

Abstract
Our group has recently developed a compact, universal protein binding microarray (PBM) that can be used to determine the binding preferences of transcription factors (TFs). This design represents all possible sequence variants of a given length k (i.e., all k-mers) on a single array, allowing a complete characterization of the binding specificities of a given TF. Here, we present the mathematical foundations of this design based on de Bruijn sequences generated by linear feedback shift registers. We show that these sequences represent the maximum number of variants for any given set of array dimensions (i.e., number of spots and spot lengths), while also exhibiting desirable pseudo-randomness properties. Moreover, de Bruijn sequences can be selected that represent gapped sequence patterns, further increasing the coverage of the array. This design yields a powerful experimental platform that allows the binding preferences of TFs to be determined with unprecedented resolution.
Key words: combinatorics, DNA arrays, genomics, linear algebra, strings
1. Introduction
Detailed knowledge of the DNA binding specificities of TFs is crucial for both genomic studies attempting to map TFs to their target genes (Bulyk, 2003), as well as biophysical investigations of protein–DNA interactions (Benos et al., 2002). Despite the importance of this data type, the binding preferences of the vast majority of TFs remain unknown, largely due to a historical lack of suitable experimental technologies. While chromatin immunoprecipitation (ChIP) experiments (Das et al., 2004), and, more recently, ChIP-chip experiments (Wyrick and Young, 2002), give specific examples of sequences bound by a TF in vivo, they do not provide an exhaustive characterization of the sequences that a TF can and (just as importantly) cannot bind. Similarly, approaches such as in vitro selection (Oliphant et al., 1989) typically identify only a limited number of high-affinity binding sites, making a direct quantification of relative binding preferences difficult.
To address this challenge, our group has developed the protein binding microarray (PBM) technology for high-throughput characterization of the in vitro binding specificities of protein-DNA interactions (Berger et al., 2006; Bulyk et al., 1999, 2001; Mukherjee et al., 2004). Briefly, a DNA-binding protein of interest is expressed with an epitope tag, then purified and applied to a double-stranded DNA microarray. The washed, protein-bound microarray is labeled with a fluorophore-conjugated anti-GST antibody. By scanning the array, quantitative information is generated regarding the preferences of the TF for each of the sequences on the array. Prior work by our group and others has demonstrated that this is an effective technology that allows rapid and high-quality determination of the DNA binding specificities of TFs (Bulyk et al., 2001; Linnell et al., 2004; Mukherjee et al., 2004; Warren et al., 2006).
A limitation of previous PBM studies, however, has been the lack of a universal array that can be used for the majority of TFs, regardless of their structural class or genome of origin. Earlier studies have utilized either microarrays containing a limited number of binding site variants chosen for the TF under consideration (Bulyk et al., 2001; Linnell et al., 2004), or large genomic fragments obtained from the same genome as the TF (specifically, S. cerevisiae) (Mukherjee et al., 2004). The former approach has the twofold disadvantage of requiring a new microarray for each additional TF assayed and also requiring some a priori knowledge of the DNA binding specificities of the TF; the latter approach suffers from the limitation that longer sequences can contain several binding sites for a given TF, making it difficult to acquire quantitative information on protein-DNA interactions. Thus, a single microarray is desired that represents all possible binding sites of a given width k (i.e., all k-mers), in order to provide a complete survey of all candidate binding sites.
Our group has recently developed such a universal array (Berger et al., 2006). The key to our design is twofold. First, we have selected our double-stranded DNA probes to have a length (L) significantly longer than the motif widths (k) that we intend to inspect, so that each spot contains L − k + 1 potential binding sites of width k. For a microarray composed of N spots, this increases the total number of k-mers represented from N (in the naïve construction where there is one k-mer per spot, as has been previously utilized (Warren et al., 2006)) to N(L − k + 1). Second, we have designed these spots to completely cover all k-mer sequence variants, so that a maximal number of distinct k-mers are represented. Consider the circular sequence shown in Figure 1a that contains all sixteen 2-mer variants exactly once. Such sequences containing all 4k overlapping k-mers one time are named de Bruijn sequences (De Bruijn, 1946; Gross and Yellen, 2004) of order k, and the spots of our universal array are obtained by computationally segmenting appropriately chosen de Bruijn sequences, leaving an overlap between adjacent sequences in order to not omit any k-mers. With this design, we are able to represent a maximal number of sequence variants in a minimum amount of sequence.
FIG. 1.

De Bruijn sequence of order 2 (a) and its associated de Bruijn graph (b).
The implementation of this design, along with generated data for five TFs, has been presented in the work of Berger et al. (2006). Here, we give an exposition of the underlying combinatorial and algebraic theory utilized in designing the array. Specifically, we provide a mathematical treatment of (1) the motivation for and utilization of linear feedback shift registers (LFSRs) to generate de Bruijn sequences; (2) theoretical developments made by our group in order to design de Bruijn sequences that not only contain contiguous k-mers, but also k-mers with biologically relevant gaps (to our knowledge, this is the first work to mathematically address the coverage of gapped k-mers in de Bruijn sequences); (3) methods for selecting de Bruijn sequences that are optimized for determining TF binding site motifs that are wider than the order of the utilized de Bruijn sequence; and (4) the utilization of complementary, independently generated de Bruijn sequences for use in replicate PBM experiments. Finally, we note that de Bruijn sequences have previously been utilized in cryptography (Golomb, 1967; Joyner et al., 2004), random number generation (Golomb, 1967; Joyner et al., 2004) and the design of tags for DNA microarrays (Ben-Dor et al., 2000). Recently, another group has independently suggested the use of de Bruijn sequences for use in PBM experiments, although that work did not consider the coverage of gapped k-mers, did not utilize LFSRs, and did not address the questions of examining TFs whose binding site motif was wider than the order of the utilized de Bruijn sequence or choosing complementary de Bruijn sequences for replicate experiments (Mintseris and Eisen, 2006). We hope that our approach will be useful to individuals either seeking to design sequences for PBM experiments or analyzing data generated by a PBM utilizing de Bruijn sequences. Additionally, we hope that the novel mathematical methods developed for this application will lead to other, un-anticipated biological applications.
