

Abstract
Infections by multiple genotypes are common in nature and are known to select for higher levels of virulence for some parasites. When parasites produce public goods (PGs) within the host, such co-infections have been predicted to select for lower levels of virulence. However, this prediction is based on simplifying assumptions regarding epidemiological feedbacks on the multiplicity of infections (MOI). Here, we analyse the case of parasites producing a PG (for example, siderophore-producing bacteria) using a nested model that ties together within-host and epidemiological processes. We find that the prediction that co-infection should select for less virulent strains for PG-producing parasites is only valid if both parasite transmission and virulence are linear functions of parasite density. If there is a trade-off relationship such that virulence increases more rapidly than transmission, or if virulence also depends on the total amount of PGs produced, then more complex relationships between virulence and the MOI are predicted. Our results reveal that explicitly taking into account the distribution of parasite strains among hosts could help better understand the selective pressures faced by parasites at the population level.
Keywords: virulence, co-infections, epidemiology, kin selection, cooperation
1. Introduction
Host co-infections by several genotypes of the same parasite species are ubiquitous, and are found in several human infections [1]. Epidemiological models show that co-infections by different genotypes can select for more virulent strains in the host population provided that virulence confers a within-host competitive advantage to the parasite [2–5]. Such a correlation between the virulence of a strain and its within-host competitiveness has been demonstrated experimentally for parasites such as Plasmodium chabaudi in mice [6] or microsporidia in Daphnia [7].
The generality of the result that co-infections select for more virulent strains has been challenged by theoretical and experimental studies based on parasites producing public goods (PGs). The rationale is that, if high levels of parasite cooperation translate into high levels of virulence, then diverse infections should favour ‘cheating’ strains that do not cooperate, and therefore lower levels of virulence should evolve. Support for this idea has come from both empirical [8] and theoretical models of ‘collective action’ between viruses [9,10].
Many prokaryote species can synthesize siderophores, which are diffusible iron-chelating compounds that bind to iron with a high affinity and allow iron to be imported into the cell [11]. Because siderophores are costly to produce but free to use, bacterial strains that cheat and do not produce siderophores should have a fitness advantage when competing with siderophore-producing strains, and it should not pay to cooperate and produce siderophores. This scenario has been studied theoretically by West & Buckling [12]. Using a kin selection model in which siderophore production is positively linked to parasite virulence, they find that the optimal level of siderophore production decreases with the number of bacterial strains per host. In other words, they predict that low within-host parasite relatedness selects for lower levels of parasite cooperation (and therefore lower levels of virulence). This pattern was found experimentally by Harrison et al. [13] for the bacterium Pseudomonas aeruginosa.
Based on these studies, it has been suggested that co-infections generally select for less virulent strains when host exploitation depends on cooperation by parasites [10,14]. Here, we argue that the situation can be more complex than previously expected because of epidemiological processes. Indeed, in natural populations, the optimal level of virulence will be determined by the interplay between the selective pressures at both the host and population levels [3,15]. The two need not necessarily agree. First, natural populations are characterized by a heterogeneous distribution of parasite strains (some hosts are uninfected, others are singly or doubly infected, and so on), which has been shown to be shaped by parasite traits (e.g. a virulent strain that causes short infections should yield a distribution with few co-infections [16]) and to alter the selective pressures experienced by parasites (e.g. different strains can be selected for depending on the frequency of susceptible hosts in the population [17]). Second, evolutionary trade-offs at the epidemiological level have a crucial effect on virulence evolution. For instance, theory predicts that a saturating trade-off between transmission rate and virulence should lead to the existence of an intermediate level of virulence [3,5,18–20]. Such a trade-off has been documented empirically (for instance for HIV in humans [21] and for a protozoan parasite of monarch butterflies [22]). In all these cases, intermediate levels of virulence maximize parasite fitness.
To explore in more detail the evolutionary consequences of epidemiological feedbacks on the evolution of PG production by parasites, we use a nested model [23] (i.e. we analyse explicitly the interplay between within-host and epidemiological dynamics). This allows us to specify the relationship between parasite density and epidemiological variables (transmission rate and virulence), and to investigate the consequences of an uneven distribution of strains across hosts. Our within-host dynamics are inspired by the example of siderophore-producing bacteria, and are directly comparable to previous models (e.g. [12]), although our general approach and message are not restricted to this particular example. Between-host dynamics are based on a co-infection model that allows coexistence between up to two strains in a host [3]. By varying the multiplicity of infection (MOI) in the host population from 1 (only single infections) to 2 (only co-infections), we show that the result that the optimal virulence increases with the MOI is only valid if there is a linear trade-off relationship between virulence and transmission. When there is a saturating trade-off or when virulence depends on the total amount of PGs produced in the host, different patterns between virulence and MOI are predicted, which depend on the feedback between MOI and epidemiology.
2. Within-host dynamics
We consider a host infected by n parasite strains. The within-host density of strain i is denoted Ni. A strain is defined by its trait xi, which corresponds to the units of PG it produces. The dynamics of the density G of PG is given by
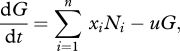 |
2.1 |
where u is the uptake rate of the PG. This equation assumes that all strains have the same uptake rate, and that the uptake of PG by strain i is proportional to its frequency Ni /N, where N = ∑i=1n Ni is the total within-host density of parasites.
The dynamics of strain i follows an ordinary differential equation with three terms reflecting, respectively, a logistic growth, a cost of PG production (which is proportional to the strategy of the strain, xi) and a benefit of PG exploitation (which is a function of the uptake rate of PG by strain i). This yields
| 2.2 |
where K/n is the maximum population size achievable by each of the n strains in the absence of PG, c is the cost of producing one unit of PG and b is the benefit of PG exploitation (figure 1a).
Figure 1.

(a) Diagram showing the three main within-host processes (parasite growth, PG production and PG uptake). (b) Within-host parasite population size at equilibrium as a function of PG production in a single infection (n = 1). Parameter values are given in the electronic supplementary material, table S1.
