoechst stains (a merged image is shown). Pgp3 was observed as discrete punctuate forms which appear to be reticulate bodies that are also positive for RpoD; however, the majority of RpoD-staining chlamydiae were negative for Pgp3, and Pgp3 was not detected outside of the inclusion. C, Enzyme-linked immunosorbent assay (ELISA) for tumor necrosis factor alpha (TNF-α) in the supernatants of peritoneal macrophages cultured from wild-type (WT) (C57BL/6J) and Toll-like receptor (TLR) 2-/- mice infected in vitro with virulent and attenuated strains. Although there was a difference in TNF-α secretion between macrophages from WT and TLR2-/- mice, there was no significant difference in TNF-α production by cells infected with plasmid-positive attenuated and plasmid-negative virulent strains. SPG, sucrose-phosphate-glutamate buffer control. D, Weight loss in infected WT and TLR2-/- mice. Mice were infected intraperitoneally with 104 inclusion-forming unit (IFU) of the virulent strain of C. psittaci 6BC. No significant difference in weight between the 2 strains was noted. E, ELISA assay for TNF-α in culture supernatants of peritoneal macrophages treated with recombinant Pgp3. Pgp3 failed to stimulate production of TNF-α. Controls: macrophages infected with virulent and attenuated strains. No statistically significant difference was observed between TNF-α levels from virulent and attenuated infected macrophage culture supernatants. F, Loss of the plasmid from the attenuated strain after 4 passages in the presence of novobiocin. Cells were stained at 48 hours after infection for Pgp3 (FITC-green), indicating the presence of plasmid in chlamydiae within inclusions, and for lipopolysaccaride (Texas red) to identify infected cells. Approximately one-half of the cells were plasmid-free (stained red), compared to <5% in the uncloned stock (not shown). Chlamydiae from passage 4 were biologically cloned to obtain 7 completely plasmid-free clones, as determined by polymerase chain reaction analysis for plasmid gene pORF2. G, Effect of curing the attenuated strain of the plasmid on weight loss in infected mice. DBA2/J mice were infected with 7 different cured clones, 2 different plasmid–containing clones, or 2 different virulent clones (plasmid-free) (n = 2 per clone) No significant difference in virulence, as measured by weight loss, was noted when the attenuated strain was cured.
Table 1.
Comparative Sequence Analysis of Virulent and Attenuated Chlamydia psittaci Strains
| Gene/locus name | GenBank accession number | Annotation | SNP virulent/attenuated strains | Amino acids | Substitution | |
| 1 | pkn5 | AEB55062.1 | serine/threonine-protein kinase | A/G | 503 | S497G |
| 2 | Intergenic | Intergenic (-55 yscC) | A/G | … | … | |
| 3 | Intergenic | Intergenic (-19 euo) | G/A | … | … | |
| 4 | gatC | AEB55318.1 | Glutamyl-tRNA(Gln) amidotransferase subunit C | A/C | 101 | K97T |
| 5 | Intergenic | Intergenic | G/A | … | … | |
| 6 | dhnA | AEB55524.1 | Fructose-biphosphate aldolase class I | A/G | 350 | I347T |
| 7 | Intergenic | Intergenic | G/A | … | … | |
| 8 | G5O_0564 | AEB55549.1 | hypothetical protein | T/C | 274 | I98T |
| 9 | Pmp20G | AEB55638.1 | Polymorphic membrane protein | C/A | 850 | P186Q |
| 10 | rsbU | AEB55977.1 | sigma regulatory protein-pp2c phosphatase (serine/threonine protein phosphatase) | A/C | 607 | S601R |
NOTE. Genome accession: CP002586.1. SNP, single-nucleotide polymorphism.
Plasmid-Positive and Plasmid-Negative C. psittaci Strains Do Not Differ in Their Ability to Stimulate TLR2
The presence or absence of the plasmid in Chlamydia muridarum strains has been associated with their ability to stimulate TLR2. Therefore, we infected macrophages from TLR2 knockout mice in vitro with plasmid-positive (attenuated) and plasmid-negative (virulent) C. psittaci. Although inflammatory cytokine production by both strains was partially dependent on TLR2, there was no significant difference between the 2 strains in TNF-α induction (Figure 5). Infection of TLR2-/- mice by the intraperitoneal route did not result in any significant difference in weight change compared to control (Figure 5).
