

Abstract
Background
Immunoreactive signal for the desmosomal protein plakoglobin (γ-catenin) is reduced at cardiac intercalated disks in patients with arrhythmogenic right ventricular cardiomyopathy (ARVC), a highly arrhythmogenic condition caused by mutations in genes encoding desmosomal proteins. Previously, we observed a “false positive” case in which plakoglobin signal was reduced in a patient initially thought to have ARVC but who actually had cardiac sarcoidosis. Sarcoidosis can masquerade clinically as ARVC, but has not previously been associated with altered desmosomal proteins.
Methods and Results
We observed marked reduction in immunoreactive signal for plakoglobin at cardiac myocyte junctions in patients with sarcoidosis and giant cell myocarditis, both highly arrhythmogenic forms of myocarditis associated with granulomatous inflammation. In contrast, plakoglobin signal was not depressed in lymphocytic (non-granulomatous) myocarditis. To determine whether cytokines might promote dislocation of plakoglobin from desmosomes, we incubated cultures of neonatal rat ventricular myocytes with selected inflammatory mediators. Brief exposure to low concentrations of IL-17, TNFα and IL-6, cytokines implicated in granulomatous myocarditis, caused translocation of plakoglobin from cell-cell junctions to intracellular sites, whereas other potent cytokines implicated in non-granulomatous myocarditis had no effect, even at much high concentrations. We also observed myocardial expression of IL-17 and TNFα, and elevated serum levels of inflammatory mediators including IL-6R, IL-8, MCP1 and MIP1β in ARVC patients (all p<0.0001 compared with controls).
Conclusions
These results suggest novel disease mechanisms involving desmosomal proteins in granulomatous myocarditis and implicate cytokines, perhaps derived in part from the myocardium, in disruption of desmosomal proteins and arrhythmogenesis in ARVC.
Keywords: plakoglobin, desmosome, sarcoidosis, giant cell myocarditis, cytokines
Introduction
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a familial disease caused by mutations in proteins that form desmosomes,1 intercellular adhesion plaques located within intercalated disks connecting cardiac myocytes. ARVC is a highly arrhythmogenic disease characterized by the early appearance of ventricular arrhythmias that often emerge prior to detectable impairment of ventricular function.1 Previously, we found that immunoreactive signal for the desmosomal protein plakoglobin (γ-catenin) is consistently reduced at myocardial intercalated disks in patients with ARVC.2 We also observed a single “false positive” case in which plakoglobin signal was virtually absent from myocardial intercalated disks in a patient who was initially suspected of having ARVC but in fact had cardiac sarcoidosis, a highly destructive form of granulomatous myocarditis that has also been associated with ventricular arrhythmias.3 Indeed, the clinical presentation of cardiac sarcoidosis may mimic ARVC.4,5 Giant cell myocarditis, an even more malignant form of granulomatous myocarditis, is also associated with ventricular arrhythmias.6 In the present study, we evaluated myocardial plakoglobin staining in patients with sarcoidosis and giant cell myocarditis and found that loss of plakoglobin signal from intercalated disks is a consistent feature. In contrast, loss of plakoglobin signal did not occur in lymphocytic (non-granulomatous) myocarditis even in the presence of extensive inflammatory infiltrates and widespread myocardial injury. We hypothesized that cytokines may play a role, and conducted in vitro studies showing that exposure of cardiac myocytes to selected cytokines involved in granulomatous inflammation7–9 leads to rapid loss of plakoglobin signal from cell-cell junctions, whereas other powerful cytokines implicated in non-granulomatous myocarditis10,11 do not affect plakoglobin distribution. To explore possible links between granulomatous myocarditis and ARVC, we measured serum levels of inflammatory cytokines in patients with ARVC. We observed high levels of selected inflammatory cytokines in ARVC patients and also observed myocardial expression of cyokines. These results suggest novel pathophysiologic links between granulomatous myocarditis, redistribution of junctional plakoglobin and arrhythmogenesis. They may also relate to the pathophysiology of ARVC and lead to new insights into understanding the clinical similarities between these two seemingly disparate entities.
Methods
Immunofluorescence analysis of junctional proteins in human myocardium
Formalin-fixed, paraffin-embedded myocardial samples obtained at autopsy or by endomyocardial biopsy from patients with ARVC, sarcoidosis, giant cell myocarditis or lymphocytic myocarditis were processed conventionally for light microscopy. Paraffin sections mounted on plain glass microscope slides were immunostained using methods validated previously.12 Primary antibodies included monoclonal mouse anti-plakoglobin (Sigma, UK), monoclonal mouse anti-N-cadherin (Sigma, UK), polyclonal rabbit anti-desmoplakin (Serotec, USA), monoclonal mouse anti-plakophilin2 (Fitzgerald, USA) and polyclonal rabbit anti-Cx43 (Sigma, UK). Immunostained preparations were analysed by laser scanning confocal microscopy (Sarastro Model 2000, Molecular Dynamics) as previously described.12 Myocardial specimens obtained at autopsy from 3 young adult individuals who had no clinical history or pathological evidence of heart disease were used as controls. A determination that signal for a given junctional protein was depressed at cell-cell junctions required substantial loss such that signal was either barely visible or absent compared to controls. The entire slide was examined and a decision about whether signal was diminished was based on the area showing the brightest signal. In myocardial specimens containing areas of inflammation, fibrosis and/or cardiac myocyte necrosis, determinations about signal intensity were made only in the most normal appearing areas devoid of inflammatory infiltrate and degenerative changes in cardiac myocytes. A specimen was eliminated from further study if such histologically normal appearing areas could not be found.
Immunoperoxidase analysis of cytokines in human myocardium
Slide-mounted paraffin sections were heated to 60° for 80 min, cooled to room temperature, deparaffinized and rehydrated. Antigen retrieval was achieved by heating the samples in citrate buffer (pH 6.1) to boiling, followed by cooling to room temperature. Immunohistochemical staining was performed using conventional methods (Universal DAKO EnVision System, Peroxidase). After endogenous peroxidase activity had been blocked, the primary antibody was applied followed by incubation with horseradish peroxidase-labeled polymer. The reaction was completed with an enzyme/substrate system with diaminobenzidine as the chromogen-substrate. Tissue was then counterstained with Mayer’s hematoxylin followed by blueing in phosphate-buffered saline at pH 7.4. Primary antibodies included rabbit polyclonal anti-human antibodies against IL-17, TNFα, IL-4 and IL-6 (Peprotech, USA). Sections of a human breast carcinoma, known to express high levels of the cytokines under investigation,13–15 were used as positive controls. In each experiment, control and patient samples were batched to ensure identical staining and signal-generating conditions. Determinations of positive staining were based on comparisons with positive (breast cancer) and negative (normal myocardium) controls. Only areas of histologically unremarkable myocardium devoid of inflammation, fibrosis and cardiac myocyte degeneration were used to determine whether staining was positive.