Our within-host model is directly comparable to earlier studies, in particular the siderophore production model of West & Buckling [12]. The main difference is that we work directly with within-host parasite population sizes, whereas West & Buckling [12] postulated a relationship between the within-host growth rate of parasites and PG production and exploitation. One advantage of our approach is that it leads to predictions in terms of parasite densities, which are in principle easier to assess than parasite growth rate. Note that, in order to allow for long-term parasite coexistence within the host, we have assumed that all strains are equally competitive to obtain baseline resources in the host (meaning that when there is no PG production, each strain can reach a maximum density of K/n). This assumption also allows us to distinguish between competition for host resources and competition for PG.
Following earlier studies [12,24], the amount of PG received by an individual parasite, uG/N, is scaled by a parameter a ≤ 1 to take into account the fact that the cost of producing the PG may increase faster than the benefit gained by the exploitation of the PG. As a consequence, when a < 1, there exists a PG production rate that maximizes parasite population size (figure 1b). In equation (2.2), we assume that the benefits and costs associated with PG uptake and production are additive, but our results also hold when they are multiplicative, as shown in the electronic supplementary material, S7.
We further assume that within-host dynamics are fast compared with between-host dynamics so that within-host densities can be assumed to be at equilibrium. The equilibrium density 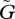 of PG is
of PG is
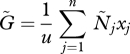 |
2.3 |
and the equilibrium density 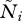 can be found by solving
can be found by solving
| 2.4 |
where
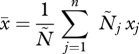 |
2.5 |
is the average rate of PG production in the population, with 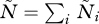 . For two strains, there are two stable equilibria: one where both strains densities are zero and one where they are strictly positive. In the following, we restrict our attention to the range of parameter values such that within-host parasite densities are non-zero.
. For two strains, there are two stable equilibria: one where both strains densities are zero and one where they are strictly positive. In the following, we restrict our attention to the range of parameter values such that within-host parasite densities are non-zero.
An important parameter in kin selection models of within-host evolution is the relatedness between parasite strains within a host. Clearly, relatedness will depend on the MOI, because it should be 1 in a host infected by a single parasite strain and less than 1 in a multiply infected host. Following Taylor & Frank [25], within-host relatedness can be defined more precisely as the regression of the average rate of PG production 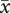 over the rate of PG production xm of a focal strain, which yields for our model
over the rate of PG production xm of a focal strain, which yields for our model
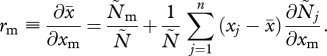 |
2.6 |
Hence, if all strains are equally abundant, or (in our model) if all strains have the same trait value, we have a simple relationship between within-host relatedness and the MOI within a host, rm = 1/n. In general, however, relatedness will also depend on how within-host dynamics shape parasite densities.
3. Epidemiological parameters
In general, the epidemiological parameters of the disease (i.e. virulence and transmission) will depend on the within-host densities of each strain [23] and on the within-host density of PG. Here, we explore two ways to link the within- and between-host models.
(a). Model 1: transmission–virulence trade-off through within-host densities
Following the majority of previous nested models [23], we assume that the transmission rate of a strain (βi) is linearly correlated with the within-host density of this strain, and that virulence (α), which is defined here as the increase in host mortality owing to the infection, is a power function of the total parasite within-host density. This gives
| 3.1 |
and
| 3.2 |
where β0 and α0 are proportionality constants, and ξ describes the shape of the transmission–virulence trade-off curve [18–20]. Note that since virulence depends on the total parasite density, it is not a linear function of the amount of PG produced (figure 1b).
If ξ = 1, transmission is a linear function of virulence and theory predicts that the parasite strain with the highest virulence will always take over the host population. If ξ > 1, transmission is a saturating function of virulence, which implies that there exists an evolutionarily stable strategy (ESS) for the parasite corresponding to an intermediate level of virulence [3,5]. In the electronic supplementary material, S4.3, we show that the predictions of our model are qualitatively similar whether transmission or virulence is assumed to be a nonlinear function of within-host parasite density.
(b). Model 2: detrimental public goods
In model 1, we have assumed that virulence depends only on parasite density, but other relationships are possible. For instance, virulence could also be affected by the total density 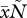 of PGs (e.g. if the PG is toxic, or if, as in the case of siderophores, it takes away resources from the host). We model this scenario with the following definition for virulence:
of PGs (e.g. if the PG is toxic, or if, as in the case of siderophores, it takes away resources from the host). We model this scenario with the following definition for virulence:
| 3.3 |
Thus, virulence is a function of the total parasite density (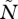 ) and of the total amount of siderophores produced (
) and of the total amount of siderophores produced (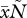 ). We do not assume that virulence depends only on the amount of siderophores produced because a strain that does not produce siderophores would then be completely avirulent.
). We do not assume that virulence depends only on the amount of siderophores produced because a strain that does not produce siderophores would then be completely avirulent.
4. From epidemiological dynamics to parasite fitness
We consider a host population at equilibrium infected by a resident parasite strain with trait xw. A new mutant strain with trait xm will increase in frequency if its epidemiological fitness, given by the basic reproductive ratio R0 (xw, xm) [26], is greater than 1. Note that this fitness depends on the mutant trait and on the resident trait. If mutations have a small phenotypic effect on the trait, the direction of selection is given by the selection gradient ∂R0/∂xm evaluated at xm = xw. Following classical invasion analyses [27,28], potential evolutionary endpoints x* can be found by solving
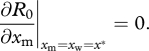 |
4.1 |
Our aim is to investigate the evolution of within-host cooperation (i.e. the value of x) and its consequences for epidemiological parameters (particularly virulence).
(a). Epidemiological dynamics
We consider an epidemiological co-infection model introduced by van Baalen & Sabelis [3], with one additional twist. Following Alizon & van Baalen [29], we introduce a parameter σI that measures the susceptibility of infected hosts to the disease. When σI = 0, infected hosts are immune to co-infection, and we recover the classical susceptible–infected (SI) model with single infections only. In the limit where σI is very large, infected hosts immediately enter the co-infected stage, and the population is entirely composed of susceptible and co-infected hosts. For completeness, we also introduce a parameter σS that measures the susceptibility of uninfected hosts. The originality of this model is that it takes into account co-infections by the same strain, which is necessary to correctly assess the invasion fitness of a rare parasite strain (for further details, see [3,30]). We further assume that the order of infection does not matter (Dmw is equivalent to Dwm). This is in line with our assumption that recovery does not occur (i.e. infections are persistent). In other words, transient dynamics within a host (when a new strain arrives) can be neglected because they are short compared with the duration of infection. The life cycle is described in figure 2, and the full dynamics are given in the electronic supplementary material, S2.1.
