

Abstract
Myotonic dystrophy type 2 (DM2) is an autosomal dominant disorder caused by the expansion of the tetranucleotidic repeat (CCTG)n in the first intron of the Zinc Finger Protein-9 gene. In DM2 tissues, the expanded mutant transcripts accumulate in nuclear focal aggregates where splicing factors are sequestered, thus affecting mRNA processing. Interestingly, the ultrastructural alterations in the splicing machinery observed in the myonuclei of DM2 skeletal muscles are reminiscent of the nuclear changes occurring in age-related muscle atrophy. Here, we investigated in vitro structural and functional features of satellite cell-derived myoblasts from biceps brachii, in the attempt to investigate cell senescence indices in DM2 patients by ultrastructural cytochemistry. We observed that in satellite cell-derived DM2 myoblasts, cell-senescence alterations such as cytoplasmic vacuolization, reduction of the proteosynthetic apparatus, accumulation of heterochromatin and impairment of the pre-mRNA maturation pathways occur earlier than in myoblasts from healthy patients. These results, together with preliminary in vitro observations on the early onset of defective structural features in DM2 myoblast derived-myotubes, suggest that the regeneration capability of DM2 satellite cells may be impaired, thus contributing to the muscular dystrophy in DM2 patients.
Key words: myotonic dystrophy type 2 - DM2, myoblasts, RNA processing, electron microscopy, immunocytochemistry.
Introduction
Myotonic dystrophies (DMs) are autosomal dominant disorders exhibiting a variety of multisystemic features among which muscular dystrophy, myotonia, dilated cardiomyopathy, cardiac conduction defects, cerebral abnormalities, insulin-resistance, and cataracts;1–3 in addition, disease-specific serological abnormalities are present, such as hyperglycemia and increased levels of gamma-glutamyltrans-ferase,4,5 hypotestosteronism, and decreased levels of IgG and IgM immunoglobulin.6 The more frequent and severe DM form is the DM1-Steinert’s disease (OMIM 160900), which affects 1:8000 individuals worldwide, and is caused by an expanded (CTG)n nucleotide sequence in the 3′ untranslated region of the Dystrophia Myotonic Protein Kinase (DMPK) gene (OMIM 605377) on chromosome 19q13.7–9 In 1999, a second DM form displaying a milder clinical phenotype has been identified:10 it has been essentially described in European Countries and has been called DM2 (OMIM 602688); it is much less frequent than DM1 since it affects 1:20000 individuals, although a recent genotypic study on Finnish patients11 suggested that DM2 frequency could be much higher than previously estimated; it is caused by the expansion of the tetranucleotidic repeat (CCTG)n in the first intron of the Zinc Finger Protein (ZNF-9) gene (OMIM 116955)12 on chromosome 3q21.13
Due to its less severe phenotype, diagnosis of DM2 is sometimes difficult and may be made relatively later, compared to DM1; to face this difficulty, the localization of ribonucleoprotein (RNP)-enriched domains (called foci) in DM2 cell nuclei has been recently proposed as a rapid and reliable marker for recognizing subjects affected by this pathology.14,15 In fact, in tissues of DM2 patients, the expanded-CCUG-containing transcripts are retained in the cell nucleus and accumulate in the form of focal aggregates.12 These nuclear foci of mutant mRNA specifically sequester splicing factors among which muscleblind-like (MBNL) proteins,14–17 which are essential for the alternative splicing for muscle cell proteins,18 and the RNP-containing complexes called snRNPs and hnRNPs:19 this leads to the depletion and/or loss of function of these nuclear regulators.20
In a recent study, the distribution of nuclear RNP-containing structures and molecular factors responsible for pre-mRNA transcription and maturation has been described in myonuclei of dystrophic skeletal muscles,21 providing evidence for alterations of post-transcriptional pre-mRNA events as much as it occurs in the myonuclei of skeletal muscles from old rats.22 These findings open interesting perspectives for comparative studies aimed at detecting common cellular mechanisms responsible for the loss of muscle mass, strength and function typical of sarcopenia in aged individuals,23,24 as well as for the muscle alterations caused by DM2, which include fibre atrophy-hypertrophy, increased number of centrally located nuclei, and the presence of fibres with nuclear clumps.3
A common hypothesis proposed to explain skeletal muscle wasting in both sarcopenia and myotonic dystrophy implies a decreased efficiency of muscle regeneration due to hindered activation, proliferation and/or differentiation capability of satellite cells.25–28
In vitro myoblast cultures derived from human satellite cells provide a suitable and unique model for studying DM2 muscular precursor cells, and can be used to elucidate the molecular and cellular mechanisms involved in the pathogenesis of this disease.29,30
In this work we have investigated some structural and functional features of myoblasts obtained from biceps brachii biopsies in the attempt to detect cell senescence traits in DM2 patients in comparison with healthy control subjects. To do this, satellite-cell-derived myoblasts were grown in vitro, and underwent several passages in culture: for sake of simplicity, myoblasts at early (2nd and 3rd) passages were defined as young in culture, while those at late (14th to 17th) passages were defined as senescing. We have applied cytochemical and immunocytochemical techniques at light and electron microscopy as proved valuable tools for studying neuromuscular diseases, especially when cell nuclear functions are involved.31–33 In fact, in eukaryotic cells, maturation of mRNA implies that the primary transcripts (pre-mRNAs) undergo several transformation steps in the spliceosome, a molecular complex composed of five small nuclear RNPs (snRNPs), many non-snRNP splicing factors, and regulating factors.34,35 The structural counterpart of such macromolecular complexes is represented by RNP-containing structures named perichromatin fibrils, perichromatin granules and interchromatin granules: the correct intranuclear distribution and molecular composition of these structures is essential for mRNA maturation, and their ectopic reorganization is a reliable sign of impaired nuclear activity.36–39
Materials and Methods
Sample collection
Samples of biceps brachii muscles were taken from male adult patients affected by DM2 (three subjects, aged 46–55) as well as from three male healthy donors (aged 46–50), after informed consent. The experimental protocols have been approved by the Ethical Committee of the IRCCS Policlinico San Donato. All the subjects were in the adulthood range, and this allowed excluding the influence of possible aging-related changes in nuclear features. The histological diagnosis was performed on serial sections processed for routine histological or histochemical staining, based on the clinical criteria set by the International Consortium for Myotonic Dystrophies.40 The biopsies were trimmed of blood vessels, fat and connective tissues, and rinsed in phosphate-buffered saline (PBS); satellite cells were isolated as reported in Cardani et al.29 and placed in HAM’s F10 medium (Sigma-Aldrich, Buchs, Switzerland) supplemented with 15% fetal bovine serum (Gibco, Invitrogen, S. Giuliano Milanese, Italy), 0.5 mg/mL albumin from bovine serum (BSA, Sigma-Aldrich), 0.5 mg/mL fetuin, 0.39 µg/mL dexamethasone, 10 ng/mL epidermal growth factor, 0.05 mg/mL insulin, 3 mg/mL glucose, 100 U/mL penicillin and 100 µg/mL streptomycin (Sigma-Aldrich). The myoblasts obtained by this procedure were propagated in plastic flasks at 37°C in a humidified 95% air / 5% CO2 atmosphere. For transmission electron microscopy, myoblasts were grown in flasks, whereas for light microscopy the myoblasts were planted on glass coverslips in multiwell plates (all the plastics were from Euroclone, Milan, Italy).