2. Results
2.1. LFSRs and the generation of de Bruijn sequences
For any alphabet Σ of size |Σ| and any word length k, there exist sequences 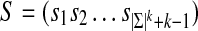 that are circular (i.e.,
that are circular (i.e., 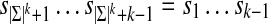 ) and of length |Σ|k containing all k-mers exactly once when words are considered in a stacked fashion. Such sequences are known as de Bruijn sequences of order k, and their existence can be confirmed by considering the directed graph whose vertices are all (k − 1)-mers and whose edges are all k-mers, where two vertices are connected by an edge if the last k − 2 letters of the first vertex are identical to the first k − 2 letters of the second. Figure 1b gives an example of such a graph, often referred to as a “de Bruijn graph” (Gross and Yellen, 2004), and we note that graphs of this form have previously been applied to the analysis of repetitive DNA (Pevzner et al., 2004) and sequence alignment (Zhang and Waterman, 2005). Observe that a de Bruijn sequence is equivalent to a walk on this directed graph that traverses every edge (i.e., an Eulerian tour) (Gross and Yellen, 2004). Since the number of edges going into each vertex is equal to the number of edges that exit it, Euler's theorem guarantees that such paths exist (Gross and Yellen, 2004). Indeed, for a given choice of |Σ| and k, the number of paths is large and equal to
) and of length |Σ|k containing all k-mers exactly once when words are considered in a stacked fashion. Such sequences are known as de Bruijn sequences of order k, and their existence can be confirmed by considering the directed graph whose vertices are all (k − 1)-mers and whose edges are all k-mers, where two vertices are connected by an edge if the last k − 2 letters of the first vertex are identical to the first k − 2 letters of the second. Figure 1b gives an example of such a graph, often referred to as a “de Bruijn graph” (Gross and Yellen, 2004), and we note that graphs of this form have previously been applied to the analysis of repetitive DNA (Pevzner et al., 2004) and sequence alignment (Zhang and Waterman, 2005). Observe that a de Bruijn sequence is equivalent to a walk on this directed graph that traverses every edge (i.e., an Eulerian tour) (Gross and Yellen, 2004). Since the number of edges going into each vertex is equal to the number of edges that exit it, Euler's theorem guarantees that such paths exist (Gross and Yellen, 2004). Indeed, for a given choice of |Σ| and k, the number of paths is large and equal to
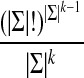 |
(Gross and Yellen, 2004); for example, for |Σ| = 4 (i.e., the DNA alphabet) and k = 9, the number of de Bruijn sequences is greater than 1090,000.
De Bruijn sequences contain a maximum density of sequence variants, as they contain all distinct k-mers within a sequence of minimum length. Moreover, for any m > k, the |Σ|k sequences of length m represented in the de Bruijn sequence will all be distinct; thus, although not all m-mers are represented on an order k de Bruijn sequence, as many distinct m-mers as possible are represented within the given sequence length. Similarly, for all m′ < k, each m′-mer is represented exactly |Σ|k−m′ times, insuring that the sampling of m′-mers is uniform. Clearly, the regularity and variability of de Bruijn sequences makes them a promising tool for designing a universal PBM. An especially facile method of generating such sequences when |Σ| = pn, for p a prime and n any integer, is through the use of linear feedback shift registers (LFSRs) (Golomb, 1967; Joyner et al., 2004). As background, recall that a Galois field GF(pn) is a set containing pn elements that is closed over the multiplication and addition {×, +} operations (one can show that such operations can be suitably defined if and only if the field contains a prime power of elements (Stewart, 1989)). For example, Figure 2 gives multiplication and addition tables over GF(4) = {0, 1, α, α + 1}.
FIG. 2.

Addition and multiplication tables over GF(4).
In order to construct a de Bruijn sequence of order k over the alphabet Σ, take an arbitrary embedding of the alphabet into GF(|Σ|), and consider the following recursive linear equation for generating the i'th element of a sequence 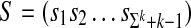 from the previous k elements, where arithmetic is performed over GF(|Σ|):
from the previous k elements, where arithmetic is performed over GF(|Σ|):
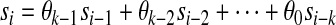 |
(1) |
If the coefficients θi ε GF(|Σ|) are chosen so that the polynomial 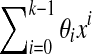 is primitive (Stewart, 1989), one can demonstrate (Golomb, 1967) that the sequence S generated by this recursive equation has periodicity |Σ|k − 1 and contains every k-mer in GF(|Σ|) except the sequence of k consecutive 0's (this word can be easily included to make S a true de Bruijn sequence by inserting an additional 0 into any of the subsequences of k − 1 0's appearing in S). Moreover, S will exhibit the following three properties characteristic of pseudo-random sequences (Golomb, 1967; Joyner et al., 2004):
is primitive (Stewart, 1989), one can demonstrate (Golomb, 1967) that the sequence S generated by this recursive equation has periodicity |Σ|k − 1 and contains every k-mer in GF(|Σ|) except the sequence of k consecutive 0's (this word can be easily included to make S a true de Bruijn sequence by inserting an additional 0 into any of the subsequences of k − 1 0's appearing in S). Moreover, S will exhibit the following three properties characteristic of pseudo-random sequences (Golomb, 1967; Joyner et al., 2004):
Balance: The number of occurrences in S of each letter differs by no more than 1.