Figure 2.

Epidemiological co-infection model. Uninfected hosts (S) can become infected by strain i at a rate that depends on the force of infection λi. Hosts singly infected by the resident (Iw) or by the mutant (Im) strain can be co-infected by another strain and become doubly infected. μ is the natural death rate of hosts and αi is the virulence (i.e. the disease-induced mortality) of hosts infected by strain i.
The invasion fitness of a mutant strain [3] (electronic supplementary material, S2.2) is
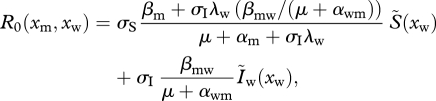 |
4.2 |
where 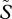 and
and 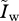 indicate the equilibrium densities of susceptible and infected hosts before the mutant appears. λw and λm are the force of infection of the resident and of the mutant strain, respectively (their full expressions are shown in the electronic supplementary material, S2.1), αwm is the overall virulence in a host co-infected by the resident and the mutant strain, βmw (respectively, βwm) is the transmission rate of the mutant (respectively, resident) strain in a host co-infected by the resident and the mutant strain.
indicate the equilibrium densities of susceptible and infected hosts before the mutant appears. λw and λm are the force of infection of the resident and of the mutant strain, respectively (their full expressions are shown in the electronic supplementary material, S2.1), αwm is the overall virulence in a host co-infected by the resident and the mutant strain, βmw (respectively, βwm) is the transmission rate of the mutant (respectively, resident) strain in a host co-infected by the resident and the mutant strain.
There are two interesting limit cases of equation (4.2). First, when σI = 0, we recover the classical expression of the basic reproductive ratio of a rare mutant parasite for an SI model with no co-infections [26] as
| 4.3 |
Second, when all hosts are infected in the population (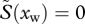 ), or when σI is very large, the evolutionary outcome will be determined solely by the ratio between parasite transmission and host mortality:
), or when σI is very large, the evolutionary outcome will be determined solely by the ratio between parasite transmission and host mortality:
| 4.4 |
In equation (4.4), parasite fitness at the between-host level is the product of the transmission rate of the focal mutant lineage and of host survival. This is similar to fitness expressions used in some previous kin-selection models [5,12], which we recover as a limit case of our model.
The comparison among equations (4.2)–(4.4) shows that, for a given trade-off between transmission and virulence, our model provides an additional level of epidemiological realism by explicitly taking into account the distribution of parasite clones among hosts. In our model, this distribution is characterized by the densities of uninfected, singly infected and doubly infected hosts, but generalizations to more complex distributions are possible. Finally, note that equation (4.2) does not depend on the host demography in the absence of infection, but only on the epidemiological processes of primary and secondary infections. In the remainder of the paper, we focus on a simplified model of host demography and assume that the total host population is constant (i.e. that every death is replaced by a new susceptible).
(b). Feedback between epidemiology and the multiplicity of infections
Variations in the susceptibility of infected hosts to secondary infections, σI, will affect the average MOI in the population, which can be measured as
| 4.5 |
where I and D are the total densities of singly and doubly infected hosts. In our model, the MOI takes values between 1 (when D = 0, i.e. all infected hosts are singly infected) and 2 (when I = 0, i.e. all infected hosts are co-infected). The quantity 1/MOI is the average within-host relatedness in the population and takes values between 1/2 and 1.
The precise relationship between MOI, σI and other epidemiological parameters will depend on the host demography in the absence of infection. For a population with a constant total size (S = 1 − I − D), we find at epidemiological equilibrium
| 4.6 |
where 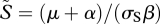 is the equilibrium density of susceptible hosts.
is the equilibrium density of susceptible hosts.
Equation (4.6) shows that epidemiology affects the MOI in the population through the susceptibility to secondary infections σI and the density of susceptible hosts. In particular, we see that the MOI is an increasing function of σI, and a decreasing function of S. When the density of susceptible hosts is low, the MOI is expected to be close to 2. When S increases as a result of variations in demographic (μ) and epidemiological traits (α and β), so does the proportion of singly infected hosts. Therefore, ultimately, there is a feedback between investment into PG and the MOI in the population. This feedback will be affected by (i) the demography of the host population, (ii) the trade-off between virulence and transmission, and (iii) the rate of secondary infections. Taken together, equations (4.2) and (4.6) show that different mortality (varying μ) or infection (varying σI) regimes may have an impact on both the optimal PG investment and the average MOI in the population.
(c). Evolutionary correlations between multiplicity of infection, public goods production and virulence
(i). Model 1: linear transmission–virulence trade-off
When the trade-off between transmission and virulence is linear (ξ = 1) and co-infections do not occur (σI = 0), selection favours strategies of host exploitation that maximize virulence [18,26]. In our model, this implies that PG production evolves until within-host parasite density is maximal (which occurs at x = 1 with our default parameter values; see figure 1b). When infected individuals become more susceptible to co-infection (σI increases), we find that the optimal level of PG production decreases (figure 3a). Because the average MOI is an increasing function of σI, this also results in a decreasing relationship between MOI and optimal PG production (figure 3b). The interpretation of this pattern is that singly infected hosts provide a refuge for cooperative strains, where they cannot be exploited by cheaters. When the proportion of singly infected hosts in the population decreases, this advantage weakens and cheaters are increasingly favoured. As a result, within-host parasite density decreases when the MOI increases, and so does evolutionarily stable (ES) parasite virulence (figure 3c). This corroborates the findings of previous models (e.g. [12]).
Figure 3.

Evolutionarily stable (ES) PG production and virulence as a function of the susceptibility of infected individuals σI and of the MOI. (d–f) Evolutionarily repellor (dashed line), the attractor for large initial investment in PG (black lines), and the attractor for low initial investment in PG, which is simply zero (grey line). This figure was obtained with a constant host population size. Parameter values are in the electronic supplementary material, table S1.