Primary cultures of neonatal rat ventricular myocytes
Primary cardiac myocyte cultures were prepared from disaggregated ventricles of 1-day old Wistar rat pups (Charles River) as previously described.16 Cell suspensions were pre-plated to reduce fibroblast content, placed in collagen-coated chamber slides at a density of 2.4 × 105cells/cm2, and grown for 4 days at 37°C in M199 (GIBCO) (supplemented with penicillin, 20U/ml; streptomycin, 20μg/ml; and 10% neonatal calf serum) in a humidified atmosphere containing 1% CO2. Epinephrine (0.01μmol/ml) was added to the medium during the first 24 hr of culture. All protocols were approved by the Harvard Medical Area Standing Committee on Animals.
Immunofluorescence and immunoblotting analysis of plakoglobin expression in cultured myocyte incubated with cytokines
Myocyte cultures were placed in serum-free M199 medium and incubated with recombinant rat IL-17, IL-12, IL-9, IL-4, IL-6, interferon-γ (INFγ) or TNFα (R&D Systems) (12.5, 50 or 400ng/ml) for 4 hr. Cultures in complete or serum-free medium without added cytokines served as controls. At the conclusion of each experimental protocol the cultures were rinsed in serum-free medium, fixed in 4% paraformaldehyde for 5 min, immunostained with a mouse monoclonal anti-plakoglobin antibody (Sigma, UK) and analyzed by laser scanning confocal microscopy (Sarastro Model 2000, Molecular Dynamics) as previously described.17 In other experiments, cultured myocytes exposed to cytokines were washed and scraped from cultures dishes in preparation for immunoblotting as previously described18 Aliquots containing10 μg of total protein were loaded on gels, electro-phoresed, transferred to nitrocellulose membranes and incubated first with a mouse monoclonal anti-plakoglobin antibody (Sigma, UK) and then with a horseradish peroxidase-linked anti-mouse IgG. The blots were developed using ECL reagents (Amersham, UK). The membranes were subsequently stripped, washed and re-probed with a mouse monoclonal anti-glyceraldehyde-3-phosphate dehydrogenase antibody (Invitrogen) for use as loading controls.
Microimmunoassay cytokine analysis
Cytokine levels were measured in serum samples from 18 patients who fulfilled Task Force criteria for ARVC.19 Disease expression and severity varied in these individuals but at the time the samples were obtained, none of them were in the hospital and all were being followed in outpatient clinics. Cytokines were also measured in serum samples from 28 healthy volunteers who served as controls. Cytokines were measured in a highly sensitive and accurate multiplex assay that uses matched antibody pairs to detect each specific cytokine.20 Briefly, antibodies were spotted on a 96-well plate and a cocktail of biotin-conjugated secondary antibodies was used for detection. The final step of the assay involved application of streptavidin coupled to a near infrared dye for detection and quantification. In effect, this assay is similar to performing multiple ELISAs within a single well. The physical location within the well of the individual dots is used to determine which cytokine is being detected. In separate wells of the plate, standard curves are run for each of the cytokines to be detected and quantified.20 The measurements in ARVC patients were performed in a single assay in which a specific limit of detection was established using the standard curve for each cytokine. When the level for a specific cytokine in a patient sample was below the limit of detection, a value of one half of the detection limit was assigned for subsequent statistical analysis. Cytokines levels in control samples were measured in several independent assays, each including determination of a lower limit of detection for each cytokine. Within these assays, the limits of detection varied only modestly from one assay to another. For statistical purposes, any control value that fell below the average limit of detection for all control assays was assigned a value of one half of the average value.
Statistical analysis
Cytokines levels in serum in ARVC patients and controls were compared using the Wilcoxon Rank Sum test with the Bonferroni adjustment for multiple comparisons. The cut-off for statistical significance was 0.05/17 or p<0.0029.
Results
Plakoglobin signal is diminished at myocardial intercalated disks in sarcoidosis
Supplemental Table 1 shows clinical features in 23 patients with sarcoidosis analyzed by immunohistochemistry and confocal microscopy. We first studied myocardial tissue from 4 patients with sarcoidosis (patients 1–4 in supplemental Table 1) provided by experienced clinicians (DJW, FIM) who had originally diagnosed ARVC based on imaging and electrocardiographic criteria but then changed the diagnosis to sarcoidosis because of definitive pathological evidence of non-necrotizing granulomas and giant cells associated with myocardial injury. In each case, we observed marked reduction or absence of plakoglobin signal at cell-cell junctions (a representative case is shown in Figure 1). To determine whether loss of plakoglobin signal occurred generally in sarcoidosis, we analyzed 19 additional cases of pathologically documented cardiac sarcoidosis. As shown in a representative example in Figure 1, reduced or absent junctional expression of plakoglobin was observed in 17 of 19 cases. Only patients 9 and 18 in supplemental Table 1 showed apparently normal plakoglobin signal. The distribution and severity of granulomatous inflammation varied in these cases from focal isolated granulomas with minimal myocardial damage and fibrosis to broad areas of myocardial destruction and extensive granulomas (Figure 1). In analyzing plakoglobin signal intensity and distribution, however, we focused only on histologically unremarkable myocardium distant from areas showing inflammatory and degenerative changes. In each case, reduced signal occurred diffusely in otherwise unaffected myocardium (Figure 1). These findings indicate that loss of plakoglobin signal from cell-cell junctions is a consistent finding in sarcoidosis.
Figure 1.