Light microscopy
For fluorescence cytochemistry, cells on glass coverslips were fixed with 4% formaldehyde in PBS for 15 min at 4°C, post-fixed with 70% ethanol at −20°C for 30 min, and rehydrated in PBS at room temperature (RT).
To specifically label myoblasts, the slides were incubated for 60 min at RT with a monoclonal antibody recognizing desmin (Dako, Glostrup, Denmark) diluted 1:100 in PBS containing 0.1% BSA and 0.05% Tween-20, and then incubated for 60 min at RT with an Alexa 488-conjugated anti-mouse IgG (Molecular Probes, Invitrogen), washed in PBS, counterstained for DNA with Hoechst 33258 (Sigma-Aldrich; 1 µg/mL for 5 min), and mounted in Mowiol (Calbiochem, Milan, Italy). 500 desmin-positive cells per patient and passage were scored for BrdU-positivity.
To estimate the S-phase fraction of myoblasts in the cell cultures, experiments of BrdU incorporation were performed. Briefly, cells were pulse-labeled with 40 µM BrdU (Sigma) for 2 h at 37°C, fixed with 70% ethanol for 30 min at −20°C, and incubated for 20 min at RT in 2 N HCl, to denature DNA partially; after neutralization with 0.1 M sodium tetraborate (pH 8.2) for 3 min, the slides were washed in PBS, permeabilized for 15 min in PBS containing 0.1% BSA and 0.05% Tween-20, and incubated with the monoclonal antibody recognizing the myoblast-specific desmin, revealed with an Alexa 568-conjugated anti-mouse IgG (Molecular Probes), as reported above; after two washings with PBS, the slides were then incubated for 1 h with an FITC-conjugated anti-BrdU mouse monoclonal antibody (Clone b44; BD Biosciences, Beckton Dickinson Italia, Milan, Italy) diluted 1:20. The slides were finally counterstained for DNA with Hoechst 33258 (0.1 µg/mL for 10 min), to be finally mounted with Mowiol (Calbiochem).
For fluorescence microscopy, an Olympus BX51 microscope equipped with a 100W mercury lamp was used, under the following conditions: 330–385 nm excitation filter (excf), 400 nm dichroic mirror (dm), and 420 nm long-pass filter, for Hoechst 33258; 450–480 nm excf, 500 nm dm, and 515–550 nm band-pass filter for FITC; 540 nm excf, 580 nm dm, and 620 nm long-pass filter, for Alexa 568. Digital images were recorded with an Olympus Magnifire digital camera system, and stored on a PC by the Olympus software, for processing and printing.
The same Olympus BX51 microscope previously described was also used for phase contrast microscopy, under the appropriate conditions; in this case, micrographs were taken with an Olympus Camedia 5050 digital camera. The percentage of myoblasts containing vacuoles was estimated on 25 randomly selected microscope fields per sample using a 40x objective lens; at least 500 cells per sample were considered.
In order to provide in situ evidence for the presence of senescent myoblasts, β-galactosidase was detected according to Dimri et al.41 In brief, samples were washed in PBS and fixed with 2% paraformaldehyde and 0.2% glutaraldehyde in 0.1 M PBS, pH 7.4, for 7 min at RT. After, cells were incubated for 18 h at 37°C in fresh senescence associated SA-β-gal staining solution [1 mg/mL 5-bromo-4-chloro-3-inolyl-b-D-galactoside (X-gal) in dimethylformamide (20 mg/mL stock); 40 mM citric acid/sodium phosphate, pH 6.0; 5 mM potassium ferrocyanide; 150 mM NaCl; 2 mM MgCl2]. After 2 washings in PBS, cells were mounted in a drop of Mowiol.
Transmission electron microscopy
Myoblasts at early (2nd – 3rd) and late (14th to 17th) passages (as defined after BrdU-incorporation experiments, see Results) were fixed in the flasks and then collected by scraping. For conventional morphology, cells were fixed with 2.5% glutaraldehyde and 2% paraformaldehyde in 0.1 M Sörensen phosphate buffer at 4°C for 1 h, washed, post-fixed with 1% OsO4 at 4°C for 2 h, dehydrated with acetone and embedded in Epon. For immunoelectron microscopy, cells were fixed with a mixture of 4% paraformaldehyde and 0.2% glutaraldehyde in 0.1 M Sörensen phosphate buffer at 4°C for 1 h, washed, treated with NH4Cl 0.5 M in PBS, dehydrated with ethanol and embedded in LR White resin.
Ultrathin sections from Epon-embedded samples were conventionally stained with uranyl acetate and lead citrate.
Ultrathin sections from LR White-embedded samples were placed on Formvar-carbon coated nickel grids and processed for immunocytochemistry. The sections were treated with the following probes: mouse monoclonal antibodies directed against the phosphorylated, activated form of RNA polymerase II (Research Diagnostic Inc., Flanders, NJ, USA) or the (Sm)snRNP (small nuclear RNP) core protein (Abcam, Cambridge, MA, USA); rabbit polyclonal antibodies directed against the cleavage stimulation factors CStF;42 chicken antibodies directed against the heterogeneous nuclear RNP (hnRNP) core protein.43 Sections were floated for 3 min on normal goat serum (NGS) diluted 1:100 in PBS and then incubated for 17 h at 4°C with the primary antibodies diluted with PBS containing 0.1% bovine serum albumin (Fluka) and 0.05% Tween 20. After rinsing, sections were floated on NGS and were then allowed to react for 30 min at RT with the appropriate secondary (either 6nm- or 12nm-gold-conjugated) antibodies (Jackson ImmunoResearch Laboratories Inc., Suffolk, UK) diluted 1:10 in PBS. Finally, the sections were rinsed and air-dried. As controls, some grids were incubated without the primary antibody and then processed as described above. To reduce chromatin contrast and selectively reveal nuclear RNP constituents, the sections were bleached by the EDTA method44 and observed in a Philips Morgagni TEM operating at 80kV and equipped with a Megaview II camera for digital image acquisition.
Quantitative assessment of the immunolabelling was carried out by estimating the gold grain density over the nucleoplasmic region on sections treated in the same run. As for anti-snRNP antibody, the labelling density was evaluated also on interchromatin granule clusters. The grain densities were measured on fifteen randomly selected electron micrographs (x 22,000) of myonuclei from each patient by using a computerized image analysis system (AnalySIS Image processing, Soft Imaging System GmbH, Munster, Germany). For background evaluation, samples treated in the absence of primary antibody were considered. Gold grains present over the selected compartments were counted and the labelling density was expressed as the number of gold grains/µm2.
In addition, the percentage of the nuclear surface occupied by heterochromatin and the perichromatin granules density (PG number/µm2 of nucleoplasm) were calculated by using the same image analysis system.
Statistical comparisons were performed by the two-way ANOVA test to evaluate the influence of two factors: the presence of the dystrophic condition (we called this factor dystrophy, in Tables 1 and 2) and the increasing passages in culture (cell senescing in vitro, in Tables 1 and 2); in addition, the interaction term between these two factors was also considered. The one-way ANOVA test was used to compare the immunolabelling density on interchromatin granules between myoblasts samples from dystrophic or healthy patients at early and late passage. Statistical significance was set at P<0.05.