Low autocorrelation: There is low correlation between pairs of letters separated by a distance j, for any j.
Proportional runs: The number of j consecutive occurrences of the same letter ω is nk−j if ω ≠ 0 and nk−j − 1 if ω = 0.
Thus, de Bruijn sequences generated by LFSRs resemble random sequences, an advantageous property as it guarantees that any trends observed in the data are not an artifact of the method of sequence generation. Moreover, unlike random sequence, LFSRs represent a maximal number of sequence variants (a truly random sequences of equivalent length would represent only 1 − e−1 ≈ 63% of k-mers on average) (Berger et al., 2006). Since the DNA alphabet contains a prime power of elements (4 = 22), LFSRs are available for use in generating de Bruijn sequences. Indeed, there are (at least) two approaches for using LFSRs to generate de Bruijn sequences over the DNA alphabet. In the first and more natural approach, one takes an arbitrary embedding of {a, c, g, t} into GF(4) = {0, 1, α, α + 1} where α2 = α + 1, and one then picks a primitive polynomial of degree k over GF(4) to use as a LFSR generating a sequence of length 4k − 1. This is schematized in Figure 3a, using the embedding 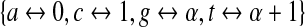 (again, under this embedding the generated sequences do not contain the sequence of k consecutive a's). Alternatively, one can pick a polynomial of degree 2k over GF(2) = {0,1} and use it to generate a sequence of length 22k − 1 over the 0-1 alphabet. Here, one associates each element of the DNA alphabet with a pair of elements in GF(2), and one must traverse this sequence over GF(2) twice, considering both reading frames. This is schematized in Figure 3b, where we have used the embedding
(again, under this embedding the generated sequences do not contain the sequence of k consecutive a's). Alternatively, one can pick a polynomial of degree 2k over GF(2) = {0,1} and use it to generate a sequence of length 22k − 1 over the 0-1 alphabet. Here, one associates each element of the DNA alphabet with a pair of elements in GF(2), and one must traverse this sequence over GF(2) twice, considering both reading frames. This is schematized in Figure 3b, where we have used the embedding 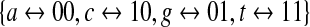 . Henceforth, we shall refer to the embeddings
. Henceforth, we shall refer to the embeddings 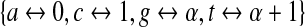 and
and 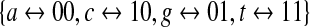 of the DNA alphabet into GF(4) and GF(2), respectively, as the standard embeddings (note that both methods of utilizing LFSRs to generate de Bruijn sequences can be generalized to arbitrary number fields with a prime power of elements). In the next section, we show that de Bruijn sequences generated by primitive polynomials over GF(2) and GF(4) actually behave differently with respect to the coverage of gapped k-mers.
of the DNA alphabet into GF(4) and GF(2), respectively, as the standard embeddings (note that both methods of utilizing LFSRs to generate de Bruijn sequences can be generalized to arbitrary number fields with a prime power of elements). In the next section, we show that de Bruijn sequences generated by primitive polynomials over GF(2) and GF(4) actually behave differently with respect to the coverage of gapped k-mers.
FIG. 3.

Generation of de Bruijn sequences over 4-letter (a) and 2-letter (b) alphabets.
Our basic design, then, is to utilize LFSRs to generate de Bruijn sequences of order k, where k is as large as possible for a given set of array dimensions and spot lengths. The generated de Bruijn sequence is then computationally segmented into shorter sequences of length l corresponding to spots on the array, leaving an overlap of k − 1 letters between adjacent spots so as not to omit any k-mers. For example, consider an array composed of spots of length 30; then all 9-mers could be represented using fewer than 12,000 spots, well within the range of current array dimensions. Such an array would also contain nearly 1/4 of all 10-mers, 1/16 of all 11-mers, etc., and thus could be expected to yield substantial information about TFs having motif widths greater than 9.
2.2. LFSRs and the coverage of spaced seeds
In Berger et al. (2006), we performed a survey of known TFBS motifs in order to determine what value of k is required in order to design an array suitable for most TFs. There, we observed that a majority of motifs contained <=9 informative positions. We also observed, however, that for nearly 25% of motifs, the pattern of informative positions was not contiguous and contained one or more gaps (i.e., positions with ≤0.3 bits of information when using the standard position-weight-matrix representation) (Stormo, 2000). While de Bruijn sequences of order k will, by definition, contain all contiguous k-mers, they do not necessarily contain all gapped k-mers. Therefore, we inspected the representation of gapped k-mers in de Bruijn sequences.
We define a seed to be the set of all possible words over the DNA alphabet with a given (possibly gapped) pattern of positions, and we represent the seed with a 0-1 string where 1's are placed at the informative positions. For example, the seed “11” corresponds to the set 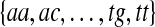 that contains all contiguous 2-mers, and the seed “1001” corresponds to the set
that contains all contiguous 2-mers, and the seed “1001” corresponds to the set 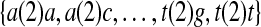 where the numbers in parentheses denote the gap size. Words with gaps will be said to be elements of spaced seeds, and those without gaps will be said to be elements of contiguous seeds. We shall use the variable ζ to represent an arbitrary seed and, for a seed ζ containing k informative positions, we say the order of ζ is k. Finally, a given LFSR L is said to cover a seed ζ if all its elements except the string composed of all a's (e.g., aa and a(2)a for the order 2 seeds 11 and 1001, respectively) appear in the sequence generated by L (the reasons for ignoring the elements composed of only the letter a will be explained shortly). Similarly, we shall refer to the coverage of ζ by L with the variable χ(L, ζ), which takes the value of 1 if the seed ζ is covered by the LFSR and 0 otherwise (again ignoring the element composed of only the letter a).
