

Abstract
The Ras/Raf/MEK/ERK pathway is primarily responsible for mitogenesis in metazoans, and mutational activation of this pathway is common in cancer. A variety of selective chemical inhibitors directed against the MAPK pathway are now available for clinical investigation and thus the determination of the importance of each of the kinases in oncogenesis is paramount. Here, we investigated the role of two Raf kinases, B-Raf and C-Raf, in Ras oncogenesis, and find that while B-Raf and C-Raf have overlapping functions in primary mesenchymal cells, C-Raf but not B-Raf is required for the proliferative effects of K-RasG12D in primary epithelial cells. Furthermore, in a lung cancer mouse model, C-Raf is essential for tumor initiation by oncogenic K-RasG12D, while B-Raf is dispensable for this process. Our findings reveal that K-RasG12D elicits its oncogenic effects primarily through C-Raf and suggest that selective C-Raf inhibition could be explored as a therapeutic strategy for K-Ras dependent cancers.
INTRODUCTION
Activating mutations in Ras genes are observed in approximately 30% of human cancers (1). Ras stimulates multiple downstream effector pathways, which are constitutively activated in a growth factor-independent fashion in cancer cells expressing oncogenic Ras (2). Despite extensive efforts, targeted therapies against Ras were largely unsuccessful in clinical trials (3), and thus pharmacological inhibition of Ras effector pathways may represent a more feasible approach to eradicate Ras mutant cancers. The contribution of Ras effector pathways to Ras-mediated transformation has been studied in some detail in human and murine cells (4-6). These studies found that the Ras effectors required for transformation depend on the species, the cell type and the tumor stage. However, these findings may be confounded by the fact that they were mainly conducted in cultured cells and by using H-Ras, the least commonly mutated Ras gene, to transform cells. K-Ras is the predominantly mutated Ras family member and activating mutations in K-Ras are associated primarily with malignancies of the lung, pancreas and colon. An in vivo evaluation of the role of Ras effectors in these cancer types is limited and has only been studied in the lung thus far. For example, the importance of the Ras-Rac axis has been validated in a K-Ras mutant mouse model of lung cancer using conditional Rac knock-out mice. Rac deficiency strongly impaired Ras tumorigenesis in the lung and developing tumors invariably retained wild-type Rac (7), suggesting an essential contribution of Rac to K-RasG12D-initiated lung cancer. In addition, the Ras-PI3K-AKT pathway has been demonstrated to be critical for Ras-mediated tumorigenesis. Here, mice expressing a Ras-binding-deficient form of the p110alpha subunit of PI3K were insensitive to oncogenic K-Ras-driven lung tumorigenesis (8). However, the contribution of the Ras-MAPK axis, a critical regulatory pathway that is often perturbed in cancer, to K-Ras-mediated transformation has been insufficiently addressed. The potential importance of MAPK pathway deregulation for transformation by Ras is emphasized by the prevalence of B-Raf mutations. These mutants exquisitely activate the MAPK pathway and have been observed in cancer types that frequently carry Ras mutations, e.g. in melanoma (N-Ras) as well as lung, colon and ovary (K-Ras) (9). Furthermore, the occurrence of activated ERK1/2 in lung cancer specimens is associated with a poor prognosis (10). Importantly, several distinct MEK inhibitors showed efficacy in the context of Ras-driven carcinoma cells in culture and xenografts, and are currently being assessed in clinical trials. In addition, pharmacological inhibition of both MEK and PI3K/TOR (11) in a K-Ras mutant mouse model of early lung cancer (12) resulted in tumor regression, indicating that concurrent targeting of these pathways may have clinical benefits. Potent B-Raf-specific and pan-Raf inhibitors are being tested in clinical trials for cancers that carry B-Raf mutations (13). Such Raf inhibitors could also be clinically beneficial in malignancies harboring activating genetic lesions in K-Ras, provided that the Raf-MEK-ERK pathway critically contributes to tumorigenesis induced by oncogenic K-Ras. To evaluate the suitability of Raf inhibitors for the treatment of K-Ras mutant cancers, the role of Raf proteins in K-Ras oncogenesis needs to be established.
In this study, we genetically ablated B-Raf or C-Raf expression in K-RasG12D mutant primary cells in vitro as well as in lung epithelial cells in vivo to evaluate the contribution of individual Raf proteins to K-RasG12D-mediated transformation. Our genetic analysis reveals that C-Raf is uniquely required for K-RasG12D-driven transformation of epithelial cells in vitro and in vivo.
RESULTS
Effect of Raf loss on K-RasG12D-mediated transformation of MEFS
Endogenous expression of oncogenic K-RasG12D partially transforms MEFs as indicated by immortality, elevated proliferation rates and the ability of forming foci under confluent culture condition (14). To analyze the role of B-Raf and C-Raf in transformation by oncogenic K-Ras, B-Raffl/fl; K-RasLSL (BBK) and C-Raffl/fl; K-RasLSL (CCK) mouse embryonic fibroblasts (MEFs) were infected with Cre recombinase-expressing adenovirus (Ad-Cre) to produce B-RafΔ/Δ; K-RasG12D and C-RafΔ/Δ; K-RasG12D cells, respectively. First, proliferation was assessed in 10% and 2% fetal calf serum. Similar to K-RasG12D (K) cells, both B-RafΔ/Δ; K-RasG12D and C-RafΔ/Δ; K-RasG12D MEFs displayed increased proliferation compared to Ad-Mock infected isogenic control cells under normal and low-serum conditions (Figure 1A, Suppl. Figure 1A, 2). Moreover, focus formation of K-RasG12D mutant MEFs was not diminished by loss of either B-Raf or C-Raf (Suppl. Figure 1C), indicating that these Raf proteins are dispensable for several characteristics of oncogenic Ras biology in MEFs.