Table 1. Two-way ANOVA test of the morphometric evaluation of heterochromatin percentage and perichromatin granule density (PG number/µm2 of nucleoplasm) in myoblasts: factor Dystrophy (healthy and DM2), factor Cell senescing in vitro (early and late passage), and interaction term (Dystrophy - Cell senescing in vitro).
| Heterochromatin % | |
|---|---|
| Dystrophy | P<0.001 |
| Cell senescing in vitro | P=0.031 |
| Interaction term | P=0.042 |
| PG density | |
|---|---|
| Dystrophy | P=0.063 |
| Cell senescing in vitro | P=0.001 |
| Interaction term | P=0.001 |
Table 2. Two-way ANOVA test of the immunolabelling densities (gold grains/µm2) of polymerase II, snRNPs, hnRNPs and CStF in myoblast nucleoplasm: factor Dystrophy (healthy and DM2), factor Cell senescing in vitro (early and late passage), and interaction term (Dystrophy - Cell senescing in vitro).
| Polymerase II | |
|---|---|
| Dystrophy | P<0.001 |
| Cell senescing in vitro | P<0.001 |
| Interaction term | P=0.001 |
| hnRNPs | |
|---|---|
| Dystrophy | P<0.001 |
| Cell senescing in vitro | P=0.002 |
| Interaction term | P<0.043 |
| snRNPs | |
|---|---|
| Dystrophy | P<0.001 |
| Cell senescing in vitro | P<0.001 |
| Interaction term | P<0.001 |
| CStF | |
|---|---|
| Dystrophy | P<0.001 |
| Cell senescing in vitro | P=0.027 |
| Interaction term | P=0.017 |
Results
In primary culture from skeletal muscles, a fraction of fibroblasts is always present and myoblasts can be recognized by their immunopositivity for the muscle specific protein, desmin (Figure 1a, a′). In the cultures used in the present investigation, the myogenic index was always fairly high and ranged from about 60% at the 2nd–3rd passage to more than 90% at the 14th–17th passage, without significant difference between control and DM2 cell populations.
Figure 1.

Phase contrast and fluorescence micrographs of myoblasts from healthy (a,a′ and b,b′) and DM2 patients (c,c′ and d,d′) at early (a and c) and late (b and d) passages in culture, after immunolabeling for desmin. In a′, the myoblast (arrow) may be distinguished from the fibroblast for its thin and elongated shape, and the immunopositivity for desmin (green fluorescence). Nuclear DNA was counterstained with Hoechst 33258 (blue fluorescence). Note the cytoplasmic vacuoles (arrowheads) in DM2 myoblasts (c,d). Scale bar: 20 µm.
At low (2nd –3rd) passages from the explant, desmin-positive myoblasts had an S-phase fraction ranging from 20–26% in both control and DM2 cultures, as estimated after BrdU-incorporation experiments. Myoblasts were considered as senescing at late (14th to 17th) passages when the percentage of BrdU-positive, S-phase cells dropped to less than 5% and 2%, in control and DM2 myoblasts respectively. This suggests that the decrease in the S-phase fraction is faster in DM2 than in control myoblasts. Consistently, the percentage of beta-galactosidase positive cells was higher, with a much stronger intensity of the final reaction products, in senescing DM2 than in healthy myoblasts (about 90 and 80% of positive cells, respectively).
At light microscopy (Figure 1), myoblasts from both control and DM2 patients appeared as thin elongated cells with a centrally located nucleus. Phase contrast microscopy showed that about 15% of young DM2 myoblasts exhibited small roundish vesicles in their cytoplasm (Figure 1c), which were on the contrary rarely found in the controls (less than 4%) (Figure 1a). At late passages, vacuolization of senescing DM2 myoblasts became much more apparent (about 30% of myoblasts contained vacuoles), with large portions of the cytoplasm often occupied by huge vacuoles and devoid of desmin (Figure 1d); about 10% of the myoblasts in senescing controls (Figure 1b) also showed small vacuoles in the cytoplasm, although there was never evidence of the extensive cytoplasm degeneration found in senescing DM2 myoblasts. At electron microscopy, myoblasts were identified by their elongated shape and the organelle features already described in literature.45–49
In the cytoplasm of young control myoblasts (Figure 2 a–c), ovoid or elongated mitochondria, large amounts of free ribosomes, well developed rough endoplasmic reticulum and numerous Golgi complexes were observed; conversely, lysosomes, residual bodies and vacuoles were only seldom found. Myoblast nuclei were generally characterised by a thin heterochromatin rim just beneath the nuclear envelope and one reticular nucleolus with many fibrillar centres and abundant dense fibrillar component (the latter component is the site of pre-rRNA transcription and splicing) (for a recent review on nucleolar components, see50). Senescing control myoblasts (Figure 2 d–f) were even more elongated than at early passages. In the cytoplasm, the organelles belonging to the proteosynthetic apparatus and the mitochondria were abundantly present; in addition, glycogen clusters, residual bodies and vacuoles - even of large size - were frequently observed. Myoblast nuclei showed increased heterochromatin amounts along the nuclear border, while the nucleoli appeared unchanged in comparison to early passages. Young DM2 myoblasts (Figure 3 a–c) were quite similar to the controls, apart from the occurrence of differently sized vacuoles in the cytoplasm, and larger amounts of heterochromatin and higher density of perichromatin granules in the nucleus (Figure 4 and Table 1, for the statistical analysis).
Figure 2.

Young (a–c) and senescing (d–f) healthy control myoblasts. (a) The myoblast shows an elongated shape and one ovoid nucleus with a reticular nucleolus (Nu) and scarce heterochromatin. In the cytoplasm, rough endoplasmic reticulum (arrows) and a Golgi complex (g) are visible. Scale bar: 1 µm. (b,c) Cytoplasmic details showing free ribosomes (asterisk), well developed rough endoplasmic reticulum (arrows), numerous Golgi complexes (g) and ovoid mitochondria (m). Vacuoles (v) are quite scarce. Bars: 2 µm. (d) The myoblast shows a very elongated shape and one elongated nucleus with scarce heterochromatin. In the cytoplasm, rough endoplasmic reticulum (arrows), residual bodies (r) and vacuoles (v) are visible. Scale bar: 2 µm. (e,f) Cytoplasmic details showing free ribosomes (asterisk), ovoid mitochondria (m), a Golgi complex (g), a residual body (r) and clustered glycogen granules (star). Scale bars: 1 µm.
Figure 3.

Young (a–c) and senescing (d–f) DM2 myoblasts. (a) The myoblast shows an elongated shape and one ovoid nucleus with a reticular nucleolus (Nu) and scarce heterochromatin. In the cytoplasm, rough endoplasmic reticulum (arrows), a Golgi complex (g) and many vacuoles (v) are visible. Scale bar: 2 µm. (b,c) Cytoplasmic details showing well developed rough endoplasmic reticulum (arrows), Golgi complexes (g), free ribosomes, elongated mitochondria (m), and many vacuoles with heterogeneous content (v). Cytoplasmic electron lucent areas (stars) sometimes occur. Scale bars: 1 µm. (d) The myoblast shows a very elongated shape and one irregularly shaped nucleus with evident heterochromatin. In the cytoplasm, well developed Golgi complexes (g), and vacuoles (v) are visible. Scale bar: 2 µm. (e) Large cytoplasmic regions are filled with heterogeneous vacuoles (v) or appear as electron lucent areas (stars). Scale bar: 2 µm. (f) Cytoplasmic detail showing rough endoplasmic reticulum (arrow), Golgi complexes (g), glycogen clusters (star) and numerous vacuoles (v). Scale bar: 1 µm.