C. psittaci Protein Pgp3 Fails to Stimulate Macrophages
The plasmid-encoded protein Pgp3 has recently been described as a secreted protein that induces a proinflammatory response in macrophages [31]. We expressed and purified the recombinant Pgp3 protein through multiple passes through the polymyxin B columns until the preparation no longer reacted to the limulus amebocyte lysate kit. Our recombinant Pgp3 failed to stimulate macrophages (Figure 5). We also used macrophages derived from TLR4-negative C3H/HeJ mice and applied low-passage Pgp3 but found no evidence of macrophage stimulation (data not shown). Furthermore, direct administration of the recombinant Pgp3 protein in vivo in C3H/HeJ mice did not impact inflammatory response or C. psittaci disease in mice (data not shown).
Chlamydia Plasmid Does Not Affect Disease Severity of C. psittaci Infection
We cured the plasmid from the attenuated strain using the DNA gyrase inhibitor novobiocin (Figure 5), obtaining 7 plasmid-cured clonal strains. Less than 5% of inclusions were negative for plasmid-encoded Pgp3 by immunofluoresence but 30%–40% were negative after 4 passages through novobiocin-treated cell culture. These plasmid-cured strains were intraperitoneally injected into mice. Surprisingly, none of the mice infected with the plasmid-cured strains lost a significant amount of weight or became ill, establishing that the presence or absence of the plasmid is unrelated to virulence in this systemic infection model of C. psittaci (Figure 5).
Coinfection With the Virulent C. psittaci Strain and Plasmid-Positive or Plasmid-Negative Attenuated Strains Results in Survival of Mice
Given the apparent difference in induction of inflammation by the 2 strains, we attempted to modify the inflammation caused by the virulent strain by the coinfection with the attenuated strain. We hypothesized that the specific inflammation triggered by the attenuated strain would override the nonspecific acute inflammation caused by the virulent strain. As hypothesized, coinfection with the virulent and attenuated strains resulted in survival of the mice in a dose-dependent manner, with the attenuated strain enhancing survival (Figure 6). We repeated this experiment using the plasmid-cured attenuated strain and found similar results, confirming that the plasmid does not play a significant role in the attenuated virulence or inflammatory response in this model (Figure 6).
Figure 6.

Rescue of virulent-strain infected mice by coinfection with the attenuated strain. A, Survival of DBA2/J mice that were infected intraperitoneally (102 inclusion-forming unit [IFU]) with virulent only or coinfected with 3 different inocula (102, 103, and 104 IFU) of the attenuated, plasmid-containing Chlamydia psittaci strain. B, Changes in weight in DBA2/J mice that were infected intraperitoneally (102 IFU) with virulent only or coinfected with 3 different inocula (102, 103, 104 IFU) of a plasmid-cured attenuated stain. The experiments demonstrate that coinfection with an attenuated strain, with or without the plasmid, protects mice from weight loss and death induced by the virulent strain.
DISCUSSION
Microbial virulence is defined as the capacity to cause disease and is mediated by factors that promote microbial survival and inflict host damage. In our model of systemic C. psittaci infection, the growth kinetics of the virulent and attenuated strains was similar, despite the qualitative difference in the inflammation and pathology observed. We characterized the inflammatory responses at the primary site of infection and found that infection with the attenuated strains was followed by a gradual increase in activated macrophages and a relatively benign disease course. In contrast, following infection with the virulent strain, the macrophage population failed to fully activate and led to an increase in neutrophil infiltrates and dramatic systemic disease characterized by weight loss, inflammatory changes in multiple organs, and death. The importance of activated macrophage recruitment in attenuation of systemic C. psittaci disease was reinforced by the observation that mice depleted of macrophages succumbed to infection with the attenuated strain. Interestingly, we were also able to reverse the disease phenotype of virulent strain infection by coinfection with the attenuated strain. These findings suggest that the difference in disease severity is due to the ability of the strains to differentially activate macrophages. Other studies have associated differential effects of virulent and attenuated C. psittaci strains on macrophage morphology and inflammatory responses in vitro, but to our knowledge, this is the first in vivo correlation [32, 33].