(ii). Model 1: saturating transmission–virulence trade-off
Assuming a saturating transmission–virulence trade-off (ξ = 2; figure 3d–f) leads to a radically different outcome. For high values of the susceptibility of infected individuals to secondary infections (σI), we find that PG production is never selected for. In this case, there is no correlation between virulence and MOI. By contrast, for low values of σI, evolutionary bistability is observed. If the population starts from a low investment into PG production, PG production is always counter-selected and the MOI has no effect on virulence evolution. If the initial PG investment is above a threshold, selection favours high investment into PG production, and this optimal level of PG production decreases as infected hosts become more susceptible to secondary infections. In the limit where only single infections are possible (σI = 0), we show in the electronic supplementary material, S4 that, if 0 < a < 1, the threshold xc above which PG production is favoured is xc = (c/(ba))1/(a−1), which yields xc = 1 with our parameter values. Hence, two evolutionary outcomes are selected for depending on the initial investment in cooperation: either pure defection (no PG production) or an intermediate investment into PG production.
Understanding what happens in the limit where only single infections occur sheds light on the processes underpinning this result. For a saturating trade-off, selection favours intermediate levels of virulence. Hence, strains maximizing within-host parasite density (x = 1) are counter-selected, and this strategy actually becomes an evolutionary repellor. Because virulence is a bell-shaped function of PG production, an intermediate level of virulence can be achieved through either a low or a large investment into PG production, depending on which side of the repellor the population starts off (figure 4). Now, as the susceptibility to secondary infections increases, so does the advantage to cheaters. This has two effects. First, a zero investment into PG production becomes attractive for a wider range of initial conditions (the repellor moves to the right in figure 4b). Second, the non-zero ESS decreases because it pays less to be a cooperator, which increases virulence (the ESS moves to the left in figure 4). At a critical value of σI, the two points collide and 0 becomes the sole evolutionary endpoint.
Figure 4.
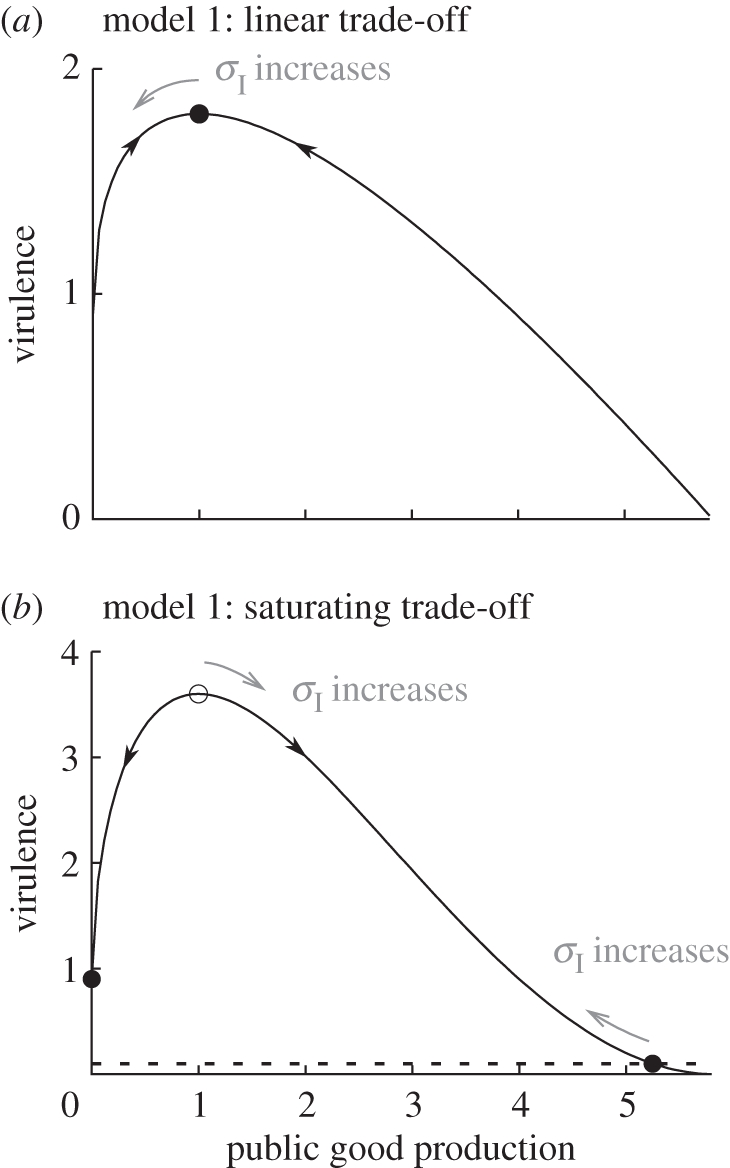
Virulence as a function of PG production in model 1. Filled circles indicate evolutionarily stable levels of PG production and virulence in the absence of multiple infections (σI = 0). The open circle at x = 1 in (b) indicates the intermediate evolutionary repellor (dashed line in figure 3d). The grey arrows indicate in what directions the evolutionary singularities move when the susceptibility to secondary infections increases. When the trade-off is nonlinear, there is a value of σI at which the repellor and the non-zero ESS collide, and a zero PG production becomes the sole evolutionary endpoint. Parameter values are in the electronic supplementary material, table S1.
Because the MOI and the optimal PG production both depend on the susceptibility parameter σI, the correlations between MOI and virulence are more complex under this scenario (figure 3f). Our model predicts either no effect of MOI on virulence, or that virulence increases with average MOI despite selection for lower PG production rates. In particular, populations can display identical levels of MOI, but very different levels of virulence, depending on the value of σI. Hence, different epidemiological dynamics can lead to similar levels of MOI at evolutionary equilibrium, but opposite predictions for virulence evolution.
In the limit where σI → ∞, our model converges to the limiting case where all hosts are co-infected. This is the scenario investigated by previous models (e.g. [12]). These models used a linear trade-off and thus predicted an intermediate optimal PG production, but we further show that the evolutionary outcome actually depends on the shape of the epidemiological trade-off. In the electronic supplementary material, S5.1, we show that if ξ > 1 + μ /α0 (i.e. if the trade-off is sufficiently concave), siderophore production cannot evolve from a low initial investment into siderophore production. If ξ is below this threshold value (in particular if the trade-off is linear), selection selects for a low, but non-zero investment in PG production, as predicted by West & Buckling [12]. In the electronic supplementary material, S6, we further show that this result holds qualitatively for the whole range of susceptibility values. Note that the predictions of a linear trade-off are not robust for even slightly nonlinear trade-offs (e.g. ξ ≈ 1.1).