Figure 4.
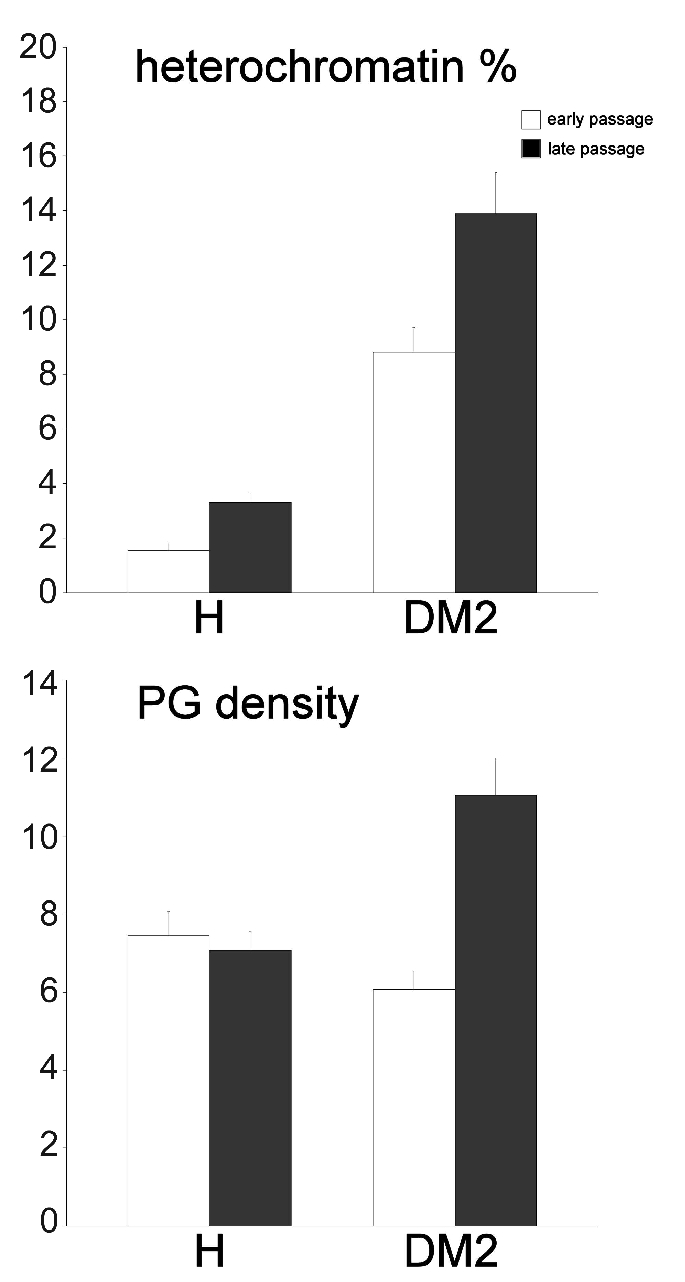
Mean ± SE values of heterochromatin percentage and perichromatin granule density (PG number/µm2 of nucleoplasm) in myoblasts from healthy and DM2 patients (n=45).
Senescing DM2 myoblasts at late passages (Figure 3 d–f) had very elongated shapes and one elongated nucleus with an often irregular border. In the cytoplasm many vacuoles and large electron lucent areas were abundantly present; mitochondria, free ribosomes, rough endoplasmic reticulum and Golgi complexes were markedly reduced in comparison to the early passages, while glycogen clusters and residual bodies were abundant and ubiquitously distributed. Myoblast nuclei frequently showed irregular shapes and evident heterochromatin clumps, and most of the nucleoli showed a reticular structure.
Observation of EDTA-stained samples showed that, in all patients, the usual RNP structural constituents involved in pre-mRNA transcription and processing were evident in the nucleoplasm: perichromatin fibrils and perichromatin granules were mostly distributed in the perichromatin region, and interchromatin granules formed clusters in the interchromatin space.36,37 Immunolabelling experiments showed a similar intranuclear distribution of polymerase II, hnRNPs, snRNPs and cleavage factor CStF in control and DM2 myoblasts at both early and late passages. Activated polymerase II, hnRNPs and CStF were specifically associated with perichromatin fibrils (Figures 5 and 6), which represent the form in situ of pre-mRNA transcription, splicing and 3′-end processing,36,37 while snRNPs were restricted to perichromatin fibrils and interchromatin granules, which represent the storage, assembly and phosphorylation sites for transcription and splicing factors (Figure 6).51,52
Figure 5.

Myoblasts from healthy (a,b) and DM2 (c,d) patients at early (a,c) and late (b,d) passages. Anti-polymerase II (6 nm) and anti-CStF (12 nm) antibodies: both probes specifically label perichromatin fibrils (arrowheads), while the interchromatin granules (ig) are devoid of signal. Arrows indicate perichromatin granules. Ch: bleached heterochromatin. Scale bars: 0.25 µm.
Figure 6.

Myoblasts from healthy (a,b) and DM2 (c,d) patients at early (a,c) and late (b,d) passages. Anti-snRNP (6 nm) and anti-hnRNP (12 nm) antibodies: both probes label perichromatin fibrils (arrowheads); in addition, the anti-snRNP antibody strongly labels interchromatin granules (ig). Arrows indicate perichromatin granules. Ch: bleached heterochromatin. Scale bars: 0.25 µm.
Quantitative evaluation of the immunolabelling (Figure 7) revealed that both factors, dystrophy and cell senescing in vitro, significantly influence the labelling values for all the parameters analysed; moreover, the interaction term was always significant indicating a relationship between the changes during in vitro senescence and the healthy or diseased origin of the cells; in fact, DM2 was always associated with lower levels for all the parameters analysed (Table 2). Anti-snRNP labelling density on interchromatin granule clusters was significantly higher in young than in senescing control myoblasts (120.53±10.04 vs 48.49±3.01 gold grains/µm2, respectively; P<0.001); conversely, in DM2 myoblasts the values were similar at early and late passages (67.86±7.98 vs 69.59±6.92 gold grains/µm2, respectively; P=0.889). Background values were negligible in all immunolabelling experiments (not shown).
Figure 7.