Macrophages recognize pathogens via pattern recognition receptors and are activated according to morphologic and functional criteria [34]. Our findings suggest that infection with the attenuated strain actively modulates the host response, leading to macrophage activation. In contrast, the virulent strain is not capable of recruiting activated macrophages or inhibiting macrophage activation; thus, the degree of disease appears to be augmented as a result of the acute neutrophil-based response.
We attempted to identify genetic factors that might account for differences in virulence. Because the original virulent and attenuated C. psittaci stocks differed in the presence or absence of the plasmid, the plasmid appeared to be a strong virulence factor candidate in our model. Furthermore, plasmid-deficient strains of C. muridarum were shown to cause less pathology in the genital tract of mice and to be correlated with the inability to stimulate host TLR2 [35]. In addition, C.trachomatis L2 strains deficient in the plasmid were found to have decreased ability to colonize and infect the mouse genital tract [16]. This is clearly not the case in the C. psittaci intraperitoneal infection model, where attenuation was found to be associated with a specific clonal strain, irrespective of the presence or absence of the plasmid.
Multiple studies have demonstrated that induction of proinflammatory cytokines by Chlamydia species is mediated through the TLR2/TLR1/TLR6 pathway [36–39]. Additional roles of TLR4 and CD14 have also been described [39]. We verified that macrophage stimulation with C. psittaci is partially dependent on TLR2, but the presence or absence or the plasmid had no effect on this response. In addition, TLR2 had little or no effect on disease severity in our systemic infection model, suggesting that activation of macrophages can be achieved through alternative routes. The plasmid-encoded Pgp3 protein has been reported to be secreted into the cytoplasm of infected cells and to exhibit direct macrophage-activating properties in vitro [31]. In our hands we found that recombinant C. psittaci Pgp3 did not stimulate macrophages. We also did not find evidence of detectable distribution of Pgp3 in the cytoplasm or membrane fraction of C. trachomatis as described previously [40], although secretion of small amounts cannot be ruled out by our immunofluorescence studies.
Our studies with plasmid-cured attenuated Chlamydia strains suggest that 1 or more of the 11 genomic SNPs are responsible for the difference in virulence between the attenuated C. psittaci 6BC/H and virulent 6BC/B strains. It is unclear whether these variations evolved over time during passage through cell culture or an existing variant was selected over the other. One of our candidate genes is a homologue of the eukaryotic-like serine/threonine protein kinase gene pkn5, which encodes a candidate effector protein of the Chlamydia type III secretion system (TTSS) [41]. Sequence analysis revealed a nonsynonomous change in the attenuated strain at a potential phosphorylation site in the C-terminus of Pkn5 that alters a conserved serine residue to a glycine. The pkn5 gene is a part of an operon encoding a conserved TTSS [41, 42], and secretion of C. trachomatis Pkn5 via TTSS has been demonstrated in an orthologous Salmonella typhimurium system [43]. Additionally, C. pneumoniae Pkn5 has been shown to localize to the inclusion membrane and may interface with the host and serves as a potential candidate [44]. Although C. trachomatis Pkn5 (CT673) lacks the activation domain I as well as the critical arginine residue in domain XI and has not been functionally characterized as a kinase [45], Herrmann et. al report that Pkn5 homologues of C. pneumoniae (Cpn0703) and C. psittaci retain this arginine residue on domain XI [44]. Still, we also found several other nonsynonomous changes in potential host signaling genes, such as ORF643 (polymorphic membrane protein) and ORF982 (serine/threonine phosphatase), and further evaluation is needed to define a virulence factor.
In summary, our results suggest that a remarkably small number of point mutations in the chromosome can drastically alter the inflammatory response and virulence caused by C. psittaci.
Funding
This work was supported by grants from Children’s Infection Defense Center/St Jude Children’s Research Hospital to I. M., National Institutes of Health/National Institute of Allergy and Infectious Diseases (NIH/NIAID) (AI081050 to I. M., AI19570 to T. P. H., and AI019782 to G. I. B.); American Heart Association (09GBIA2050135 to I. M.); and Department of Defense (W81XWH-1-0391 to G. I. B. and I. M.).