A. Microscopic appearance of the myocardium from a patient with cardiac sarcoidosis showing fibrosis and multinucleated giant cells within granulomatous lesions (Masson’s trichrome stain, x20). B. Normal appearing myocardium from the same patient showing no apparent inflammatory or degenerative changes (Masson’s trichrome stain, x20). C. Representative confocal immunofluorescence images of control myocardium and myocardium from a patient with cardiac sarcoidosis clinically masquerading as ARVC and another patient with pathologically documented cardiac sarcoidosis. Specific immunoreactive signal for plakoglobin was significantly depressed in both cases compared to controls as was signal for the major gap junction protein, Cx43. Expression of other desmosomal proteins including desmoplakin and plakophilin-2 varied but signal for the non-desmosomal adhesion protein N-cadherin was always present and indistinguishable from controls.
To determine whether other desmosomal proteins were similarly affected, we immunostained 8 randomly selected cases for desmoplakin and plakophilin-2 (identified in supplemental Table 1). Signals were clearly depressed in 6 and indistinguishable from controls in the remaining 2 cases (Figure 1). Similar variability in the amount of junctional signal for desmoplakin and plakophilin-2 has been reported in ARVC.2
Diminished junctional signal for Cx43, the major ventricular gap junction protein, has also been reported in ARVC2 leading to speculation that gap junction remodeling may play a role in conduction abnormalities and arrhythmogenesis. Accordingly, we immunostained all 23 sarcoidosis cases for Cx43 and found clearly depressed or absent signal in 11 including 2 of the 4 cases initially mistaken for ARVC, and in 3 of the 5 cases in which patients initially presented with arrhythmias (Figure 1 and supplemental Table 1).
Plakoglobin signal is also reduced in giant cell myocarditis but not in lymphocytic myocarditis
We next sought to determine whether plakoglobin signal was altered in in giant cell myocarditis which, like sarcoidosis, is a highly arrhythmogenic form of inflammatory heart disease characterized by the presence of granulomas, multi-nucleated giant cells and extensive myocardial necrosis.6,21 Accordingly, we examined the distribution of selected intercalated disk proteins in myocardial samples from 16 patients with documented giant cell myocarditis. Clinical information about these patients is included in supplemental Table 2. Junctional plakoglobin signal was absent or clearly diminished in 14 of the 16 samples (a representative case is shown in Figure 2). Only patients 9 and 13 in supplemental Table 2 showed apparently normal plakoglobin signal. Although most of these cases showed severe myocardial inflammation and fibrosis, we were able to examine histologically normal appearing tissue and observed reduced plakoglobin signal throughout these areas. Immunoreactive signals for desmoplakin and plakophilin2 were depressed in 4 of 7 randomly selected cases of giant cell myocarditis, and Cx43 signal was diminished in 8 of the entire set of 16 cases (representative images are shown in Figure 2 and specific cases are identified in supplemental Table 2). Six patients with giant cell myocarditis presented with significant arrhythmias or sudden cardiac death (patients 6, 8–12 in supplemental Table 2). Cx43 signal was depressed in 3 (patients 6, 11 and 12) but also in 5 patients in whom arrhythmias were apparently not prominent.
Figure 2.

A. Microscopic appearance of the myocardium from a patient with giant cell myocarditis showing severe destruction of cardiac myocytes (hematoxylin/eosin stain, x20). B. Normal appearing myocardium from the same patient showing no apparent inflammatory or degenerative changes (hematoxylin/eosin stain, x20). C. Representative confocal immunofluorescence images of control myocardium and myocardium from two patients with giant cell myocarditis (GCM). Specific immunoreactive signal for plakoglobin was significantly depressed in both cases compared to controls as was signal for the major gap junction protein, Cx43. Expression of other desmosomal proteins including desmoplakin and plakophilin-2 varied but signal for the non-desmosomal adhesion protein N-cadherin was present in all GCM cases analyzed and indistinguishable from controls.
To determine whether loss of plakoglobin signal occurs in all forms of myocarditis, we next examined 25 cases of lymphocytic myocarditis. Clinical features of these patients are listed in supplemental Table 3. Each of these cases fulfilled accepted pathological criteria22 including mononuclear inflammation (mainly lymphocytes and macrophages) and myocyte necrosis without evidence of giant cells or granulomatous inflammation. PCR documentation of the presence of viral genomes was available in 10 cases (4 cases of enterovirus and 1 each of parvovirus B19, Coxsackie virus B, adenovirus, Herpes simplex, Epstein-Barr and influenza A, identified in supplemental Table 3). PCR was either negative or had not been performed in the remaining 15 cases. The severity of inflammation and myocyte necrosis varied widely and some cases showed extensive damage. However, plakoglobin signal was equivalent to control levels in 22 of 25 cases (Figure 3 and supplemental Table 3). This was true both in cases of viral myocarditis and in other cases of lymphocytic (non-granulomatous, non-giant cell) myocarditis of unknown etiology. Signals for desmoplakin and plakophilin2 were normal in 6 of 8 randomly selected cases of lymphocytic myocarditis, and signal for Cx43 was depressed in 8 of the entire set of 25 cases (representative images are shown in Figure 3 and specific patients are identified in supplemental Table 3). Thus, although gap junction remodeling occurs in some cases of myocarditis and desmosomal protein distribution may also be affected, diminished plakoglobin signal does not appear to be a consistent feature in lymphocytic myocarditis.
Figure 3.

A. Microscopic appearance of myocardium from a patient with lymphocytic myocarditis of viral etiology (influenza A) showing inflammatory infiltrates and myocyte necrosis (hematoxylin/eosin stain, x10). B. Normal appearing myocardium from the same patient showing no inflammatory infiltrate or degenerative changes (hematoxylin/eosin stain, x20). C. Representative confocal immunofluorescence images of control myocardium and myocardium from a patient with lymphocytic myocarditis. Specific immunoreactive signals for N-cadherin, plakoglobin, desmoplakin and plakophilin-2 were indistinguishable from controls. Signal for Cx43 at cell-cell junctions appeared diminished compared to controls.