where the numbers in parentheses denote the gap size. Words with gaps will be said to be elements of spaced seeds, and those without gaps will be said to be elements of contiguous seeds. We shall use the variable ζ to represent an arbitrary seed and, for a seed ζ containing k informative positions, we say the order of ζ is k. Finally, a given LFSR L is said to cover a seed ζ if all its elements except the string composed of all a's (e.g., aa and a(2)a for the order 2 seeds 11 and 1001, respectively) appear in the sequence generated by L (the reasons for ignoring the elements composed of only the letter a will be explained shortly). Similarly, we shall refer to the coverage of ζ by L with the variable χ(L, ζ), which takes the value of 1 if the seed ζ is covered by the LFSR and 0 otherwise (again ignoring the element composed of only the letter a).
For a given sequence S over {a, c, g, t}, let Ak,S denote the set of all (potentially gapped) subsequences of k a's that occur in S; for example, in the sequence shown in Figure 3a 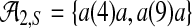 , and in Figure 3b
, and in Figure 3b 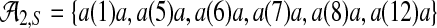 . For ζ a seed of order k and S a sequence generated by a LFSR over GF(q), where q equals either 2 or 4, one can demonstrate the following facts concerning the coverage of spaced seeds by LFSRs.
. For ζ a seed of order k and S a sequence generated by a LFSR over GF(q), where q equals either 2 or 4, one can demonstrate the following facts concerning the coverage of spaced seeds by LFSRs.
Proposition 1
a) Under the standard embeddings of the DNA alphabet, ζ is covered by S if and only if 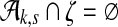 . b) There exists a j ε N such that every element of ζ not in Ak,S occurs either 0 times or exactly qj times in S. Also, the element of Ak,S in ζ occurs qj − 1 times.
. b) There exists a j ε N such that every element of ζ not in Ak,S occurs either 0 times or exactly qj times in S. Also, the element of Ak,S in ζ occurs qj − 1 times.
Proof
Consider the case where q = 4. Because our sequence 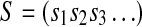 is generated by a LFSR, we know that for any i
is generated by a LFSR, we know that for any i
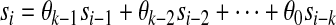 |
(2) |
Given values of i and m where m ≥ k, let (Si,m) denote the subsequence in S of m letters beginning at the letter si; also, let this same notation denote the vector of dimension m over GF(q) 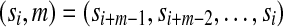 . Consider the matrix
. Consider the matrix
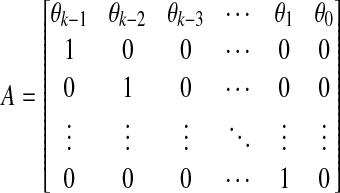 |
It is clear that for any 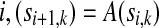 and, by induction, for any
and, by induction, for any 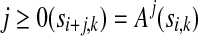 . Also, observe that for any m ≥ k, (si,m) can be constructed from (si,k) by applying the m × k matrix
. Also, observe that for any m ≥ k, (si,m) can be constructed from (si,k) by applying the m × k matrix
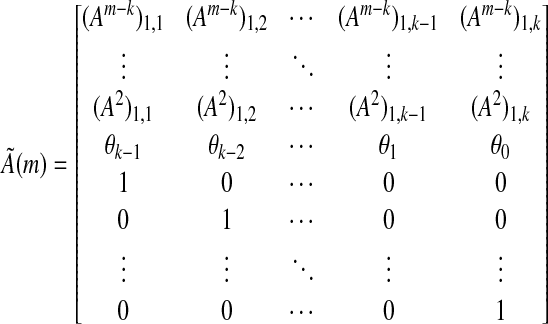 |
as 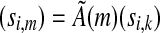 (note that in
(note that in 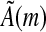 , the entries (An)i,j refer to the matrix element in row i and column j in the n'th power of A). Consider a seed ζ having width m and containing k informative positions. Let
, the entries (An)i,j refer to the matrix element in row i and column j in the n'th power of A). Consider a seed ζ having width m and containing k informative positions. Let 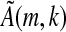 be the k × k sub-matrix of
be the k × k sub-matrix of 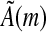 when restricting to only those rows corresponding to the informative positions of ζ. Consider the set
when restricting to only those rows corresponding to the informative positions of ζ. Consider the set 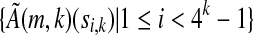 (i.e., the set of elements of ζ that occur in S).