Acknowledgments
The authors thank Dr Gary Myers at the University of Maryland for providing the original sequence and annotations.
References
- 1.Peipert JF. Clinical practice. Genital chlamydial infections. N Engl J Med. 2003;349:2424–30. doi: 10.1056/NEJMcp030542. [DOI] [PubMed] [Google Scholar]
- 2.Burton MJ, Mabey DC. The global burden of trachoma: a review. PLoS Negl Trop Dis. 2009;3:e460. doi: 10.1371/journal.pntd.0000460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalayoglu MV, Libby P, Byrne GI. Chlamydia pneumoniae as an emerging risk factor in cardiovascular disease. JAMA. 2002;288:2724–31. doi: 10.1001/jama.288.21.2724. [DOI] [PubMed] [Google Scholar]
- 4.Longbottom D, Coulter LJ. Animal chlamydioses and zoonotic implications. J Comp Pathol. 2003;128:217–44. doi: 10.1053/jcpa.2002.0629. [DOI] [PubMed] [Google Scholar]
- 5.Beeckman DS, Vanrompay DC. Zoonotic Chlamydophila psittaci infections from a clinical perspective. Clin Microbiol Infect. 2009;15:11–7. doi: 10.1111/j.1469-0691.2008.02669.x. [DOI] [PubMed] [Google Scholar]
- 6.Gregory DW, Schaffner W. Psittacosis. Semin Respir Infect. 1997;12:7–11. [PubMed] [Google Scholar]
- 7.Buttery RB, Wreghitt TG. Outbreak of psittacosis associated with a cockatiel. Lancet. 1987;2:742. doi: 10.1016/s0140-6736(87)91101-9. [DOI] [PubMed] [Google Scholar]
- 8.Gaede W, Reckling KF, Dresenkamp B, et al. Chlamydophila psittaci infections in humans during an outbreak of psittacosis from poultry in Germany. Zoonoses Public Health. 2008;55:184–8. doi: 10.1111/j.1863-2378.2008.01108.x. [DOI] [PubMed] [Google Scholar]
- 9.Heddema ER, van Hannen EJ, Duim B, et al. An outbreak of psittacosis due to Chlamydophila psittaci genotype A in a veterinary teaching hospital. J Med Microbiol. 2006;55:1571–5. doi: 10.1099/jmm.0.46692-0. [DOI] [PubMed] [Google Scholar]
- 10.Matsui T, Nakashima K, Ohyama T, et al. An outbreak of psittacosis in a bird park in Japan. Epidemiol Infect. 2008;136:492–5. doi: 10.1017/S0950268807008783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weston VC, Mansell P, Allison SP. Family outbreak of psittacosis. Lancet. 1990;335:1226–7. doi: 10.1016/0140-6736(90)92753-5. [DOI] [PubMed] [Google Scholar]
- 12.Williams J, Tallis G, Dalton C, et al. Community outbreak of psittacosis in a rural Australian town. Lancet. 1998;351:1697–9. doi: 10.1016/S0140-6736(97)10444-5. [DOI] [PubMed] [Google Scholar]
- 13.Byrne GI. Chlamydia trachomatis strains and virulence: rethinking links to infection prevalence and disease severity. J Infect Dis. 2010;201(Suppl 2):S126–33. doi: 10.1086/652398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caldwell HD, Wood H, Crane D, et al. Polymorphisms in Chlamydia trachomatis tryptophan synthase genes differentiate between genital and ocular isolates. J Clin Invest. 2003;111:1757–69. doi: 10.1172/JCI17993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kari L, Whitmire WM, Carlson JH, et al. Pathogenic diversity among Chlamydia trachomatis ocular strains in nonhuman primates is affected by subtle genomic variations. J Infect Dis. 2008;197:449–56. doi: 10.1086/525285. [DOI] [PubMed] [Google Scholar]