(iii). Model 2: detrimental public goods
We now turn to model 2 and assume that parasite virulence depends both on within-host parasite density and on the total amount of PG produced (equation (3.3)). We find a non-monotonous relationship between the optimal level of PG production and the susceptibility to secondary infections (figure 3g). As a result, selection leads to both PG production and virulence being maximal at intermediate MOI (figure 3h,i). The reason why this happens is that the direct contribution of PG to virulence yields an additional cost in terms of parasite survival (compared with the scenario where the transmission–virulence trade-off is linear, virulence increases faster). In the electronic supplementary material, S4.2, we show that this additional cost becomes stronger when the MOI decreases, whereas the benefits of cooperating go in the opposite direction. This explains why the net benefit-to-cost ratio of cooperators peaks at an intermediate MOI.
(iv). Effect of background host mortality
Equation (4.6) shows that the MOI is also a function of other epidemiological traits such as host mortality μ. Everything else being constant, the MOI is a decreasing function of μ. In the electronic supplementary material, figure S4, we show the resulting correlations between MOI, PG production and virulence when varying host background mortality and keeping the susceptibility to secondary infections σI constant. Comparing figure 3 with electronic supplementary material, figure S4, reveals that different evolutionary correlations may be expected between MOI and virulence in the population depending on which epidemiological parameter varies across populations or treatments. Although the prediction that virulence decreases with the MOI when the trade-off is linear is qualitatively preserved, variations in host mortality or in susceptibility do not in general lead to the same patterns for other trade-off functions. Note that the complexity of this result is not surprising since the value of μ affects at the same time the optimal PG production rate within a host (by determining the duration of the infection) and the MOI (which itself affects the optimal PG production through epidemiological feedbacks). This suggests that, to fully understand the relationship between virulence and MOI at the population level, a detailed knowledge of the mechanisms underpinning the change in MOI is required.
5. Discussion
The current paradigm is that, for parasites producing PGs in their hosts, such as phages [8] or siderophore-producing bacteria [12,31], multiple infections should select for lower levels of virulence [10,14]. Indeed, if virulence is associated with the amount of PGs parasites produce, co-infections can favour less virulent (‘cheater’) strains because they allow them to exploit more virulent (‘cooperative’) strains. In other words, when within-host relatedness decreases, parasite cooperation should decrease, and so should virulence [9,12,13,32]. Here, we show that these predictions need not always hold because epidemiological feedbacks and host demography can alter the selective pressures on virulence through changes in the distribution of parasite strains among hosts.
Using a mechanistic within-host model of PG production embedded in an epidemiological framework allowing for co-infections, we explore the evolutionary correlations between the MOI and parasite virulence under different assumptions regarding the relationship between virulence, transmission, PG production and parasite within-host densities.
With a linear trade-off between transmission and virulence (as assumed by West & Buckling [12]), selection favours ever-increasing levels of virulence, and therefore population-level feedbacks do not constrain within-host investment in PG production. Hence, when only single infections are permitted, the ESS is the level of PG production that maximizes within-host parasite density. By contrast, when the fraction of multiple infections among infected hosts increases, the optimal investment in PG production will be lower because defectors have an advantage in co-infected hosts.
The evolutionary outcomes are different when we assume a saturating transmission–virulence trade-off, which is at the core of many models in evolutionary epidemiology [20]. In particular, the optimal level of virulence can be higher when the MOI is maximal than when it is minimal, which contrasts with earlier predictions. This is due to the fact that, when virulence increases faster than transmission, population-level feedbacks favour intermediate levels of virulence because when virulence is too high, parasites do not transmit sufficiently before the host dies [18–20]. Here, such intermediate levels of virulence are achieved when within-host parasite density is not maximal, which occurs at both low and high (but not intermediate) levels of PG production. When the MOI in the population is sufficiently high, only a zero investment in PG production is evolutionarily stable, and there is no correlation between MOI and virulence. By contrast, when the MOI in the population is low to moderate, assuming a saturating trade-off introduces a bistability, and non-zero levels of PG production can evolve if the initial level of PG production is above a threshold. Otherwise, PG production is counter-selected. This results in a negative relationship between MOI and PG production, but a positive relationship between MOI and virulence.
In general, the exact shape of the trade-off between transmission and virulence will affect evolutionary predictions. For instance, assuming that virulence depends both on parasite and on PG within-host densities, we find that PG production and virulence can peak at intermediate levels of the MOI, resulting in a bell-shaped relationship between MOI, PG production and virulence. A possibility for a sequel study would be to explore trade-off shapes more extensively by resorting to a critical function analysis [33].
Our analysis shows that the relationship between the MOI, PG production and parasite virulence may be less general than expected from previous studies that showed that, for PG-producing parasites, multiple infections should always select for lower PG production and lower levels of virulence. There are two main reasons for this. First, some studies (e.g. [12]) assumed a linear transmission–virulence trade-off, which is unrealistic for many infectious diseases and leads to more virulent strains always having an advantage at the between-host level. Hence, when the trade-off is linear, virulence evolution is only constrained by within-host parasite interactions, while parasite between-host fitness is known to be shaped by population-level processes that can alter and, as we show, even reverse within-host selective pressures [26]. Second, the MOI in previous studies (reviewed by Brown et al. [10] and Buckling & Brockhurst [14]) was usually defined as a fixed parameter n that can be varied freely (‘open models’ [34]), whereas what empiricists generally have access to is a population average that will be affected by epidemiological and demographic processes (e.g. the MOI generally depends on transmission, virulence, and other host and disease life-history traits).
(a). Kin selection
In our study, the MOI is measured as the population average of the within-host relatedness between parasites, and is therefore affected by epidemiological and demographic feedbacks. It is therefore not surprising that our study has strong links with demographic kin selection models of the evolution of social traits. Indeed, an alternative way to view our epidemiological setting would be to consider the population of hosts as an infinite island model [35,36], each host being an island that can be infected by 0, 1, … , n parasite strains. This is analogous to Alizon & Taylor's [37] model of fecundity helping, with the important differences that in our model parasite clones play the role of individuals, and transition rates are affected by within-host densities, not only by the number of clones sharing a host. Our model is actually a limiting case of this process where we set n = 2 for simplicity. Indeed, this allows us to derive a simple analytical expression for the R0 of a mutant parasite. Moreover, previous studies have also shown that models with at most two strains per host already capture much of the effect of multiple infections on virulence evolution [3,29,38].