(i.e., the set of elements of ζ that occur in S). 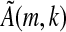 is either invertible or it is not. If
is either invertible or it is not. If 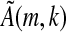 is invertible, then its image is all 4k elements of ζ, and every element of the seed occurs in the LFSR with the exception of the sequence that contains a 0 (equivalently, an “a” under the standard embedding) at every informative position, as the kernel of
is invertible, then its image is all 4k elements of ζ, and every element of the seed occurs in the LFSR with the exception of the sequence that contains a 0 (equivalently, an “a” under the standard embedding) at every informative position, as the kernel of 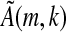 is trivial. Thus, ζ will be covered if the sequence with a's at the informative positions of the spaced seed (which is an element of Ak,s) does not appear in S. Similarly, this argument is reversible and so the converse holds; thus, Proposition 1a is proven. If
is trivial. Thus, ζ will be covered if the sequence with a's at the informative positions of the spaced seed (which is an element of Ak,s) does not appear in S. Similarly, this argument is reversible and so the converse holds; thus, Proposition 1a is proven. If 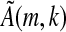 is not invertible, then its kernel will contain 4j elements for j ε N, its image will contain 4k−j elements, and each of these elements will be the image of 4j vectors (si,k). Since every contiguous k-mer (si,k) except the sequence of k consecutive a's occurs in S, Proposition 1b holds and the proof is completed for the case q = 4. The proof for q = 2 is nearly identical. Now, however, our matrix A will have dimension 2k × 2k. Note that here, the kernel for the matrix analogous to
is not invertible, then its kernel will contain 4j elements for j ε N, its image will contain 4k−j elements, and each of these elements will be the image of 4j vectors (si,k). Since every contiguous k-mer (si,k) except the sequence of k consecutive a's occurs in S, Proposition 1b holds and the proof is completed for the case q = 4. The proof for q = 2 is nearly identical. Now, however, our matrix A will have dimension 2k × 2k. Note that here, the kernel for the matrix analogous to 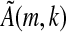 will contain 2j elements for some j. ▪
will contain 2j elements for some j. ▪
Thus, the spaced seeds that are missed correspond exactly to gapped patterns of a's occurring within the LFSR and, for any spaced seed, the fraction of elements that are represented will be approximately either 2−j when using a polynomial over GF(2), or 4−j when using a polynomial over GF(4). We inspected the coverage of seeds most likely to correspond to known motifs for LFSRs over GF(2) and GF(4), in order to see if some polynomials empirically provided better coverage than others. Here, it is known that the number of primitive polynomials of degree k over a field with q elements is given by the formula
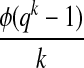 |
where ϕ is Euler's totient function (Terras, 1999) that returns the number of integers relatively prime to the input value (Golomb, 1967; Stewart, 1989); also it is easily seen that the number of spaced seeds of width up to m and order k is given by the formula
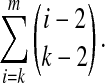 |
Because we could not see how to determine Ak,S (and thus the set of covered seeds) for a given LFSR other than by explicit computation, we focused our analysis on the 7776 polynomials over GF(2) of order 18 and the 15,552 polynomials over GF(4) of order 9. For each of the de Bruijn sequences generated by these polynomials, we inspected whether each of the 44 seeds of widths 9 ≤ m ≤ 11 and order k = 9 was covered. For a given LFSR L, let 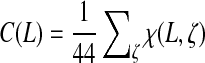 (i.e., the average coverage), where the summation is over all of the spaced seeds ζ with widths between 9 and 11. We were pleased to observe that, by a judicious choice of LFSR, it is possible to completely cover over ∼86% (38/44) of these seeds when considering polynomials over GF(4) and ∼82% (36/44) of these polynomials over GF(2). Also, the mean coverage of polynomials over GF(4) was ∼74 ± 12%, significantly higher than average coverage of ∼44 ± 12% for polynomials over GF(2).
(i.e., the average coverage), where the summation is over all of the spaced seeds ζ with widths between 9 and 11. We were pleased to observe that, by a judicious choice of LFSR, it is possible to completely cover over ∼86% (38/44) of these seeds when considering polynomials over GF(4) and ∼82% (36/44) of these polynomials over GF(2). Also, the mean coverage of polynomials over GF(4) was ∼74 ± 12%, significantly higher than average coverage of ∼44 ± 12% for polynomials over GF(2).
2.3. Sampling k-mers larger than the order of the de Bruijn sequence
In this section, we demonstrate that the representation of spaced seeds is connected to the uniform sampling of words longer than the order of the shift register. As stated previously, the fraction of m-mers represented in an order k de Bruijn sequence is 4k–m (where m ≥ k). In this section, we demonstrate that if the sequence covers all spaced seeds of width ≤m and order k, then the sampled m-mers are well-spaced and regularly sampled (this will be made precise momentarily), facilitating interpolation to m-mers not represented on the array. Thus, a suitable selection of de Bruijn sequence to cover spaced seeds is valuable to determining TFBSs whose width is greater than the order of the generating de Bruijn sequence.
Let d be the Hamming metric on words of length k over the DNA alphabet (Terras, 1999) (i.e., the metric that counts the number of mismatches between pairs of words). For a de Bruijn sequence of order k, let m be an integer such that m > k. We say that the sampling of m-mers is m,k-spaced if for each word w of width m occurring in the de Bruijn sequence, there does not exist a distinct word w′ in the de Bruijn sequence such that d(w, w′) ≤ m − k. Also, we say that the sampling of m-mers is m,k-equidistant if (1) for any choice of k − 1 positions in w there exists a w′ occurring in the de Bruijn sequence that agrees with w at these k − 1 positions and such that d(w, w′) = m − k + 1, and (2) the number of words w′ appearing in the de Bruijn sequence such that d(w, w′) = m − k + 1 is constant over the choice of w.
Intuitively, m-mers are regularly sampled if they are m,k-spaced and m,k-equidistant. This is cartooned by the graphs in Figure 4, where nodes represent the 4m possible m-mers, and the black nodes represent the 4k m-mers that are represented within a given de Bruijn sequence of order k. In this graph, two m-mers are adjacent in the graph if they differ at only one position. A randomly chosen de Bruijn sequence will sample a random collection of m-mers (Fig. 4a), yet an auspiciously chosen de Bruijn sequence (i.e., m,k-spaced and m,k-equidistant) will regularly sample m-mers (Fig. 4b).
FIG. 4.

Cartoon depicting all m-mers (gray vertices) and m-mers samples by an order k < m de Bruijn sequence (black vertices). Vertices are connected by an edge if they are 1 mismatch away. (a) de Bruijn sequence that samples m-mers randomly. (b) de Bruijn sequence that samples m-mers regularly.
One can then prove the following two propositions regarding m,k-spacing and m,k-equidistance. We note that they apply to general de Bruijn sequences, and not only those de Bruijn sequences generated by an LFSR.