Short-term exposure of cardiac myocytes to selected cytokines causes loss of junctional plakoglobin signal
To determine whether dislocation of plakoglobin from myocardial cell-cell junctions might be mediated by specific cytokines associated with giant cell inflammation, we incubated primary cultures of neonatal rat ventricular myocytes with varying concentrations (12.5, 50 or 400ng/ml) of exogenous cytokines for 4 hr. Cultures were then immunostained for plakoglobin and examined by confocal microscopy. Potent cytokines including IL-9, IL-12, IL-4 and INFγ, which have been implicated in non-giant cell inflammation10,11 had no apparent effect on the distribution of plakoglobin even at the highest concentration of 400ng/ml (Figure 4). In contrast, IL-17 and TNFα, both of which are thought to mediate granulomatous myocarditis and are elevated in circulating blood in patients and animal models of human giant cell myocarditis,7–9 caused a marked loss of plakoglobin signal from myocardial cell-cell junctions even at the lowest concentration of 12.5ng/ml (Figure 4). IL-6, which has been found to be elevated in the blood of patients with myocardial infarction and heart failure, and is associated with poor outcomes,23–25 also caused loss of plakoglobin signal at the lowest concentration (Figure 4). To determine whether loss of plakoglobin immunoreactive signal was due to protein degradation, we repeated experiments in which cells were exposed to cytokines and then performed western blots to measure total plakoglobin content in the cells. As shown in Figure 4, there was no apparent change in plakoglobin levels despite the loss of signal from cell-cell junctions. These results are consistent with intracellular translocation rather than protein degradation to explain the loss of junctional signal.
Figure 4.
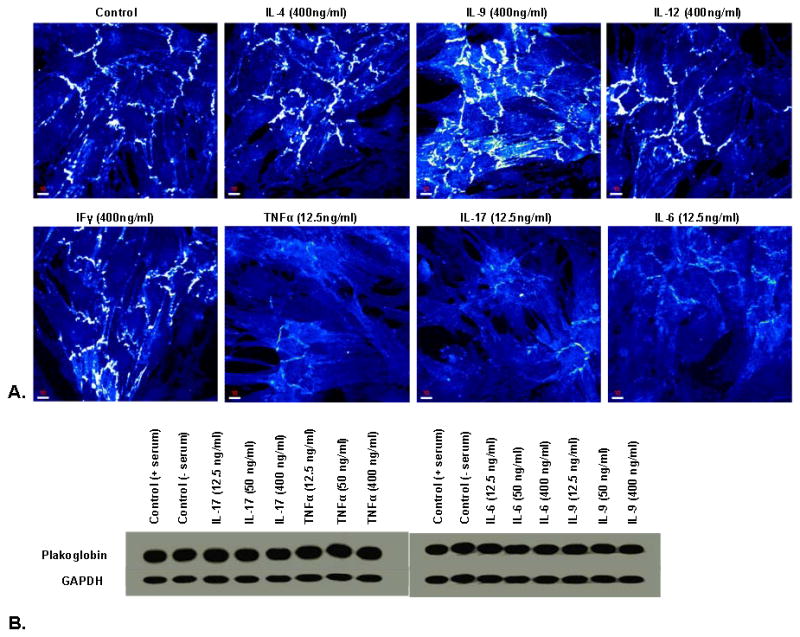
A. Representative confocal immunofluorescence images showing the distribution of junctional plakoglobin in cardiac myocytes incubated with varying concentrations of selected cytokines. IL-17, TNFα and IL-6 caused obvious loss of plakoglobin signal even at the lowest concentration of 12.5ng/ml of medium while other cytokines including IL-4, IL-9, IL-12 and interferonγ (IFγ) had no effect even at a concentration of 400ng/ml medium. B. Representative immunoblots showing no apparent change in total cellular content of plakoglobin in myocytes incubated with varying concentrations of IL-17, TNFα and IL-6. Gluceraldehyde-3-phosphate dehydrogenase (GAPDH) served as a loading control.
Myocardial expression of cytokines in ARVC
Although granulomatous inflammation has not been reported in ARVC, varying amounts of mononuclear inflammation of the myocardium are frequently seen in this condition.26 It is also known that cardiac myocytes can express various cytokines in response to injury or disease.24,27–29 Accordingly, as a first step to investigate whether locally produced cytokines might mediate changes in plakoglobin distribution in ARVC, we used immunohistochemistry to determine if various cytokines were expressed in ARVC myocardium. We used immunoperoxidase to take advantage of the signal amplification in this enzyme-based method to enhance the sensitivity of detection in ARVC patient samples. We analyzed samples from 9 ARVC patients, 5 with documented desmosomal gene mutations (1 in DSG2 and 2 each in PKP2 and DP) and 4 with no mutation identified during genetic screening but whose clinical evaluation and pathological findings were diagnostic of ARVC (see supplemental Table 4). Immunofluorescent signal for plakoglobin at cardiac intercalated disks was virtually absent in all 9 cases (data not shown). We immunostained these samples for IL-17, TNFα and IL-6, all of which caused loss of plakoglobin signal from myocardial cell-cell junctions in vitro. Myocardial samples obtained at autopsy from 3 individuals with no history or pathological evidence of heart disease were used as negative controls. Sections of a human breast carcinoma, known to produce multiples cytokines,13–15 were used as a positive control. Myocardial samples from 2 cases of sarcoidosis and 1 of giant cell myocarditis were also used as positive controls for IL-17 and TNFα expression. We were careful to examine regions in ARVC samples devoid of inflammatory infiltrates and fibrofatty infiltration. Increased signals for IL-17 and TNFα were seen in 9 of 9, and 8 of 9 cases of ARVC, respectively (representative images are shown in Figure 5). However, no apparent IL-6 expression was detected in ARVC myocardium (Figure 5).
Figure 5.

Representative immunoperoxidase staining for IL-17, TNFα and IL-6 expression in myocardial samples from ARVC patients. A human breast carcinoma sample and myocardial samples from patients with sarcoidosis and giant cell myocarditis were used as positive controls. Myocardium obtained at autopsy from an individual with no history or pathological evidence of heart disease was used as a negative controls. The intensity of immunoreactive signal indicated by the brown reaction product was increased in ARVC myocardium compared with control myocardium for IL-17 and TNFα, but not IL-6.
Circulating levels of cytokines are altered in ARVC patients
To further explore potential mechanistic links between inflammation and ARVC, we used a sensitive microimmunoassay20 to measure the levels of various cytokines in the serum of 18 patients who fulfilled Task Force criteria for the diagnosis of ARVC (clinical features are listed in supplemental Table 5). These values were compared to measurements made in 28 healthy controls. As shown in Table 1 and Figure 6, ARVC patients had significantly increased levels of multiple pro-inflammatory cytokines including IL-6 receptor (IL-6R), IL-8, monocyte chemoattractant protein1 (MCP1) and macrophage inflammatory protein1b (MIP1β). Levels of TNFα receptor types 1 and 2 (TNFαR1 and TNFαR2), known to track with adverse outcomes in heart failure,30–32 were also elevated in ARVC patients. In addition, levels of the anti-inflammatory cytokine IL-1 receptor 2 (IL-1R2) were significantly depressed in ARVC patients compared to controls.