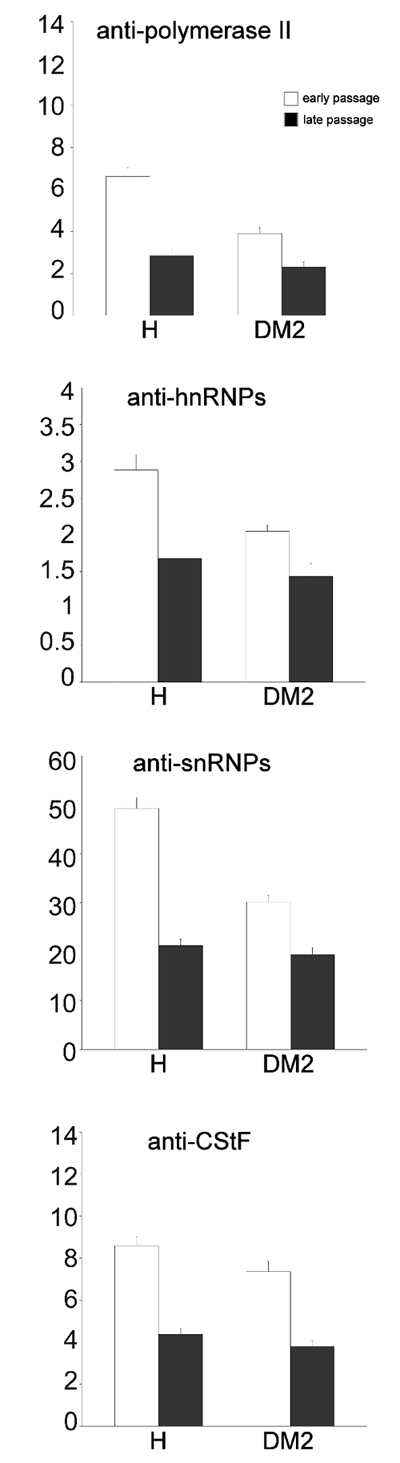
Quantitative immunoelectron microscopy of myoblast nuclei from healthy and DM2 patients (n=45). Each bar represents the mean ± SE values of the labelling density (gold grains/µm2) for different pre-mRNA processing factors in the nucleoplasmic region.
Discussion
The multifactorial phenotype of DM2, consisting of muscle weakness and atrophy, and the wide spectrum of extramuscular manifestations strongly support the hypothesis of a multisystemic syndrome caused by a general alteration of the pre-mRNA post-transcriptional pathway.53–55 However, the cellular mechanisms leading to the highly variable clinical manifestations in the different tissues of DM2 patients have not been clarified yet. In particular, at the skeletal muscle level, no explanation for the muscle wasting has so far been found. It has been hypothesised that, in dystrophic muscle tissue, the myogenic potential is altered, probably in relation to a hindered regenerative capability of satellite cells.25,28,56–59 Interestingly, a similar hypothesis has been also proposed to explain sarcopenia, i.e. the age-related loss of muscle mass and function;23,24 in addition, recent data on dystrophic skeletal muscle myonuclei21 have demonstrated alterations of mRNA pathways reminiscent of those observed during aging.22 This strongly suggests a common mechanistic basis for both skeletal muscle aging and dystrophy.
Consistent with this hypothesis, the morpho-functional features of myoblasts from healthy and DM2 patients at different passages in culture provided experimental evidence for the more pronounced occurrence of senescence-related events in satellite cells of DM2-affected muscles. At early culture passages, when cell proliferation rate is high, both control and DM2 myoblasts show the typical morphological features of growing myoblasts previously described by several authors, i.e. cells characterised by elongated shapes, cytoplasm rich in free ribosomes, rough endoplasmic reticulum and Golgi complexes, nuclei with scarce heterochromatin and prominent nucleoli.45–49 At this culture stage, the only distinctive morphological feature of DM2 myoblasts is the presence of several vacuoles in the cytoplasm which could be related to a dysregulation of the protein degradation systems in these cells.60 Therefore, cytoplasmic and nuclear features clearly indicate a high proteosynthetic rate in both control and DM2 myoblasts at early compared to late passages; in particular, the large amount of euchromatin and the presence of reticular nucleoli with a large dense fibrillar component and numerous fibrillar centres are suggestive of a high metabolic activity.50 However, the ultrastructural immunocytochemical analyses revealed that DM2 myoblasts are relatively less active than the controls: the immunolabeling for activated polymerase I (which is necessary to synthesize pre-mRNA), for hnRNPs and snRNPs (which are essential for pre-mRNA co-transcriptional splicing34,35), and for CStF (involved in the maturation of 3′ ends61), were all significantly lower in DM2 than in control myoblasts. Accordingly, morphometrical evaluations demonstrated an increase in the amount of heterochromatin in DM2 myoblasts, consistent with the already described reduction in protein synthesis.60
A similar decrease in the factors involved in pre-mRNA transcription and maturation, and a parallel increase in the amount of heterochromatin have been found in senescing control myoblasts, when the proliferation rate decreased. However, no significant change in perichromatin granule density has been found in senescing control myoblasts: these granules represent storage and/or transport sites for spliced (pre-)mRNA36 and their steady number indicates unperturbed RNA pathways. Moreover, these control myoblasts are characterised by the presence of residual bodies, many vacuoles and glycogen clusters: these are typical features of aging cells, where the cytoplasmic degradation systems undergo dysfunction.62 Thus, DM2 myoblasts at early culture passages (when they proliferate at a similar rate as the controls) already show some morphological and functional features found in healthy myoblasts at late culture passage only.
The cytoplasmic and nuclear features typical of cells senescing in culture are definitely more pronounced in DM2 myoblasts at late culture passage than in the corresponding healthy controls. The amount of vacuoles and residual bodies is remarkably high, and large cytoplasmic regions appear as empty, likely due to degradation phenomena; the cell nuclei are often irregular in shape and contain many heterochromatin clumps; the factors involved in pre-mRNA transcription and processing are further reduced in comparison to senescing healthy myoblasts. It should be also noted that snRNPs, usually occurring on perichromatin fibrils and interchromatin granules, show an accumulation in the interchromatin granules which has not been observed in senescing healthy myoblasts. Intranuclear accumulation of factors involved in pre-mRNA maturation has been previously observed in different aging tissues63–65 including skeletal muscle,22,66 and this has been considered as a consequence of altered pre-mRNA processing and/or impaired intranuclear or nucleus-to-cytoplasm transport. Accordingly, in senescing DM2 myoblasts a significant increase in perichromatin granule amount has been found; this phenomenon is known to occur when RNA processing is physiologically or experimentally impaired39,67,68 as well as during aging.22,63–66,69 Taken together, the results of the present study demonstrate that satellite cell-derived DM2 myoblasts are characterised by senescence-related features mainly consisting in the early appearance of cytological alterations and impairment of the pre-mRNA maturation pathways. It is possible that the sequestration of splicing factors in the foci of DM2 myoblasts14–19 may hamper the functionality of the whole splicing machinery; this would consequently reduce, in dystrophic muscles, the capability of satellite cells to positively respond to anabolic stimuli. Interestingly, experimental therapeutic approaches for DM1 aimed at reducing intranuclear sequestration of splicing factors demonstrated that the restoration of normal splicing processes represents a key passage for alleviating disease symptoms.70,71
It has been reported that alteration of the RNA pathways does not affect the fusion process, so that cultured DM2 myoblasts are able to differentiate into myotubes as much as myoblasts from healthy subjects do.29,72 Similarly, cultured myoblasts from aged healthy subjects are able to form myotubes in culture.49 This would suggest that the capability of cultured myoblasts to proliferate and differentiate is maintained in aged and dystrophic subjects. However, the present ultrastructural and cytochemical data demonstrate that significant differences exist for some cytological and functional characteristics in the myoblasts from healthy and DM2 patients, despite their similarities in proliferation rate and age in culture. Interestingly, recent preliminary observations on the in vitro differentiation of DM2 myoblasts showed an early structural disorganization of the derived myotubes, especially the cytoskeletal apparatus, which may affect their functionality and survival.73
Further studies are however needed to investigate in vitro myoblast differentiation and myotube survival in DM2 patients using both morphological and biomolecular analyses such as those previously used to characterise the differentiation process of C2C12 myoblast cell line.74 Moreover, studies on the occurrence of senescence traits in both satellite cells and myofibres in DM2 muscle biopsies75 would provide important clues in the attempt to characterize the cellular mechanisms at the basis of muscle wasting in DM2 myotonic dystrophy.