This analogy allows us to clarify the key difference between our model and previous studies of PG production in parasites, which typically consider that mutations have no effect on the distribution of parasite strains across individuals. This amounts to neglecting selection through the effects of the trait on the demographic state of the population, which can be an important factor influencing the evolution of social traits such as cooperation and virulence [39]. By contrast, our model allows us to consider how such demographic or epidemiological processes affect the distribution of parasite strains across hosts, and how the resulting variation in MOI alters selection on PG production and virulence.
(b). Empirical implications
Harrison et al. [13] have shown experimentally that, when co-infecting a host with two strains with different levels of virulence, the less virulent strain was favoured. However, their experimental set-up follows closely the model assumptions of West & Buckling [12] and focuses on isolated hosts. Our analysis suggests that to fully understand how multiple infections affect the evolution of virulence, population-level processes (such as transmission between hosts) need to be considered in future studies. This has been done in other empirical studies that take into account demographic and epidemiological feedbacks (e.g. [40,41]).
A further empirical implication of our results is that the relationship between PG production and virulence is not necessarily linear (figure 4). Hence, comparing two strains with different levels of virulence may not be sufficient to analyse the selective pressures on virulence in PG-producing parasites and to determine the evolutionary outcome. To gain more insight, future experiments should first test the link between PG production rate and host virulence. This could be achieved by generating a collection of bacterial strains that vary in the siderophore production rate (as in [42]) and assessing parasite density and virulence for each strain. This would allow empiricists to test the bell-shaped within-host trade-off between parasite density and PG production that is typically assumed in theoretical models, and also shed light on the range of biologically feasible levels of PG production. Our model reveals that, owing to epidemiological feedbacks, selection could favour overproduction of PG beyond the value that maximizes within-host parasite density. An important question is whether such levels of PG production are biologically feasible. Arguably, this overshooting may be too costly at the within-host level and prevented by selection for regulatory mechanisms. Such mechanisms have been studied in P. aeruginosa and data show that regulation can be facultative, or lost in some mutants [43,44]. This calls for further empirical and theoretical investigations of the joint evolution of siderophore production and regulatory mechanisms.
Testing the effect of multiple infections on virulence evolution is notoriously difficult. One of the main challenges is that it is technically complicated to prevent multiple infections from occurring. A possibility would be to use the setting developed by Ebert & Mangin [40], which simulates a large host baseline mortality by removing a fraction of hosts and replacing it with the same amount of susceptible hosts. This marginalizes multiple infections but it can also generate complicated short-term evolutionary dynamics [45]. Here, we show that in addition to this issue, the way the MOI varies (e.g. host susceptibility to co-infections versus host background mortality rate) is likely to affect the evolutionary outcome of the model.
(c). Perspectives
Few co-infection models consider within-host dynamics explicitly (but see [46]), which means that theoretical predictions often cannot be interpreted in terms of within-host cooperative or competitive interactions. An exception is provided by Alizon & van Baalen [29], who analysed a model of within-host competition between parasites mediated by the immune system. Here, we focus on PG-producing parasites, but there exists a huge variety of within-host interactions between parasites, which deserve further investigation [47].
Throughout most of our analysis, we limited the number of strains per host to two. Classical open models do not have this limitation, but they also ignore epidemiological feedbacks. Our simplification allows for analytical tractability, while capturing the important features of the co-infection process [3]. Moreover, because we assume rare mutations, evolution proceeds through the replacement of one strain by another and the parasite population is at most dimorphic. However, the interplay of mutation and infection processes could generate a higher between-host variance in the distribution of parasite strains. To take into account the fact that hosts can harbour more than two parasite genotypes, alternative evolutionary models would be required (e.g. models of co-infection by different species [38] or population-genetic models [45]). This would provide some interesting insights into the robustness of our result to a higher genetic variance and higher mutation rates within the parasite population.
Another simplifying assumption of our study is that the host population is well mixed. Yet the spatial or social structure of the host population has been shown to have a strong impact on disease evolution [5,48–50]. In a spatially structured host population, virulence evolution is known to be determined by a balance between epidemiological structuring and genetic structuring [50]. It will be interesting to investigate in more detail how the interplay between the within-host and between-host structures of the parasite population affects the evolution of virulence in PG-producing parasites.
Acknowledgements
We thank S. Gandon for discussions, and T. Day, Y. Michalakis and three reviewers for helpful comments. S.A. is funded by the CNRS and the IRD. S.L. was supported by a Marie Curie Intra-European Fellowship FP7-220080 (Mobvir) from the Seventh Framework Programme of the European Commission, by CNRS and by an ANR grant RPDOC 2010 (SpatEvolEpid).