Table 1.
Cytokine levels (pg/ml) in the sera of healthy control subjects and patients with ARVC
| Name | Control | ARVC | P-value | ||||
|---|---|---|---|---|---|---|---|
| N | Mean | Median | N | Mean | Median | ||
|
| |||||||
| IL-1R2 | 28 | 3966.91 | 3731.5 | 18 | 1004.91 | 893.8 | <0.0001* |
| IL-4 | 28 | 1592.38 | 610.35 | 18 | 3723.96 | 879.85 | 0.77 |
| IL-6 | 28 | 63.95 | 20.35 | 18 | 817.47 | 101.25 | 0.01 |
| IL-6R | 28 | 2395.43 | 2004.95 | 18 | 7779.34 | 7936.25 | 0.0005* |
| IL-8 | 28 | 33.95 | 21.15 | 18 | 634.67 | 555.85 | <0.0001* |
| IL-10 | 28 | 217.75 | 66.7 | 18 | 416.48 | 8 | 0.04 |
| IL-12p70 | 28 | 150.75 | 14.02 | 18 | 161.72 | 42.7 | 0.94 |
| GROα | 27 | 98.87 | 2.25 | 18 | 337.01 | 18.75 | 0.05 |
| IFNγ | 28 | 1320.07 | 233.39 | 18 | 6644.39 | 40 | 0.44 |
| MCP1 | 28 | 25.92 | 16.55 | 18 | 134.47 | 132.55 | <0.0001* |
| MIP1α | 28 | 41.76 | 14.25 | 18 | 162.31 | 79.4 | 0.06 |
| MIP1β | 28 | 261.82 | 176.95 | 18 | 4865.01 | 3338.85 | <0.0001* |
| TNFαR1 | 28 | 1386.65 | 1119.6 | 18 | 4288.54 | 4064.7 | <0.0001* |
| TNFαR2 | 28 | 2132.84 | 1647.15 | 18 | 54585.22 | 53288.75 | <0.0001* |
| TNFα | 28 | 164.95 | 109.75 | 18 | 258.42 | 53.8 | 0.22 |
| IL-1β | 28 | 60.50 | 25.85 | 18 | 60.1 | 24 | 0.99 |
IL-1R2, interleukin-1 receptor type 2; IL-4, interleukin-4; IL-6, interleukin-6, IL-6R, interleukin-6 receptor; IL-8, interleukin 8; IL-10, interleukin-10; IL-12p70, interleukin-12 (active form); GROα, growth regulated protein-α; IFNγ, interferon-γ; MCP1, monocyte chemoattractant protein-1; MIP1α, macrophage inflammatory protein1α; MIP1β, macrophage inflammatory protein1β; TNFαR1, tumor necrosis factor-α receptor type 1; TNFαR2, tumor necrosis factor-α receptor type 2; TNFα, tumor necrosis factor-α; IL-1β, interleukin-1β.
Significant at p<0.05 with Bonferroni adjustment for multiple comparisons (cut off = 0.05/17= 0.0029)
Figure 6.

Graphs showing the relative proportion of individuals within various concentration ranges for serum cytokines in which there were significant differences between ARVC patients and healthy controls. IL6R, interleukin-6 receptor; MIP1β, macrophage inflammatory protein 1beta; MCP1, monocyte chemoattractant protein-1; IL-8, interleukin-8; TNFαR1, tumor necrosis factor-α receptor type 1; TNFαR2, tumor necrosis factor-α receptor type 2.
Discussion
In this study, we report 2 major new findings: 1) disruption of desmosomal proteins in patients with granulomatous myocarditis, and 2) local myocardial production of selected cytokines and alterations in the balance between circulating pro-inflammatory and anti-inflammatory cytokines in patients with ARVC. Both of these findings implicate new disease mechanisms and establish potential new links between ARVC and selected forms of myocarditis.
To our knowledge, this is the first evidence to implicate disruption of desmosomal proteins in 2 forms of granulomatous myocarditis. We also observed that brief exposure of cardiac myocytes in vitro to exogenous cytokines implicated in granulomatous inflammation can cause redistribution of plakoglobin from junctional to intracellular sites, thus providing a potential mechanistic link. While there has been no previous demonstration of altered desmosomes in granulomatous myocarditis, it has been shown that desmogleins are targeted by auto-antibodies in pemphigus and by an exfoliative toxin from S. aureus in bullous impetigo and staphylococcal scalded skin syndrome.33 It is also known that pro-inflammatory cytokines can disrupt tight junctions in intestinal epithelial cells.34 It is plausible to speculate, therefore, that cytokines produced locally or systemically in granulomatous myocarditis could affect desmosomes in the heart and contribute to cardiac myocyte injury and functional derangements. Because plakoglobin (γ-catenin) can participate in Wnt signaling pathways,35 our observations raise the possibility that altered Wnt signaling might also play a role in granulomatous myocarditis. Extensive additional work will be required to explore these possibilities, but our observations open new avenues of investigation for future studies.
Remodeling of gap junctions appears to be a consistent feature in ARVC.2,36,37 In the present study, we observed loss of immunoreactive signal for Cx43, the major ventricular gap junction protein, in some but not all cases of sarcoidosis and giant cell myocarditis, and there was not a strong correlation between loss of Cx43 signal and clinical expression of arrhythmias in these patients. Gap junction remodeling occurs in many forms of heart disease38,39 and was also seen in some patients with lymphocytic myocarditis in which loss of plakoglobin signal was not observed. Thus, the specific role of gap junction remodeling in arrhythmogenesis in ARVC and myocarditis remains unknown and further work is required to prove a causal relationship.