Acknowledgments:
the authors would like to thank Mrs. Paola Veneroni for her skilful technical assistance. Marzia Giagnacovo is a PhD student in receipt of a fellowship from the Dottorato di Ricerca in Biologia Cellulare (University of Pavia). The authors would also express their gratitude to Prof. T.E. Martin for kindly providing the anti-hnRNP core protein antibody.
References
- 1.Meola G, Moxley RT., 3rd Myotonic dystrophy type 2 and related myotonic disorders. J Neurol. 2004;251:1173–82. doi: 10.1007/s00415-004-0590-1. [DOI] [PubMed] [Google Scholar]
- 2.Bachinski LL, Udd B, Meola G, Sansone V, Bassez G, Eymard B, et al. Confirmation of the type 2 myotonic dystrophy (CCTG)n expansion mutation in patients with proximal myotonic myopathy/proximal myotonic dystrophy of different European origins: a single shared haplotype indicates an ancestral founder effect. Am J Hum Genet. 2003;73:835–48. doi: 10.1086/378566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vihola A, Bassez G, Meola G, Zhang S, Haapasalo H, Paetau A, et al. Histopathological differences of myotonic dystrophy type 1 (DM1) and PROMM/DM2. Neurology. 2003;60:1854–7. doi: 10.1212/01.wnl.0000065898.61358.09. [DOI] [PubMed] [Google Scholar]
- 4.Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nature Genet. 2001;29:40–7. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- 5.Savkur RS, Philips AV, Cooper TA, Dalton JC, Moseley ML, Ranum LP, et al. Insulin receptor splicing alteration in myotonic dystrophy type 2. Am J Hum Genet. 2004;74:1309–13. doi: 10.1086/421528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Day JW, Ricker K, Jacobsen JF, Rasmussen LJ, Dick KA, Kress W, et al. Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology. 2003;60:657–64. doi: 10.1212/01.wnl.0000054481.84978.f9. [DOI] [PubMed] [Google Scholar]
- 7.Brook JD, McCurrach ME, Harley HG, Buckler AJ, Church D, Aburatani H, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992:385–7. doi: 10.1016/0092-8674(92)90418-c. [DOI] [PubMed] [Google Scholar]
- 8.Fu YH, Pizzuti A, Fenwick RG, Jr, King J, Rajnarayan S, Dunne PW, et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science. 1992;255:1256–8. doi: 10.1126/science.1546326. [DOI] [PubMed] [Google Scholar]
- 9.Mahadevan M, Tsilfidis C, Sabourin L, Shutler G, Amemiya C, Jansen G, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 1992;255:1253–5. doi: 10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- 10.Day JW, Roelofs R, Leroy B, Pech I, Benzow K, Ranum LP. Clinical and genetic characteristics of a five-generation family with a novel form of myotonic dystrophy (DM2) Neuromuscul Disord. 1999;9:19–27. doi: 10.1016/s0960-8966(98)00094-7. [DOI] [PubMed] [Google Scholar]
- 11.Suominen T, Bachinski LL, Auvinen S, Hackman P, Baggerly KA, Angelini C, et al. Population frequency of myotonic dystrophy: higher than expected frequency of myotonic dystrophy type 2 (DM2) mutation in Finland. Eur J Hum Genet. 2011;19:776–82. doi: 10.1038/ejhg.2011.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–7. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 13.Ranum LP, Rasmussen PF, Benzow KA, Koob MD, Day JW. Genetic mapping of a second myotonic dystrophy locus. Nature Genet. 1998;19:196–8. doi: 10.1038/570. [DOI] [PubMed] [Google Scholar]
- 14.Cardani R, Mancinelli E, Rotondo G, Sansone V, Meola G. Muscleblind-like protein 1 nuclear sequestration is a molecular pathology marker of DM1 and DM2. Eur J Histochem. 2006;50:177–82. [PubMed] [Google Scholar]
- 15.Cardani R, Mancinelli E, Giagnacovo M, Sansone V, Meola G. Ribonuclear inclusions as biomarker of myotonic dystrophy type 2, even in improperly frozen or defrozen skeletal muscle biopsies. Eur J Histochem. 2009;53:107–12. doi: 10.4081/ejh.2009.e13. [DOI] [PubMed] [Google Scholar]
- 16.Fardaei M, Rogers MT, Thorpe HM, Larkin K, Hamshere MG, Harper PS, et al. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded-repeat transcripts in DM1 and DM2 cells. Human Mol Genet. 2002;11:805–14. doi: 10.1093/hmg/11.7.805. [DOI] [PubMed] [Google Scholar]
- 17.Pascual M, Vicente M, Monferrer L, Artero R. The Muscleblind family of proteins: an emerging class of regulators of developmentally programmed alternative splicing. Differentiation. 2006;74:65–80. doi: 10.1111/j.1432-0436.2006.00060.x. [DOI] [PubMed] [Google Scholar]
- 18.Llorian M, Smith CW. Decoding muscle alternative splicing. Curr Opin Genet Dev. 2011;21:380–7. doi: 10.1016/j.gde.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 19.Perdoni F, Malatesta M, Cardani R, Giagnacovo M, Mancinelli E, Meola G, et al. RNA/MBNL1-containing foci in myoblast nuclei from patients affected by myotonic dystrophy type 2: an immunocytochemical study. Eur J Histochem. 2009;53:151–8. doi: 10.4081/ejh.2009.e18. [DOI] [PubMed] [Google Scholar]
- 20.Mankodi A, Urbinati CR, Yuan QP, Moxley RT, Sansone V, Krym M, et al. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum Mol Genet. 2001;10:2165–70. doi: 10.1093/hmg/10.19.2165. [DOI] [PubMed] [Google Scholar]
- 21.Malatesta M, Giagnacovo M, Cardani R, Meola G, Pellicciari C. RNA processing is altered in skeletal muscle nuclei of patients affected by myotonic dystrophy. Histochem Cell Biol. 2011;135:419–25. doi: 10.1007/s00418-011-0797-z. [DOI] [PubMed] [Google Scholar]
- 22.Malatesta M, Perdoni F, Muller S, Zancanaro C, Pellicciari C. Nuclei of aged myofibres undergo structural and functional changes suggesting impairment in RNA processing. Eur J Histochem. 2009;53:97–106. doi: 10.4081/ejh.2009.e12. [DOI] [PubMed] [Google Scholar]
- 23.Thompson LD. Age-related muscle dysfunction. Exp Gerontol. 2009;44:106–11. doi: 10.1016/j.exger.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cruz-Jentoft AJ, Baeyens JP, Bauer JM, Boirie Y, Cederholm T, Landi F, et al. European Working Group on Sarcopenia in Older People, Sarcopenia: European consensus on definition and diagnosis: Report of the European Working Group on Sarcopenia in Older People. Age Aging. 2010;39:412–23. doi: 10.1093/ageing/afq034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harper PS. 3rd ed. Saunders; London, UK: 2001. Myotonic dystrophy. [Google Scholar]