Proposition 2
The sampling of m-mers is m,k-spaced in an order k de Bruijn sequence if and only if all spaced seeds of width m′ ≤ m and order k are covered.
Proof
Assume that the de Bruijn sequence covers all spaced seeds of width m′ and order k. Assume (for contradiction) that there are words w and w′ such that d(w, w′) ≤ m − k; then w and w′ will agree at at least k letters. Consider the spaced seed that contains 1's at the positions where w and w′ agree, and let m′ be the distance between the first and last 1 in this spaced seed (note that m′ may be less than m if w and w′ do not agree at the first or last positions). Then w and w′ will map to the same element of this spaced seed, and so the seed cannot be covered by the pigeonhole principle, giving a contradiction. Conversely, assume that a given de Bruijn sequence is m,k-spaced. Let ζ be a spaced seed of width m′ ≤ m and order k. Every element of ζ that appears in the de Bruijn sequence must occur only once. To see this, assume (for contradiction) that there is some element of ζ that occurs more than once. Then there are m-mers w and w′ appearing in the de Bruijn sequence that agree at the k informative positions of the spaced seed. Then 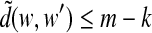 , in violation of our assumption that the sampling of m-mers is m,k-spaced. Thus, ζ must be covered since the number of its elements that occur in the de Bruijn sequence is 4k, all of which are distinct. ▪
, in violation of our assumption that the sampling of m-mers is m,k-spaced. Thus, ζ must be covered since the number of its elements that occur in the de Bruijn sequence is 4k, all of which are distinct. ▪
Proposition 3
If the sampling of m-mers is m′,k-spaced for all k ≤ m′ ≤ m, it is m,k-equidistant.
Proof
For any m′ ≤ m, we know that all spaced seeds of width m′ and order k are covered, by Proposition 2. Let w be an m-mer and pick any k − 1 informative positions in w. Since all spaced seeds of width m′ ≤ m and order k are covered, there will be exactly three distinct m-mers w′ such that w′ ≠ w and that occur in the de Bruijn sequence and agree with w at these k − 1 informative positions (call this set W). The elements of W will all be at a distance of 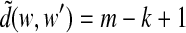 (so condition 1 is satisfied). Also, take a different choice of k − 1 informative positions in w, and consider the set of three words W′ agreeing with w at these k − 1 informative positions. W and W′ must be disjoint, since if there is a word in common between them, then it would agree with w at at least k informative positions, and then the de Bruijn sequence could not cover all spaced seeds of width m′ ≤ m and order k. This implies that every element has a constant number of m-mers at a distance of m − k + 1, and so Condition 2 holds. ▪
(so condition 1 is satisfied). Also, take a different choice of k − 1 informative positions in w, and consider the set of three words W′ agreeing with w at these k − 1 informative positions. W and W′ must be disjoint, since if there is a word in common between them, then it would agree with w at at least k informative positions, and then the de Bruijn sequence could not cover all spaced seeds of width m′ ≤ m and order k. This implies that every element has a constant number of m-mers at a distance of m − k + 1, and so Condition 2 holds. ▪
Finally, for the special case of m = k + 1, one can state the following proposition giving analytic conditions relating m,k-spacing and m,k-equidistance to the choice of polynomial used for the LFSR:
Proposition 4
(a) A de Bruijn sequence of order k generated by a LFSR over GF(4) is k + 1,k-spaced and k + 1,k-equidistant if and only if none of the coefficients θi of the generating polynomial 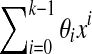 are equal to 0. (b) A de Bruijn sequence of order k generated by a LFSR over GF(2) is k + 1,k-spaced and k + 1,k-equidistant if and only if it does not contain three consecutive coefficients
are equal to 0. (b) A de Bruijn sequence of order k generated by a LFSR over GF(2) is k + 1,k-spaced and k + 1,k-equidistant if and only if it does not contain three consecutive coefficients 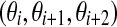 for even i such that
for even i such that 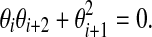 .
.
Proof
Assume the case where the de Bruijn sequence is generated by the polynomial 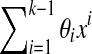 over GF(4). By Propositions 2 and 3 it is sufficient to prove that all seeds of width k + 1 and order k are covered if and only if all θi are non-zero. Coverage of order k width k + 1 seeds is equivalent to asserting that for any (k + 1)-mer
over GF(4). By Propositions 2 and 3 it is sufficient to prove that all seeds of width k + 1 and order k are covered if and only if all θi are non-zero. Coverage of order k width k + 1 seeds is equivalent to asserting that for any (k + 1)-mer 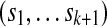 and any 1 ≤ i ≤ k, there exists a value
and any 1 ≤ i ≤ k, there exists a value 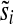 such that
such that 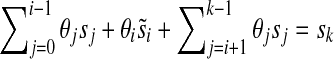 . Clearly, this can occur if and only if for all i θi ≠ 0. The proof for polynomials over GF(2) is nearly identical, but involves solving two such equations simultaneously. ▪
. Clearly, this can occur if and only if for all i θi ≠ 0. The proof for polynomials over GF(2) is nearly identical, but involves solving two such equations simultaneously. ▪
2.4. Complementary de Bruijn sequences and replicate experiments
An additional advantage of our design is that, for any given value of k and desired set of represented gapped k-mers, if one acceptable de Bruijn sequence exists, there will in general be several that could be used (this is easily seen by, for example, permuting the letters or taking the reversal of the de Bruijn sequence). In Berger et al. (2006), we exploited this fact by doing replicate experiments on distinct de Bruijn sequences, both of which were 11,10-spaced and 11,10-equidistant (i.e., they covered all 10-mers containing a single gapped position). There, we observed that performing replicate experiments on distinct de Bruijn sequences, rather than the same de Bruijn sequence, improved our ability to correctly quantify the binding preferences of the well-characterized TF Zif268. We anticipate that this approach of performing replicate experiments on distinct de Bruijn sequences will be a valuable means for improving PBM experiments. In this section, we inspect some formal aspects of this experimental strategy.