Pathologists have long recognized inflammation as a common feature of ARVC29 but it has not been shown whether it plays a primary pathogenic role or develops as a secondary response to myocyte injury and degeneration. Furthermore, inflammatory mechanisms in heart disease do not depend solely on the accumulation of inflammatory cells within the myocardium. Cardiac myocytes themselves can produce inflammatory mediators24,27–29 and circulating levels of various cytokines have been correlated with the development of atrial fibrillation40 or adverse outcomes in patients with heart failure.30–32 In the present studies, we found that selected cytokines, implicated in granulomatous inflammation but not other cytokines, were capable of promoting rapid intracellular translocation of junctional plakoglobin in cultured neonatal rat ventricular myocytes. Thus, our observations suggest that inflammatory mediators may play a role in ARVC even in the absence of infiltrating inflammatory cells in the heart. While this simple “proof-of-principle” study is of potential interest and worth exploring in future studies, its relevance in terms of disease mechanisms in granulomatous myocarditis and/or ARVC remains undefined. However, we also observed immunoreactive signal for TNFα and IL-17 in ARVC myocardium, thus raising the possibility that local production of cytokines could play a role in redistribution of plakoglobin and contribute to myocyte injury. Myocardial production of TNFα and its receptors has been reported in human heart failure.27,41 We also observed significant elevations in selected pro-inflammatory factors and a significant reduction in the level of IL-1 receptor 2, an anti-inflammatory protein that acts as a decoy receptor to bind IL-1 but not lead to downstream signaling.42 A recent study has reported elevated levels of IL-1β, IL-6 and TNFα in 8 ARVC patients.43 This study used a commercially available ELISA assay that has far lower sensitivity than the assay used in the present study,20 and reported values of <10 pg/ml for all measurements. Taken together, however, this previous study and our observations indicate that patients with ARVC exhibit significant increases in circulating inflammatory mediators, perhaps reflecting an imbalance between pro-inflammatory and anti-inflammatory proteins. Neither study was sufficiently powered to correlate patterns of cytokine expression with disease severity or clinical outcomes. Furthermore, we do not mean to suggest that elevated cytokine levels necessarily carry any diagnostic significance. Indeed, increased circulating inflammatory mediators have been reported in a variety of heart diseases including sarcoidosis and lymphocytic myocarditis.44,45 However, our observations provide a foundation for future studies in which analysis of serum inflammatory biomarkers in ARVC can be assessed in risk-stratification and correlated with arrhythmias or other manifestations of disease. These observations also raise the possibility of anti-inflammatory therapy in ARVC.
A limitation in the present study is the relative lack of mechanistic insights that could be gained from studying formalin-fixed, paraffin-embedded tissue samples. Nevertheless, we discovered potentially important new links between ARVC and some forms of myocarditis. We have also provided new information implicating cytokines in ARVC and new evidence that the myocardium itself may be the source of some of these mediators. Thus, disease pathways activated by mutations in desmosomal proteins may stimulate expression and possible secretion of inflammatory mediators which, in turn, could promote myocyte injury and/or arrhythmogenesis. Perhaps the most important strength of our observations is that they come from the actual human disease. In turn, they will stimulate and inform experimental studies and prospective clinical trials to test hypotheses about disease mechanisms, patient risk-stratification and new therapeutic targets.
Supplementary Material
Acknowledgments
We thank Drs. Andrew Lichtman and Richard Mitchell from Brigham and Women’s Hospital for helpful discussions about this work.
Funding Sources: Work from the senior author’s laboratory was supported by fellowship grants from the Heart Rhythm Society and the American Heart Association (AA), and by a research grant from NIH (HL102361) (JES). Collection of giant cell myocarditis tissue was supported by a grant from the US Food and Drug Administration (FD-R-001861-01) (LTC).
Footnotes
Conflict of Interest Disclosures: None
References
- 1.Basso C, Corrado D, Marcus FI, Nava A, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Lancet. 2009;373:1289–1300. doi: 10.1016/S0140-6736(09)60256-7. [DOI] [PubMed] [Google Scholar]
- 2.Asimaki A, Tandri H, Huang H, Halushka MK, Gautam S, Basso C, Thiene G, Tsatsopoulou A, Protonotarios N, McKenna WJ, Calkins H, Saffitz JE. A new diagnostic test for arrhythmogenic right ventricular cardiomyopathy. N Engl J Med. 2009;360:1075–1084. doi: 10.1056/NEJMoa0808138. [DOI] [PubMed] [Google Scholar]
- 3.Soejima K, Yada H. The work-up and management of patients with apparent or subclinical cardiac sarcoidosis: with emphasis on the associated heart rhythm abnormalities. J Cardiovasc Electrophysiol. 2009;20:578–583. doi: 10.1111/j.1540-8167.2008.01417.x. [DOI] [PubMed] [Google Scholar]
- 4.Vasaiwala SC, Finn C, Delpriore J, Leya F, Gagermeier J, Akar JG, Santucci P, Dajani K, Bova D, Picken MM, Basso C, Marcus F, Wilber DJ. Prospective study of cardiac sarcoid mimicking arrhythmogenic right ventricular dysplasia. J Cardiovasc Electrophysiol. 2009;20:473–476. doi: 10.1111/j.1540-8167.2008.01351.x. [DOI] [PubMed] [Google Scholar]