- 26.Machida S, Narusawa M. The roles of satellite cells and hematopoietic stem cells in impaired regeneration of skeletal muscle in old rats. Ann N Y Acad Sci. 2006;1067:349–53. doi: 10.1196/annals.1354.049. [DOI] [PubMed] [Google Scholar]
- 27.Verdijk LB, Koopman R, Schaart G, Meijer K, Savelberg HH, van Loon LJ. Satellite cell content is specifically reduced in type II skeletal muscle fibers in the elderly. Am J Physiol Endocrinol Metab. 2007;292:E151–7. doi: 10.1152/ajpendo.00278.2006. [DOI] [PubMed] [Google Scholar]
- 28.Bigot A, Klein AF, Gasnier E, Jacquemin V, Ravassard P, Butler-Browne G, et al. Large CTG repeats trigger p16-dependent premature senescence in myotonic dystrophy type 1 muscle precursor cells. Am J Pathol. 2009;174:1435–42. doi: 10.2353/ajpath.2009.080560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cardani R, Baldassa S, Botta A, Rinaldi F, Novelli G, Mancinelli E, et al. Ribonuclear inclusions and MBNL1 nuclear sequestration do not affect myoblast differentiation but alter gene splicing in myotonic dystrophy type 2. Neuromuscul Disord. 2009;19:335–43. doi: 10.1016/j.nmd.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 30.Loro E, Rinaldi F, Malena A, Masiero E, Novelli G, Angelini C, et al. Normal myogenesis and increased apoptosis in myotonic dystrophy type-1 muscle cells. Cell Death Diff. 2010;17:1315–24. doi: 10.1038/cdd.2010.33. [DOI] [PubMed] [Google Scholar]
- 31.Meola G. Advanced microscopic and histochemical techniques: diagnostic tools in the molecular era of myology. Eur J Histochem. 2005;49:93–6. [PubMed] [Google Scholar]
- 32.Tews DS, Goebel HH. Diagnostic immunohistochemistry in neuromuscular disorders. Histopathol. 2005;46:1–23. doi: 10.1111/j.1365-2559.2005.02025.x. [DOI] [PubMed] [Google Scholar]
- 33.Malatesta M, Meola G. Structural and functional alterations of the cell nucleus in skeletal muscle wasting: the evidence in situ. Eur J Histochem. 2010;54:e44. doi: 10.4081/ejh.2010.e44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lührmann R, Kastner B, Bach M. Structure of spliceosomal snRNPs and their role in pre-mRNA splicing. Biochim Biophys Acta. 1990;1087:265–92. doi: 10.1016/0167-4781(90)90001-i. [DOI] [PubMed] [Google Scholar]
- 35.Madhani HD, Guthrie C. Dynamic RNA-RNA interactions in the spliceosome. Annu Rev Genet. 1994;28:1–26. doi: 10.1146/annurev.ge.28.120194.000245. [DOI] [PubMed] [Google Scholar]
- 36.Fakan S. Ultrastructural cytochemical analyses of nuclear functional architecture. Eur J Histochem. 2004;48:5–14. [PubMed] [Google Scholar]
- 37.Biggiogera M, Fakan S. Visualization of nuclear organization by ultrastructural cytochemistry. Methods Cell Biol. 2008;88:431–49. doi: 10.1016/S0091-679X(08)00422-6. [DOI] [PubMed] [Google Scholar]
- 38.Biggiogera M, Cisterna B, Spedito A, Vecchio L, Malatesta M. Perichromatin fibrils as early markers of transcriptional alterations. Differentiation. 2008;76:57–65. doi: 10.1111/j.1432-0436.2007.00211.x. [DOI] [PubMed] [Google Scholar]
- 39.Malatesta M, Biggiogera M, Baldelli B, Barabino SML, Martin TE, Zancanaro C. Hibernation as a far-reaching program for the modulation of RNA transcription. Microsc Res Tech. 2008;71:564–72. doi: 10.1002/jemt.20587. [DOI] [PubMed] [Google Scholar]
- 40.Moxley RT, 3rd, Meola G, Udd B, Ricker K. Report of the 84th ENMC workshop: PROMM (proximal myotonic myopathy) and other myotonic dystrophy-like syndromes: 2nd workshop 13–15th October 2000, Loosdrecht, The Netherlands. Neuromuscul Disord. 2002;12:306–17. doi: 10.1016/s0960-8966(01)00284-x. [DOI] [PubMed] [Google Scholar]
- 41.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–7. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Veraldi KL, Arhin GK, Martincic K, Chung-Ganster LH, Wilusz J, Milcarek C. HnRNP F influences binding of a 64-kilodalton subunit of cleavage stimulation factor to mRNA precursors in mouse B cells. Mol Cell Biol. 2001;21:1228–38. doi: 10.1128/MCB.21.4.1228-1238.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones RE, Okamura CS, Martin TE. Immunofluorescent localization of the proteins of nuclear ribonucleoprotein complexes. J Cell Biol. 1980;86:235–43. doi: 10.1083/jcb.86.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bernhard W. A new staining procedure for electron microscopic cytology. J Ultrastruct Res. 1969;27:250–65. doi: 10.1016/s0022-5320(69)80016-x. [DOI] [PubMed] [Google Scholar]
- 45.Shimada Y, Fischman DA, Moscona AA. The fine structure of embryonic chick skeletal muscle cells differentiated in vitro. J Cell Biol. 1967;35:445–53. doi: 10.1083/jcb.35.2.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schultz E, McCormick KM. Skeletal muscle satellite cells. Rev Physiol Biochem Pharmacol. 1994;123:213–57. doi: 10.1007/BFb0030904. [DOI] [PubMed] [Google Scholar]
- 47.Yablonka-Reuveni Z, Anderson SK, Bowen-Pope DF, Nameroff M. Biochemical and morphological differences between fibroblasts and myoblasts from embryonic chicken skeletal muscle. Cell Tissue Res. 1988;252:339–48. doi: 10.1007/BF00214376. [DOI] [PubMed] [Google Scholar]
- 48.Danisovic L, Varga I, Polak S, Ulicna M, Bohmer D, Vojtassak J. Morphology of in vitro expanded human muscle-derived stem cells. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2008;152:235–8. doi: 10.5507/bp.2008.036. [DOI] [PubMed] [Google Scholar]
- 49.Corbu A, Scaramozza A, Badiali-DeGiorgi L, Tarantino L, Papa V, Rinaldi R, et al. Satellite cell characterization from aging human muscle. Neurol Res. 2010;32:63–72. doi: 10.1179/174313209X385725. [DOI] [PubMed] [Google Scholar]
- 50.Cisterna B, Biggiogera M. Ribosome biogenesis: from structure to dynamics. Int Rev Cell Mol Biol. 2010;284:67–111. doi: 10.1016/S1937-6448(10)84002-X. [DOI] [PubMed] [Google Scholar]
- 51.Puvion E, Puvion-Dutilleul F. Ultrastructure of the nucleus in relation to transcription and splicing: roles of perichromatin fibrils and interchromatin granules. Exp Cell Res. 1996;229:217–25. doi: 10.1006/excr.1996.0363. [DOI] [PubMed] [Google Scholar]