The following proposition implies that all pairs of order k de Bruijn sequences generated by LFSRs will share a constant number of (k + 1)-mers.
Proposition 5
Let S and S′ be two distinct de Bruijn sequences of order k, both generated by an LFSR over either GF(2) or GF(4). Then exactly 4k−1 (k + 1)-mers will be commonly represented on both S and S′.
Proof
Assume that S and S′ are generated by the polynomials 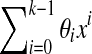 and
and 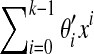 , respectively, over GF(4). Then S and S′ will share a (k + 1)-mer
, respectively, over GF(4). Then S and S′ will share a (k + 1)-mer 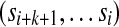 if and only if
if and only if 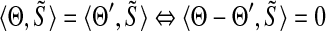 where
where 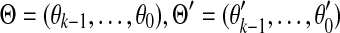 and
and 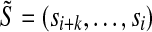 . Since the null space of a linear form must always be of dimension k − 1, there will be exactly 4k−1 values that satisfy this equation. The proof for GF(2) is nearly identical, but involves finding the null space for two linear forms simultaneously. ▪
. Since the null space of a linear form must always be of dimension k − 1, there will be exactly 4k−1 values that satisfy this equation. The proof for GF(2) is nearly identical, but involves finding the null space for two linear forms simultaneously. ▪
Thus, it is not in general possible to optimize the coverage of words longer than the order of the de Bruijn sequences in performing replicate experiments, some (k + 1)-mers will appear in both sequences. Note that Proposition 5 also answers a natural question regarding the selection of the optimal order (k) to use for a given set of array dimensions (either on a single array or multiple arrays). It is not immediately clear whether it is better to have four different de Bruijn sequences of order 4k−1 or one de Bruijn sequence of order 4k, as each requires an equal number of spots of the same length. Proposition 5 implies that a de Bruijn sequence of order 4k is preferable, as de Bruijn sequences of order 4k−1 will have overlap with respect to the k-mers that they represent.
Finally, we note that, although it does not seem that complementary order k primitive polynomials can be utilized in order to maximize the coverage of m-mers, m > k, we have observed that suitable sets of complementary polynomials can be selected for the coverage of gapped k-mers. Here, we have found by empirical inspection that if one polynomial misses a given spaced seed, then another polynomial can be identified that covers it. Thus, this parameter can be optimized.
2.5. Open questions
We see (at least) two broad areas in which further mathematical/algorithmic efforts could lead to improvements in array design. First, assuming the use of LFSRs for generating de Bruijn sequences, there is need for an improved mathematical theory relating the coverage of spaced seeds to the generating polynomial. In this work, we have presented an explicit formula for determining whether a given polynomial represents all k-mers with a single gapped position (i.e., k + 1,k-spaced and k + 1,k-equidistant de Bruijn sequences), but we have not yet been able to extend this theory to k-mers with multiple gaps.
Second, only a small fraction of de Bruijn sequences correspond to sequences generated by an LFSR, and the utility of such non-LFSR-generated de Bruijn sequences remains largely unexplored. In current applications we have utilized LFSRs as they provably satisfy pseudo-randomness properties that are advantageous, since they guarantee that there are no confounding correlations in the experimental data that are an artifact of the methods utilized to generate the de Bruijn sequences. Additionally, LFSRs allow for the complete coverage of certain gapped k-mer patterns, which we have observed to be useful for determination of the binding specificities of TFs. However, it may well be the case that there are additional families of de Bruijn sequences that cover even more gapped k-mers while still resembling random sequence. Similarly, there may be additional desirable properties of de Bruijn sequences that we have not yet considered, and for which LFSRs might not be optimal.
3. Conclusion
We have presented the combinatorial design of a protein binding microarray. Importantly, this design has been optimized in several key aspects: (1) All k-mers are represented in a minimum amount of sequence, permitting a maximum number of binding site variants to be represented in a cost-efficient manner. This allows the binding specificities of TFs to be assayed with word-by-word resolution. (2) The unbiased nature of the construction provides a design that can be used for TFs from any species and/or structural class, making it a universal platform. (3) Our design is flexible, allowing ever greater binding site coverage as array technology improves and feature density increases. For example, all 11-mers can already be represented on Agilent arrays (Berger et al., 2006), and all 12-mers on NimbleGen arrays (Singh-Gasson et al., 1999); moreover, this number is expected to grow quickly. Similarly, our design allows replicate experiments to be performed with distinct de Bruijn sequences, resulting in reduced experimental noise and greater coverage of sequence space. (4) We have utilized de Bruijn sequences generated by LFSRs which provably resemble random sequence. This guarantees that any statistical trends observed in data generated by a PBM experiment are not an artifact of how the sequences were constructed. (5) Our design not only maximizes the coverage of contiguous k-mers, but also gapped k-mers. This simultaneously allows an interrogation of the binding specificities of TFs with gapped motifs and also improves the ability of the design to approximate the binding specificities of TFs whose width is greater than the order of the de Bruijn sequence.
Indeed, our group has already implemented this design and applied it to determine the binding specificities of five TFs from different organisms and structural classes with an unprecedented level of resolution (Berger et al., 2006). There, we demonstrated that this platform could be used to assay the binding preferences of individual binding site variants, allowing us to identify at least one case of positional interdependence in a binding site motif. Similarly, we were able to approximate a binding site motif that was 12 bases in length using a de Bruijn sequence of order 10, attesting to the advantages of a careful and thorough coverage of gapped k-mers (point 5 above). Our group is now using this technology to determine the binding specificities of many TFs from a range of organisms, providing a much needed data type for the biological community. Thus, this microarray design provides a powerful, general and robust platform, and its implementation provides an experimental tool that will allow effective determination of TF binding site specificities both now and in the future.