- 16.Carlson JH, Whitmire WM, Crane DD, et al. The Chlamydia trachomatis plasmid is a transcriptional regulator of chromosomal genes and a virulence factor. Infect Immun. 2008;76(6):2273–83. doi: 10.1128/IAI.00102-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tan C, Hsia RC, Shou H, Carrasco JA, Rank RG, Bavoil PM. Variable expression of surface-exposed polymorphic membrane proteins in in vitro-grown Chlamydia trachomatis. Cell Microbiol. 2011;12:174–87. doi: 10.1111/j.1462-5822.2009.01389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burall LS, Rodolakis A, Rekiki A, Myers GS, Bavoil PM. Genomic analysis of an attenuated Chlamydia abortus live vaccine strain reveals defects in central metabolism and surface proteins. Infect Immun. 2009;77:4161–7. doi: 10.1128/IAI.00189-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sturdevant GL, Kari L, Gardner DJ, et al. Frameshift mutations in a single novel virulence factor alter the in vivo pathogenicity of Chlamydia trachomatis for the female murine genital tract. Infect Immun. 2010;78:3660–8. doi: 10.1128/IAI.00386-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murthy AK, Guentzel MN, Zhong G, Arulanandam BP. Chlamydial protease-like activity factor—insights into immunity and vaccine development. J Reprod Immunol. 2009;83:179–84. doi: 10.1016/j.jri.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valdivia RH. Chlamydia effector proteins and new insights into chlamydial cellular microbiology. Curr Opin Microbiol. 2008;11:53–9. doi: 10.1016/j.mib.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 22.Miyairi I, Mahdi OS, Ouellette SP, Belland RJ, Byrne GI. Different growth rates of Chlamydia trachomatis biovars reflect pathotype. J Infect Dis. 2006;194:350–7. doi: 10.1086/505432. [DOI] [PubMed] [Google Scholar]
- 23.van Rooijen N, Bakker J, Sanders A. Transient suppression of macrophage functions by liposome-encapsulated drugs. Trends Biotechnol. 1997;15:178–85. doi: 10.1016/s0167-7799(97)01019-6. [DOI] [PubMed] [Google Scholar]
- 24.Biewenga J, van der Ende MB, Krist LF, Borst A, Ghufron M, van Rooijen N. Macrophage depletion in the rat after intraperitoneal administration of liposome-encapsulated clodronate: depletion kinetics and accelerated repopulation of peritoneal and omental macrophages by administration of Freund’s adjuvant. Cell Tissue Res. 1995;280:189–96. doi: 10.1007/BF00304524. [DOI] [PubMed] [Google Scholar]
- 25.Van Rooijen N, Sanders A. Liposome mediated depletion of macrophages: mechanism of action, preparation of liposomes and applications. J Immunol Methods. 1994;174:83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 26.Aziz RK, Bartels D, Best AA, et al. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Overbeek R, Begley T, Butler RM, et al. The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 2005;33:5691–702. doi: 10.1093/nar/gki866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Storni E, Donati M, Marangoni A, Accardo S, Cevenini R. Comparative PCR-based restriction fragment length polymorphism analysis of the plasmid gene orf3 of Chlamydia trachomatis and Chlamydia psittaci. FEMS Immunol Med Microbiol. 2006;48:313–8. doi: 10.1111/j.1574-695X.2006.00149.x. [DOI] [PubMed] [Google Scholar]
- 29.O’Connell CM, Nicks KM. A plasmid-cured Chlamydia muridarum strain displays altered plaque morphology and reduced infectivity in cell culture. Microbiology. 2006;152:1601–7. doi: 10.1099/mic.0.28658-0. [DOI] [PubMed] [Google Scholar]
- 30.Miyairi I, Tatireddigari VR, Mahdi OS, et al. The p47 GTPases Iigp2 and Irgb10 regulate innate immunity and inflammation to murine Chlamydia psittaci infection. J Immunol. 2007;179:1814–24. doi: 10.4049/jimmunol.179.3.1814. [DOI] [PubMed] [Google Scholar]