- 5.Ladyjanskaia GA, Basso C, Hobbelink MG, Kirkels JH, Lahpor JR, Cramer MJ, Thiene G, Hauer RN, Oosterhout MFV. Sarcoid myocarditis with ventricular tachycardia mimicking ARVD/C. J Cardiovasc Electrophysiol. 2010;21:94–98. doi: 10.1111/j.1540-8167.2009.01479.x. [DOI] [PubMed] [Google Scholar]
- 6.Granér M, Lommi J, Kupari M, Räisänen-Sokolowski A, Toivonen L. Multiple forms of sustained monomorphic ventricular tachycardia as common presentation in giant-cell myocarditis. Heart. 2007;93:119–121. doi: 10.1136/hrt.2005.079053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blauwet LA, Cooper LT. Antimicrobial agents for myocarditis: target the pathway, not the pathogen. Heart. 2010;96:494–495. doi: 10.1136/hrt.2009.173740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nash CL, Panaccione R, Sutherland LR, Meddings JB. Giant cell myocarditis, in a patient with Crohn’s disease, treated with etanercept - a tumour necrosis factor-alpha antagonist. Can J Gastroenterol. 2001;15:607–611. doi: 10.1155/2001/954340. [DOI] [PubMed] [Google Scholar]
- 9.Liu W, Feng W, Wang F, Li W, Gao C, Zhou B, Ma M. Osteoprotegerin/RANK/RANKL axis in cardiac remodeling due to immuno-inflammatory myocardial disease. Exp Mol Pathol. 2008;84:213–217. doi: 10.1016/j.yexmp.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 10.Ahuja SS, Estrada CA, Lindsey ML. Crosstalk between cytotoxic T-lymphocyte associated antigen-4 and interleukin-12 in cytotoxic T-lymphocyte-mediated myocarditis: adding another link to the chain. Circ Res. 2007;101:218–220. doi: 10.1161/CIRCRESAHA.107.158238. [DOI] [PubMed] [Google Scholar]
- 11.Fairweather D, Frisancho-Kiss S, Yusung SA, Barrett MA, Davis SE, Gatewood SJ, Njoku DB, Rose NR. Interferon-gamma protects against chronic viral myocarditis by reducing mast cell degranulation, fibrosis, and the profibrotic cytokines transforming growth factor-beta 1, interleukin-1 beta, and interleukin-4 in the heart. Am J Pathol. 2004;165:1883–1894. doi: 10.1016/s0002-9440(10)63241-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saffitz JE, Green KG, Kraft WJ, Schechtman KB, Yamada KA. Effects of diminished expression of connexin43 on gap junction number and size in ventricular myocardium. Am J Physiol (Heart Circ Physiol) 2000;278:H1662–H1670. doi: 10.1152/ajpheart.2000.278.5.H1662. [DOI] [PubMed] [Google Scholar]
- 13.Godin-Ethier J, Pelletier S, Hanafi LA, Gannon PO, Forget MA, Routy JP, Boulassel MR, Krzemien U, Tanguay S, Lattouf JB, Arbour N, Lapointe R. Human activated T lymphocytes modulate IDO expression in tumors through Th1/Th2 balance. J Immunol. 2009;183:7752–7760. doi: 10.4049/jimmunol.0901004. [DOI] [PubMed] [Google Scholar]
- 14.MacKenzie CR, González RG, Kniep E, Roch S, Däubener W. Cytokine mediated regulation of interferon-gamma-induced IDO activation. Adv Exp Med Biol. 1999;467:533–539. doi: 10.1007/978-1-4615-4709-9_66. [DOI] [PubMed] [Google Scholar]
- 15.Singer CF, Kronsteiner N, Hudelist G, Marton E, Walter I, Kubista M, Czerwenka K, Schreiber M, Seifert M, Kubista E. Interleukin-1 system and sex steroid receptor expression in human breast cancer: interleukin 1alpha protein secretion is correlated with malignant phenotype. Clin Cancer Res. 2003;9:4877–4883. [PubMed] [Google Scholar]
- 16.Pimentel RC, Yamada KA, Kleber AG, Saffitz JE. Autocrine regulation of myocyte Cx43 expression by VEGF. Circ Res. 2002;90:671– 677. doi: 10.1161/01.res.0000014823.75393.4d. [DOI] [PubMed] [Google Scholar]
- 17.Yamada K, Green KG, Samarel AM, Saffitz JE. Distinct pathways regulate expression of cardiac electrical and mechanical junction proteins in response to stretch. Circ Res. 2005;97:346–353. doi: 10.1161/01.RES.0000178788.76568.8a. [DOI] [PubMed] [Google Scholar]
- 18.Asimaki A, Syrris P, Wichter T, Matthias P, Saffitz JE, McKenna WJ. A novel dominant mutation in plakoglobin causes arrhythmogenic right ventricular cardiomyopathy. Am J Hum Genet. 2007;81:964–973. doi: 10.1086/521633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado DA, Cox MG, Daubert J, Fontaine G, Gear K, Hauer RN, Nava A, Picard MH, Protonotarios N, Saffitz JE, Sanborn DM, Steinberg JS, Tandri HM, Thiene G, Towbin JA, Tsatsopoulou A, Wichter T, Zareba W. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D); proposed modification of the Task Force criteria. Circulation. 2010;121:1533–1541. doi: 10.1161/CIRCULATIONAHA.108.840827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Knight PR, Sreekumar A, Siddiqui J, Laxman B, Copeland S, Chinnaiyan A, Remick DG. Development of a sensitive microarray immunoassay and comparison with standard enzyme-linked immunoassay for cytokine analysis. Shock. 2004;21:26–30. doi: 10.1097/01.shk.0000101668.49265.19. [DOI] [PubMed] [Google Scholar]
- 21.Rosenstein ED, Zucker MJ, Kramer N. Giant cell myocarditis: most fatal of autoimmune diseases. Semin Arthritis Rheum. 2000;30:1–16. doi: 10.1053/sarh.2000.8367. [DOI] [PubMed] [Google Scholar]
- 22.Aretz HT, Billingham ME, Edwards WD, Factor SM, Fallon JT, Fenoglio JJ, Olsen EG, Schoen FJ. Myocarditis. A histopathologic definition and classification. Am J Cardiovasc Pathol. 1987;1:3–14. [PubMed] [Google Scholar]
- 23.Eiken HG, Oie E, Damas JK, Yndestad A, Bjerkeli V, Aass H, Simonsen S, Geiran OR, Tonnessen T, Christensen G, Froland SS, Gullestad L, Attramada H, Aukrust P. Myocardial gene expression of leukaemia inhibitory factor, interleukin-6 and glycoprotein130 in end-stage human heart failure. Eur J Clin Invest. 2001;31:389–397. doi: 10.1046/j.1365-2362.2001.00795.x. [DOI] [PubMed] [Google Scholar]
- 24.Podewski EK, Hilfiker-Kleiner D, Hilfiker A, Morawietz H, Lichtenberg A, Wollert KC, Drexler H. Alterations in Janus kinase (JAK) - signal transducers and activations of transcription (STAT) signaling in patients with end-stage dilated cardiomyopathy. Circulation. 2003;107:789–802. doi: 10.1161/01.cir.0000057545.82749.ff. [DOI] [PubMed] [Google Scholar]