Acknowledgments
We thank Savina Jaeger for critical reading of the manuscript. This work was funded in part by NIH/NHGRI (grant R01 HG003985 to M.L.B.). M.F.B. was supported in part by a National Science Foundation Graduate Research Fellowship. A.A.P. was supported in part by a National Defense Science and Engineering Graduate Fellowship, a National Science Foundation Graduate Research Fellowship, and an Athinoula Martinos Fellowship.
Disclosure Statement
A.A.P. and M.L.B. are co-inventors on a pending patent on the sequence design strategy described in this paper.
References
- Ben-Dor A. Karp R. Schwikowski B., et al. Universal DNA tag systems: a combinatorial design scheme. J. Comput. Biol. 2000;7:503–519. doi: 10.1089/106652700750050916. [DOI] [PubMed] [Google Scholar]
- Benos P.V. Lapedes A.S. Stormo G.D. Is there a code for protein-DNA recognition? Probab(ilistical)ly. Bioessays. 2002;24:466–475. doi: 10.1002/bies.10073. [DOI] [PubMed] [Google Scholar]
- Berger M.F. Philippakis A.A. Qureshi A.M., et al. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat. Biotechnol. 2006;24:1429–1435. doi: 10.1038/nbt1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulyk M.L. Computational prediction of transcription-factor binding site locations. Genome Biol. 2003;5:201. doi: 10.1186/gb-2003-5-1-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulyk M.L. Gentalen E. Lockhart D.J., et al. Quantifying DNA-protein interactions by double-stranded DNA arrays. Nat. Biotechnol. 1999;17:573–577. doi: 10.1038/9878. [DOI] [PubMed] [Google Scholar]
- Bulyk M.L. Huang X. Choo Y., et al. Exploring the DNA-binding specificities of zinc fingers with DNA microarrays. Proc. Natl. Acad. Sci. USA. 2001;98:7158–7163. doi: 10.1073/pnas.111163698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das P.M. Ramachandran K. vanWert J., et al. Chromatin immunoprecipitation assay. Biotechniques. 2004;37:961–969. doi: 10.2144/04376RV01. [DOI] [PubMed] [Google Scholar]
- De Bruijn N.G. A combinatorial problem. Proc. Kon. Ned. Akad. Wetensch. 1946;49:758–764. [Google Scholar]
- Golomb S. Shift Register Sequences. Aegean Park Press; Laguna Hills, CA: 1967. [Google Scholar]
- Gross J.L. Yellen J. Handbook of Graph Theory. CRC Press; New York: 2004. [Google Scholar]
- Joyner D. Kreminski R. Turisco J. Applied Abstract Algebra. Johns Hopkins University Press; Baltimore: 2004. [Google Scholar]
- Linnell J. Mott R. Field S., et al. Quantitative high-throughput analysis of transcription factor binding specificities. Nucleic Acids Res. 2004;32:e44. doi: 10.1093/nar/gnh042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintseris J. Eisen M.B. Design of a combinatorial DNA microarray for protein-DNA interaction studies. BMC Bioinform. 2006;7:429. doi: 10.1186/1471-2105-7-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S. Berger M.F. Jona G., et al. Rapid analysis of the DNA-binding specificities of transcription factors with DNA microarrays. Nat. Genet. 2004;36:1331–1339. doi: 10.1038/ng1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliphant A.R. Brandl C.J. Struhl K. Defining the sequence specificity of DNA-binding proteins by selecting binding sites from random-sequence oligonucleotides: analysis of yeast GCN4 protein. Mol. Cell Biol. 1989;9:2944–2949. doi: 10.1128/mcb.9.7.2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevzner P.A. Tang H. Tesler G. De novo repeat classification and fragment assembly. Genome Res. 2004;14:1786–1796. doi: 10.1101/gr.2395204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh-Gasson S. Green R.D. Yue Y., et al. Maskless fabrication of light-directed oligonucleotide microarrays using a digital micromirror array. Nat. Biotechnol. 1999;17:974–978. doi: 10.1038/13664. [DOI] [PubMed] [Google Scholar]
- Stewart I. Galois Theory. Chapman & Hall; London: 1989. [Google Scholar]
- Stormo G.D. DNA binding sites: representation and discovery. Bioinformatics. 2000;16:16–23. doi: 10.1093/bioinformatics/16.1.16. [DOI] [PubMed] [Google Scholar]
- Terras A. Fourier Analysis on Finite Groups and Applications. Cambridge University Press; Cambridge, UK: 1999. [Google Scholar]
- Warren C.L. Kratochvil N.C. Hauschild K.E., et al. Defining the sequence-recognition profile of DNA-binding molecules. Proc. Natl. Acad. Sci. USA. 2006;103:867–872. doi: 10.1073/pnas.0509843102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyrick J.J. Young R.A. Deciphering gene expression regulatory networks. Curr. Opin. Genet. Dev. 2002;12:130–136. doi: 10.1016/s0959-437x(02)00277-0. [DOI] [PubMed] [Google Scholar]
- Zhang Y. Waterman M.S. An Eulerian path approach to local multiple alignment for DNA sequences. Proc. Natl. Acad. Sci. USA. 2005;102:1285–1290. doi: 10.1073/pnas.0409240102. [DOI] [PMC free article] [PubMed] [Google Scholar]