References
- 1.Brogden K. A., Guthmiller J. M., Taylor C. E. 2005. Human polymicrobial infections. Lancet 365, 253–255 10.1016/S0140-6736(05)17745-9 (doi:10.1016/S0140-6736(05)17745-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bremermann H. J., Pickering J. 1983. A game-theoretical model of parasite virulence. J. Theor. Biol. 100, 411–426 10.1016/0022-5193(83)90438-1 (doi:10.1016/0022-5193(83)90438-1) [DOI] [PubMed] [Google Scholar]
- 3.van Baalen M., Sabelis M. W. 1995. The dynamics of multiple infection and the evolution of virulence. Am. Nat. 146, 881–910 10.1086/285830 (doi:10.1086/285830) [DOI] [Google Scholar]
- 4.May R. M., Nowak M. A. 1995. Coinfection and the evolution of parasite virulence. Proc. R. Soc. Lond. B 261, 209–215 10.1098/rspb.1995.0138 (doi:10.1098/rspb.1995.0138) [DOI] [PubMed] [Google Scholar]
- 5.Frank S. A. 1996. Models of parasite virulence. Q. Rev. Biol. 71, 37–78 10.1086/419267 (doi:10.1086/419267) [DOI] [PubMed] [Google Scholar]
- 6.de Roode J. C., et al. 2005. Virulence and competitive ability in genetically diverse malaria infections. Proc. Natl Acad. Sci. USA 102, 7624–7628 10.1073/pnas.0500078102 (doi:10.1073/pnas.0500078102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ben-Ami F., Mouton L., Ebert D. 2008. The effects of multiple infections on the expression and evolution of virulence in a Daphnia-endoparasite system. Evolution 62, 1700–1711 10.1111/j.1558-5646.2008.00391.x (doi:10.1111/j.1558-5646.2008.00391.x) [DOI] [PubMed] [Google Scholar]
- 8.Turner P. E., Chao L. 1999. Prisoner's dilemma in an RNA virus. Nature 398, 441–443 10.1038/18913 (doi:10.1038/18913) [DOI] [PubMed] [Google Scholar]
- 9.Brown S. P. 2001. Collective action in RNA virus. J. Evol. Biol. 14, 821–828 10.1046/j.1420-9101.2001.00317.x (doi:10.1046/j.1420-9101.2001.00317.x) [DOI] [Google Scholar]
- 10.Brown S. P., Hochberg M. E., Grenfell B. T. 2002. Does multiple infection select for raised virulence? Trends Microbiol. 10, 401–405 10.1016/S0966-842X(02)02413-7 (doi:10.1016/S0966-842X(02)02413-7) [DOI] [PubMed] [Google Scholar]
- 11.Wandersman C., Delepelaire P. 2004. Bacterial iron sources: from siderophores to hemophores. Annu. Rev. Microbiol. 58, 611–647 10.1146/annurev.micro.58.030603.123811 (doi:10.1146/annurev.micro.58.030603.123811) [DOI] [PubMed] [Google Scholar]
- 12.West S. A., Buckling A. 2003. Cooperation, virulence and siderophore production in bacterial parasites. Proc. R. Soc. Lond. B 270, 37–44 10.1098/rspb.2002.2209 (doi:10.1098/rspb.2002.2209) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harrison F., Browning L. E., Vos M., Buckling A. 2006. Cooperation and virulence in acute Pseudomonas aeruginosa infections. BMC Biol. 4, 21–25 10.1186/1741-7007-4-21 (doi:10.1186/1741-7007-4-21) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Buckling A., Brockhurst M. A. 2008. Kin selection and the evolution of virulence. Heredity 100, 484–488 10.1038/sj.hdy.6801093 (doi:10.1038/sj.hdy.6801093) [DOI] [PubMed] [Google Scholar]
- 15.Levin B. R., Bull J. J. 1994. Short-sighted evolution and the virulence of pathogenic microorganisms. Trends Microbiol. 2, 76–81 10.1016/0966-842X(94)90538-X (doi:10.1016/0966-842X(94)90538-X) [DOI] [PubMed] [Google Scholar]
- 16.van Baalen M., Sabelis M. W. 1995. The scope for virulence management: a comment on Ewald's view on the evolution of virulence. Trends Microbiol. 3, 414–416 10.1016/S0966-842X(00)88991-X (doi:10.1016/S0966-842X(00)88991-X) [DOI] [PubMed] [Google Scholar]
- 17.Day T., Proulx S. R. 2004. A general theory for the evolutionary dynamics of virulence. Am. Nat. 163, E40–E63 10.1086/382548 (doi:10.1086/382548) [DOI] [PubMed] [Google Scholar]
- 18.Anderson R. M., May R. M. 1982. Coevolution of hosts and parasites. Parasitology 85, 411–426 10.1017/S0031182000055360 (doi:10.1017/S0031182000055360) [DOI] [PubMed] [Google Scholar]
- 19.Ewald P. W. 1994. Evolution of infectious disease. Oxford, UK: Oxford University Press [Google Scholar]
- 20.Alizon S., Hurford A., Mideo N., van Baalen M. 2009. Virulence evolution and the trade-off hypothesis: history, current state of affairs and the future. J. Evol. Biol. 22, 245–259 10.1111/j.1420-9101.2008.01658.x (doi:10.1111/j.1420-9101.2008.01658.x) [DOI] [PubMed] [Google Scholar]
- 21.Fraser C., Hollingsworth T. D., Chapman R., de Wolf F., Hanage W. P. 2007. Variation in HIV-1 set-point viral load: epidemiological analysis and an evolutionary hypothesis. Proc. Natl Acad. Sci. USA 104, 17 441–17 446 10.1073/pnas.0708559104 (doi:10.1073/pnas.0708559104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Roode J. C., Yates A. J., Altizer S. 2008. Virulence–transmission trade-offs and population divergence in virulence in a naturally occurring butterfly parasite. Proc. Natl Acad. Sci. USA 105, 7489–7494 10.1073/pnas.0710909105 (doi:10.1073/pnas.0710909105) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mideo N., Alizon S., Day T. 2008. Linking within- and between-host dynamics in the evolutionary epidemiology of infectious diseases. Trends Ecol. Evol. 23, 511–517 10.1016/j.tree.2008.05.009 (doi:10.1016/j.tree.2008.05.009) [DOI] [PubMed] [Google Scholar]
- 24.Gardner A., West S. A., Buckling A. 2004. Bacteriocins, spite and virulence. Proc. R. Soc. Lond. B 271, 1529–1535 10.1098/rspb.2004.2756 (doi:10.1098/rspb.2004.2756) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taylor P. D., Frank S. A. 1996. How to make a kin selection model. J. Theor. Biol. 180, 27–37 10.1006/jtbi.1996.0075 (doi:10.1006/jtbi.1996.0075) [DOI] [PubMed] [Google Scholar]