- 52.Bogolyubov D, Stepanova I, Parfenov V. Universal nuclear domains of somatic and germ cells: some lessons from oocyte interchromatin granule cluster and Cajal body structure and molecular composition. Bioessays. 2009;31:400–9. doi: 10.1002/bies.200800100. [DOI] [PubMed] [Google Scholar]
- 53.Wheeler TM, Thornton CA. Myotonic dystrophy: RNA-mediated muscle disease. Curr Opin Neurol. 2007;20:572–6. doi: 10.1097/WCO.0b013e3282ef6064. [DOI] [PubMed] [Google Scholar]
- 54.Ranum LP, Cooper TA. RNA-mediated neuromuscular disorders. Annu Rev Neurosci. 2006;29:259–77. doi: 10.1146/annurev.neuro.29.051605.113014. [DOI] [PubMed] [Google Scholar]
- 55.Schoser B, Timchenko L. Myotonic dystrophies 1 and 2: complex diseases with complex mechanisms. Curr Genomics. 2010;11:77–90. doi: 10.2174/138920210790886844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Timchenko NA, Iakova P, Cai ZJ, Smith JR, Timchenko LT. Molecular basis for impaired muscle differentiation in myotonic dystrophy. Mol Cell Biol. 2001;21:6927–38. doi: 10.1128/MCB.21.20.6927-6938.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Timchenko NA, Patel R, Iakova P, Cai ZJ, Quan L, Timchenko LT. Overexpression of CUG triplet repeat-binding protein, CUGBP1, in mice inhibits myogenesis. J Biol Chem. 2004;279:13129–39. doi: 10.1074/jbc.M312923200. [DOI] [PubMed] [Google Scholar]
- 58.Kim W, Sharpless N. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–75. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 59.Salisbury E, Sakai K, Schoser B, Huichalaf C, Schneider-Gold C, Nguyen H, et al. Ectopic expression of cyclin D3 corrects differentiation of DM1 myoblasts through activation of RNA CUG-binding protein, CUGBP1. Exp Cell Res. 2008;314:2266–78. doi: 10.1016/j.yexcr.2008.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Salisbury E, Schoser B, Schneider-Gold C, Wang GL, Huichalaf C, Jin B, et al. Expression of RNA CCUG repeats dysregulates translation and degradation of proteins in myotonic dystrophy 2 patients. Am J Pathol. 2009;175:748–62. doi: 10.2353/ajpath.2009.090047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wahle E, Rüegsegger U. 3′-end processing of pre-mRNA in eukaryotes. FEMS Microbiol Rev. 1999;23:277–95. doi: 10.1111/j.1574-6976.1999.tb00400.x. [DOI] [PubMed] [Google Scholar]
- 62.Jameson CW. Towards a unified and interdisciplinary model of aging. Med Hypotheses. 2004;63:83–6. doi: 10.1016/j.mehy.2004.01.021. [DOI] [PubMed] [Google Scholar]
- 63.Malatesta M, Baldelli B, Battistelli S, Fattoretti P, Bertoni-Freddari C. Aging affects the distribution of the circadian CLOCK protein in rat hepatocytes. Microsc Res Tech. 2005;68:45–50. doi: 10.1002/jemt.20221. [DOI] [PubMed] [Google Scholar]
- 64.Malatesta M, Fattoretti P, Baldelli B, Battistelli S, Balietti M, Bertoni-Freddari C. Effects of aging on the fine distribution of the circadian CLOCK protein in reticular formation neurons. Histochem Cell Biol. 2007;127:641–7. doi: 10.1007/s00418-007-0284-8. [DOI] [PubMed] [Google Scholar]
- 65.Malatesta M, Biggiogera M, Cisterna B, Balietti M, Bertoni-Freddari C, Fattoretti P. Perichromatin fibrils accumulation in hepatocyte nuclei reveals alterations of pre-mRNA processing during aging. DNA Cell Biol. 2011;29:49–57. doi: 10.1089/dna.2009.0880. [DOI] [PubMed] [Google Scholar]
- 66.Malatesta M, Perdoni F, Muller S, Pellicciari C, Zancanaro C. Pre-mRNA processing is partially impaired in satellite cell nuclei from aged muscles. J Biomed Biotechnol. 2010;2010:410405–410405. doi: 10.1155/2010/410405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Puvion-Dutilleul F, Puvion E. Relationship between chromatin and perichromatin granules in cadmium-treated isolated hepatocytes. J Ultrastruct Res. 1981;74:341–50. doi: 10.1016/s0022-5320(81)80125-6. [DOI] [PubMed] [Google Scholar]
- 68.Zancanaro C, Malatesta M, Vogel P, Osculati F, Fakan S. Ultrastructural and morphomentrical analyses of the brown adipocyte nucleus in a hibernating dormouse. Biol Cell. 1993;79:55–61. doi: 10.1016/0248-4900(93)90263-e. [DOI] [PubMed] [Google Scholar]
- 69.Malatesta M, Bertoni-Freddari C, Fattoretti P, Caporaloni C, Fakan S, Gazzanelli G. Altered RNA structural constituents in aging and vitamin E deficiency. Mech Aging Dev. 2003;124:175–81. doi: 10.1016/s0047-6374(02)00117-3. [DOI] [PubMed] [Google Scholar]
- 70.Mulders SAM, van den Broek WJAA, Wheeler TM, Croes HJE, van Kuik-Romeijn P, de Kimpe SJ, et al. Triplet-repeat oligonucleotide-mediated reversal of RNA toxicity in myotonic dystrophy. Proc Nat Acad Sci USA. 2009;106:13915–20. doi: 10.1073/pnas.0905780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Warf MB, Nakamori M, Matthys CM, Thornton CA, Berglund JA. Pentamidine reverses the splicing defects associated with myotonic dystrophy. Proc Nat Acad Sci USA. 2009;106:18551–6. doi: 10.1073/pnas.0903234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pelletier R, Hamel F, Beaulieu D, Patry L, Haineault C, Tarnopolsky M, et al. Absence of a differentiation defect in muscle satellite cells from DM2 patients. Neurobiol Dis. 2009;36:181–90. doi: 10.1016/j.nbd.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 73.Giagnacovo M, Costanzo M, Cardani R, Veneroni P, Pellicciari C, Meola G. In vitro aging of myotubes derived from myoblasts of patients affected by myotonic dystrophy type 2 (DM2): ultrastructural evidence. Microscopie. 2011;15:60–7. [Google Scholar]
- 74.Casadei L, Vallorani L, Gioacchini AM, Guescini M, Burattini S, D'Emilio A, et al. Proteomics-based investigation in C2C12 myoblast differentiation. Eur J Histochem. 2009;53:261–8. doi: 10.4081/ejh.2009.e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Giagnacovo M, Cardani R, Meola G, Pellicciari C, Malatesta M. Routinely frozen biopsies of human skeletal muscle are suitable for morphological and immunocytochemical analyses at transmission electron microscopy. Eur J Histochem. 2010;54:e31. doi: 10.4081/ejh.2010.e31. [DOI] [PMC free article] [PubMed] [Google Scholar]