- 31.Li Z, Chen D, Zhong Y, Wang S, Zhong G. The chlamydial plasmid-encoded protein pgp3 is secreted into the cytosol of Chlamydia-infected cells. Infect Immun. 2008;76:3415–28. doi: 10.1128/IAI.01377-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beeckman DS, Rothwell L, Kaiser P, Vanrompay DC. Differential cytokine expression in Chlamydophila psittaci genotype A-, B- or D-infected chicken macrophages after exposure to Escherichia coli O2:K1 LPS. Dev Comp Immunol. 2010;34:812–20. doi: 10.1016/j.dci.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 33.Beeckman DS, Vanrompay DC. Biology and intracellular pathogenesis of high or low virulent Chlamydophila psittaci strains in chicken macrophages. Vet Microbiol. 2010;141:342–53. doi: 10.1016/j.vetmic.2009.09.032. [DOI] [PubMed] [Google Scholar]
- 34.Ma J, Chen T, Mandelin J, et al. Regulation of macrophage activation. Cell Mol Life Sci. 2003;60:2334–46. doi: 10.1007/s00018-003-3020-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.O’Connell CM, Ingalls RR, Andrews CW, Jr, Scurlock AM, Darville T. Plasmid-deficient Chlamydia muridarum fail to induce immune pathology and protect against oviduct disease. J Immunol. 2007;179:4027–34. doi: 10.4049/jimmunol.179.6.4027. [DOI] [PubMed] [Google Scholar]
- 36.Nagarajan UM, Ojcius DM, Stahl L, Rank RG, Darville T. Chlamydia trachomatis induces expression of IFN-gamma-inducible protein 10 and IFN-beta independent of TLR2 and TLR4, but largely dependent on MyD88. J Immunol. 2005;175:450–60. doi: 10.4049/jimmunol.175.1.450. [DOI] [PubMed] [Google Scholar]
- 37.O’Connell CM, Ionova IA, Quayle AJ, Visintin A, Ingalls RR. Localization of TLR2 and MyD88 to Chlamydia trachomatis inclusions. Evidence for signaling by intracellular TLR2 during infection with an obligate intracellular pathogen. J Biol Chem. 2006;281:1652–9. doi: 10.1074/jbc.M510182200. [DOI] [PubMed] [Google Scholar]
- 38.Joyee AG, Yang X. Role of toll-like receptors in immune responses to chlamydial infections. Curr Pharm Des. 2008;14:593–600. doi: 10.2174/138161208783885344. [DOI] [PubMed] [Google Scholar]
- 39.Bas S, Neff L, Vuillet M, et al. The proinflammatory cytokine response to Chlamydia trachomatis elementary bodies in human macrophages is partly mediated by a lipoprotein, the macrophage infectivity potentiator, through TLR2/TLR1/TLR6 and CD14. J Immunol. 2008;180:1158–68. doi: 10.4049/jimmunol.180.2.1158. [DOI] [PubMed] [Google Scholar]
- 40.Comanducci M, Cevenini R, Moroni A, et al. Expression of a plasmid gene of Chlamydia trachomatis encoding a novel 28 kDa antigen. J Gen Microbiol. 1993;139:1083–92. doi: 10.1099/00221287-139-5-1083. [DOI] [PubMed] [Google Scholar]
- 41.Peters J, Wilson DP, Myers G, Timms P, Bavoil PM. Type III secretion a la Chlamydia. Trends Microbiol. 2007;15:241–51. doi: 10.1016/j.tim.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 42.Hefty PS, Stephens RS. Chlamydial type III secretion system is encoded on ten operons preceded by sigma 70-like promoter elements. J Bacteriol. 2007;189:198–206. doi: 10.1128/JB.01034-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ho TD, Starnbach MN. The Salmonella enterica serovar typhimurium-encoded type III secretion systems can translocate Chlamydia trachomatis proteins into the cytosol of host cells. Infect Immun. 2005;73:905–11. doi: 10.1128/IAI.73.2.905-911.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Herrmann M, Schuhmacher A, Muhldorfer I, Melchers K, Prothmann C, Dammeier S. Identification and characterization of secreted effector proteins of Chlamydophila pneumoniae TW183. Res Microbiol. 2006;157:513–24. doi: 10.1016/j.resmic.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 45.Verma A, Maurelli AT. Identification of two eukaryote-like serine/threonine kinases encoded by Chlamydia trachomatis serovar L2 and characterization of interacting partners of Pkn1. Infect Immun. 2003;71:5772–84. doi: 10.1128/IAI.71.10.5772-5784.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]