- 25.Gwechenberger M, Hülsmann M, Berger R, Graf S, Springer C, Stanek B, Pacher R. Interleukin-6 and B-type natriuretic peptide are independent predictors for worsening of heart failure in patients with progressive congestive heart failure. J Heart Lung Transplant. 2004;23:839–844. doi: 10.1016/j.healun.2003.07.023. [DOI] [PubMed] [Google Scholar]
- 26.Basso C, Thiene G, Corrado D, Angelini A, Nava A, Valente M. Arrhythmogenic right ventricular cardiomyopathy: Dysplasia, dystrophy or myocarditis? Circulation. 1996;94:983–991. doi: 10.1161/01.cir.94.5.983. [DOI] [PubMed] [Google Scholar]
- 27.Irwin MW, Mak S, Mann DL, Qu R, Penninger JM, Yan A, Dawood F, Wen WH, Shou Z, Liu P. Tissue expression and immunolocalization of tumor necrosis factor-alpha in post-infarction dysfunctional myocardium. Circulation. 1999;99:1492–1498. doi: 10.1161/01.cir.99.11.1492. [DOI] [PubMed] [Google Scholar]
- 28.Nian M, Lee P, Khaper N, Liu P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res. 2004;94:1543–1553. doi: 10.1161/01.RES.0000130526.20854.fa. [DOI] [PubMed] [Google Scholar]
- 29.Agrawal RA, Koyani CN, Singh R. Molecular targets and regulators of cardiac hypertrophy. Pharmacol Res. 2010;61:269–280. doi: 10.1016/j.phrs.2009.11.012. [DOI] [PubMed] [Google Scholar]
- 30.Deswal A, Petersen NJ, Feldman AM, Young JB, White BG, Mann DL. Cytokines and cytokine receptors in advanced heart failure: An analysis of the cytokine database from the Vesnarinone Trial (VEST) Circulation. 2001;103:2055–2059. doi: 10.1161/01.cir.103.16.2055. [DOI] [PubMed] [Google Scholar]
- 31.Bradham WS, Boskurt B, Gunasinghe H, Mann D, Spinale FG. Tumor necrosis factor-alpha and myocardial remodeling in progression of heart failure: a clinical perspective. Cardiovasc Res. 2002;53:822–830. doi: 10.1016/s0008-6363(01)00503-x. [DOI] [PubMed] [Google Scholar]
- 32.Dibbs Z, Thornby J, White BG, Mann DL. Natural variability of circulating levels of cytokines and cytokine receptors in patients with heart failure: Implications for clinical trials. J Am Coll Cardiol. 1999;33:1935–1942. doi: 10.1016/s0735-1097(99)00130-8. [DOI] [PubMed] [Google Scholar]
- 33.Amagai M. Autoimmune and infectious skin diseases that target desmogleins. Proc Jpn Acad Ser B Phys Biol Sci. 2010;86:524– 537. doi: 10.2183/pjab.86.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schulzke JD, Ploeger S, Amasheh M, Fromm A, Zeissig S, Troeger H, Richter J, Bojarski C, Schumann M, Fromm M. Epithelial tight junctions in intestinal inflammation. Ann NY Acad Sci. 2009;1165:294–300. doi: 10.1111/j.1749-6632.2009.04062.x. [DOI] [PubMed] [Google Scholar]
- 35.Schmidt A, Koch PJ. Desmosomes: just cell adhesion or is there more? Cell Adh Migr. 2007;1:28–32. doi: 10.4161/cam.1.1.4204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaplan SR, Gard JJ, Protonotarios N, Tsatsopoulou A, Spiliopoulou C, Anastasakis A, Squarcioni CP, McKenna WJ, Thiene G, Basso C, Brousse N, Fontaine G, Saffitz JE. Remodeling of myocyte gap junctions in arrhythmogenic right ventricular cardiomyopathy due to a deletion in plakoglobin (Naxos disease) Heart Rhythm. 2004;1:3–11. doi: 10.1016/j.hrthm.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 37.Fidler LM, Wilson GJ, Liu F, Cui X, Scherer SW, Taylor GP, Hamilton RM. Abnormal connexin43 in arrhythmogenic right ventricular cardiomyopathy caused by plakophilin-2 mutations. J Cell Mol Med. 2009;13:4219–4228. doi: 10.1111/j.1582-4934.2008.00438.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saffitz JE, Hames KY, Kanno S. Remodeling of gap junctions in ischemic and nonischemic forms of heart disease. J Membr Biol. 2007;218:65–71. doi: 10.1007/s00232-007-9031-2. [DOI] [PubMed] [Google Scholar]
- 39.Severs NJ, Dupont E, Thomas N, Kaba R, Rothery S, Jain R, Sharpey K, Fry CH. Alterations in cardiac connexin expression in cardiomyopathies. Adv Cardiol. 2006;42:228– 242. doi: 10.1159/000092572. [DOI] [PubMed] [Google Scholar]
- 40.Li J, Solus J, Chen Q, Rho YH, Milne G, Stein CM, Darbar D. Role of inflammation and oxidative stress in atrial fibrillation. Heart Rhythm. 2010;7:438–444. doi: 10.1016/j.hrthm.2009.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Torre-Amione G, Kapadia S, Lee J, Durant JB, Bies RD, Young JB, Mann DL. Tumor necrosis factor-α and tumor necrosis factor receptors in the failing human heart. Circulation. 1996;93:704–711. doi: 10.1161/01.cir.93.4.704. [DOI] [PubMed] [Google Scholar]
- 42.Symons JA, Young PR, Duff GW. Soluble type II interleukin 1 (IL-1) receptor binds and blocks processing of IL-1 beta precursor and loses affinity for IL-1 receptor antagonist. Proc Natl Acad Sci USA. 1995;92:1714–1718. doi: 10.1073/pnas.92.5.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campian ME, Verberne HJ, Hardziyenka M, de Groot EAA, van Moerkerken AF, van Eck-Smit BLF, Tan HL. Assessment of inflammation in patients with arrhythmogenic right ventricular cardiomyopathy/dysplasia. Eur J Nucl Med Mol Imaging. 2010;37:2079–2085. doi: 10.1007/s00259-010-1525-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fuse K, Kodama M, Okura Y, Ito M, Aoki Y, Hirono S, Kato K, Hanawa H, Aizawa Y. Levels of serum interleukin-10 reflect disease activity in patients with cardiac sarcoidosis. Jpn Circ J. 2000;64:755–759. doi: 10.1253/jcj.64.755. [DOI] [PubMed] [Google Scholar]
- 45.Matsumori A, Yamada T, Suzuki H, Matoba Y, Sasayama S. Increaed circulting cytokines in patients with myocarditis and cardiomyopathy. Br Heart J. 1994;72:561–566. doi: 10.1136/hrt.72.6.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.