- 26.Dieckmann U. 2002. Adaptive dynamics of pathogen–host interactions. In Adaptive dynamics of infectious diseases: in pursuit of virulence management (eds Dieckmann U., Metz J. A. J., Sabelis M. W., Sigmund K.), pp. 39–59 Cambridge, UK: Cambridge University Press; 10.1017/CBO9780511525728.006 (doi:10.1017/CBO9780511525728.006) [DOI] [Google Scholar]
- 27.Geritz S. A. H., Kisdi E., Meszéna G., Metz J. A. J. 1998. Evolutionarily singular strategies and the adaptive growth and branching of the evolutionary tree. Evol. Ecol. 4, 1–79 10.1023/A:1006554906681 (doi:10.1023/A:1006554906681) [DOI] [Google Scholar]
- 28.Otto S., Day T. 2007. A biologist's guide to mathematical modeling in ecology and evolution. Princeton, NJ: Princeton University Press [Google Scholar]
- 29.Alizon S., van Baalen M. 2008. Multiple infections, immune dynamics and virulence evolution. Am. Nat. 172, E150–E158 10.1086/590958 (doi:10.1086/590958) [DOI] [PubMed] [Google Scholar]
- 30.Lipsitch M., Colijn C., Cohen T., Hanage W. P., Fraser C. 2009. No coexistence for free: neutral null models for multistrain pathogens. Epidemics 1, 2–13 10.1016/j.epidem.2008.07.001 (doi:10.1016/j.epidem.2008.07.001) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buckling A., Harrison F., Vos M., Brockhurst M. A., Gardner A., West S. A., Griffin A. 2007. Siderophore-mediated cooperation and virulence in Pseudomonas aeruginosa. FEMS Microbiol. Ecol. 62, 135–141 10.1111/j.1574-6941.2007.00388.x (doi:10.1111/j.1574-6941.2007.00388.x) [DOI] [PubMed] [Google Scholar]
- 32.Brown S. P., Johnstone R. 2001. Cooperation in the dark: signalling and collective action in quorum-sensing bacteria. Proc. R. Soc. Lond. B 268, 961–965 10.1098/rspb.2001.1609 (doi:10.1098/rspb.2001.1609) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Mazancourt C., Dieckmann U. 2004. Trade-off geometries and frequency-dependent selection. Am. Nat. 164, 765–778 10.1086/424762 (doi:10.1086/424762) [DOI] [PubMed] [Google Scholar]
- 34.Gardner A., West S. A. 2006. Demography, altruism, and the benefits of budding. J. Evol. Biol. 19, 1707–1716 10.1111/j.1420-9101.2006.01104.x (doi:10.1111/j.1420-9101.2006.01104.x) [DOI] [PubMed] [Google Scholar]
- 35.Wright S. 1931. Evolution in Mendelian populations. Genetics 16, 97–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rousset F. 2004. Genetic structure and selection in subdivided populations. Princeton, NJ: Princeton University Press [Google Scholar]
- 37.Alizon S., Taylor P. 2008. Empty sites can promote altruistic behaviour. Evolution 62, 1335–1344 10.1111/j.1558-5646.2008.00369.x (doi:10.1111/j.1558-5646.2008.00369.x) [DOI] [PubMed] [Google Scholar]
- 38.Choisy M., de Roode J. C. 2010. Mixed infections and the evolution of virulence: effects of resource competition, parasite plasticity and impaired host immunity. Am. Nat. 175, E105–E118 10.1086/651587 (doi:10.1086/651587) [DOI] [PubMed] [Google Scholar]
- 39.Rousset F., Ronce O. 2004. Inclusive fitness for traits affecting metapopulation demography. Theor. Popul. Biol. 65, 127–141 10.1016/j.tpb.2003.09.003 (doi:10.1016/j.tpb.2003.09.003) [DOI] [PubMed] [Google Scholar]
- 40.Ebert D., Mangin K. 1997. The influence of host demography on the evolution of virulence of microsporidian gut parasite. Evolution 51, 1828–1837 10.2307/2411005 (doi:10.2307/2411005) [DOI] [PubMed] [Google Scholar]
- 41.Boots M., Mealor M. 2007. Local interactions select for lower pathogen infectivity. Science 315, 1284–1286 10.1126/science.1137126 (doi:10.1126/science.1137126) [DOI] [PubMed] [Google Scholar]
- 42.Jiricny N., Diggle S. P., West S. A., Evans B. A., Ballantyne G., Ross-Gillespie A., Griffin A. S. 2010. Fitness correlates with the extent of cheating in a bacterium. J. Evol. Biol. 23, 738–747 10.1111/j.1420-9101.2010.01939.x (doi:10.1111/j.1420-9101.2010.01939.x) [DOI] [PubMed] [Google Scholar]
- 43.Kümmerli R., Jiricny N., Clarke L. S., West S. A., Griffin A. S. 2009. Phenotypic plasticity of a cooperative behaviour in bacteria. J. Evol. Biol. 22, 589–598 10.1111/j.1420-9101.2008.01666.x (doi:10.1111/j.1420-9101.2008.01666.x) [DOI] [PubMed] [Google Scholar]
- 44.Kümmerli R., Brown S. P. 2010. Molecular and regulatory properties of a public good shape the evolution of cooperation. Proc. Natl Acad. Sci. USA 107, 18 921–18 926 10.1073/pnas.1011154107 (doi:10.1073/pnas.1011154107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Day T., Gandon S. 2007. Applying population-genetic models in theoretical evolutionary epidemiology. Ecol. Lett. 10, 876–888 10.1111/j.1461-0248.2007.01091.x (doi:10.1111/j.1461-0248.2007.01091.x) [DOI] [PubMed] [Google Scholar]
- 46.Coombs D., Gilchrist M. A., Ball C. L. 2007. Evaluating the importance of within- and between-host selection pressures on the evolution of chronic pathogens. Theor. Popul. Biol. 72, 576–591 10.1016/j.tpb.2007.08.005 (doi:10.1016/j.tpb.2007.08.005) [DOI] [PubMed] [Google Scholar]
- 47.Mideo N. 2009. Parasite adaptations to within-host competition. Trends Parasitol. 25, 261–268 10.1016/j.pt.2009.03.001 (doi:10.1016/j.pt.2009.03.001) [DOI] [PubMed] [Google Scholar]
- 48.Boots M., Sasaki A. 1999. ‘Small worlds’ and the evolution of virulence: infection occurs locally and at a distance. Proc. R. Soc. Lond. B 266, 1933–1938 10.1098/rspb.1999.0869 (doi:10.1098/rspb.1999.0869) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lion S., van Baalen M. 2008. Self-structuring in spatial evolutionary ecology. Ecol. Lett. 11, 277–295 10.1111/j.1461-0248.2007.01132.x (doi:10.1111/j.1461-0248.2007.01132.x) [DOI] [PubMed] [Google Scholar]
- 50.Lion S., Boots M. 2010. Are parasites ‘prudent’ in space? Ecol. Lett. 13, 1245–1255 10.1111/j.1461-0248.2010.01516.x (doi:10.1111/j.1461-0248.2010.01516.x) [DOI] [PMC free article] [PubMed] [Google Scholar]