Figure 1.
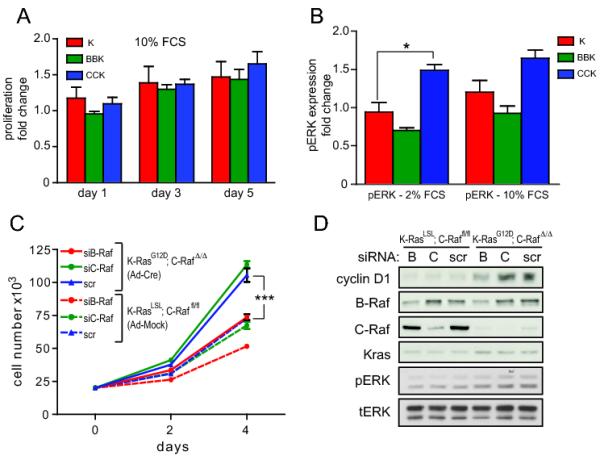
Effect of Raf deficiency on K-RasG12D-mediated transformation of MEFs. (A) Increase in cell number of Ad-Cre infected MEFs of the indicated genotypes relative to Ad-Mock infected isogenic control cells. Cells were analyzed in 10% serum. The fold increase of proliferation of Ad-Cre infected cells compared to Ad-Mock infected isogenic control cells is shown. (B) Quantification of Western blot analysis of MAPK activation in response to K-RasG12D expression in 2% and 10% fetal calf serum. pERK was normalized to total ERK and fold change compared to Ad-Mock infected isogenic control cells is shown. Fold change of pERK expression was averaged from three independent MEF lines with the same genotype. (C) Proliferation of C-Raffl/fl; K-RasLSL (Ad-Mock) or C-RafΔ/Δ; K-RasG12D (Ad-Cre) MEFs transfected with 100pmol siRNA against C-Raf, B-Raf or scrambled (scr) control. (D) Western blot for ERK phosphorylation and cyclin D1 expression in MEFs analyzed in (C). Abbreviations used are CCK, C-Raffl/fl; K-RasLSL; BBK, B-Raffl/fl; K-RasLSL; CK, C-Raffl/wt; K-RasLSL; BK, B-Raffl/wt; K-RasLSL; K, K-RasLSL and error bars represent ± SEM (ns, not significant; *, P<0.05; **, P<0.01; ***, P<0.001) here and for all figures.
Next, we analyzed MAPK signaling downstream of Ras-Raf in K-RasG12D, B-RafΔ/Δ; K-RasG12D and C-RafΔ/Δ; K-RasG12D MEFs. Western analysis confirmed recombination of the floxed Raf alleles as well as induction of K-RasG12D expression upon infection with Ad-Cre (Suppl. Figure 1C). Interestingly, under low and normal serum conditions deletion of B-Raf or C-Raf slightly decreased or elevated steady state pERK levels, respectively (Fig. 1B). However, only the increase in pERK in C-Raf-depleted cells grown in 2% serum was statistically significant. Hence, modest differences in steady-state pERK levels did not correlate with induction of proliferation in response to oncogenic K-Ras. To unequivocally investigate whether Raf deficiency alters MAPK activation, ERK phosphorylation in response to serum stimulation was examined. Notably, the kinetics of MAPK activation of serum-starved and re-stimulated K-RasG12D, B-RafΔ/Δ; K-RasG12D and C-RafΔ/Δ; K-RasG12D MEFs were comparable (Suppl. Figure 1D). These findings indicate that loss of either B-Raf or C-Raf alone does not abrogate oncogenic transformation by K-RasG12D and that B-Raf and C-Raf may have overlapping functions in MEFs.
As genetic ablation of either B-Raf or C-Raf alone had no impact, the effect of combined depletion of B-Raf and C-Raf on K-RasG12D-mediated transformation of MEFs was assessed. Accordingly, we utilized siRNA against B-Raf in C-Raffl/fl; K-RasLSL MEFs infected with Ad-Cre or empty adenovirus. Proliferation of C-Raffl/fl; K-RasLSL control cells transfected with either B-Raf siRNA or control siRNAs (scrambled or C-Raf siRNA) was comparable. In contrast, RNAi-mediated B-Raf depletion by approximately 50-60% in C-RafΔ/Δ; K-RasG12D MEFs reduced proliferation to levels comparable to C-Raffl/fl; K-RasLSL control cells (Figure 1C). Moreover, this rescue of the proliferation defect in K-RasG12D-mutant MEFs was accompanied by a modest decrease in pERK and a more substantial decrease in cyclin D1, a known target of the MAPK pathway (Figure 1D). Knock-down of C-Raf in BBK MEFs similarly rescued proliferation (data not shown). These data suggest that K-RasG12D signals through both B-Raf and C-Raf in MEFs, and combined deletion of both paralogues is necessary to suppress K-Ras driven hyperproliferation of MEFs and activation of downstream targets.
Depletion of Raf proteins in K-RasG12D mutant epithelial cells in vitro
MEFs are a commonly used experimental system to assess the oncogenicity of mutant K-Ras; however, the number of conclusions that can be drawn from experiments using MEFs is limited with regards to human cancer as K-Ras is typically mutated in epithelial cells. We therefore investigated whether B-Raf or C-Raf are critical for K-RasG12D-induced transformation in primary epithelial cells. To address this, signaling downstream of K-RasG12D and proliferation of K-RasLSL, B-Raffl/fl; K-RasLSL and C-Raffl/fl; K-RasLSL baby mouse kidney (BMK) epithelial cells were examined. BMK cells have previously been shown to exhibit increased proliferation in response to K-RasG12D activation (7, Figure 2A). Similar to MEFs, BMKs were infected either with Ad-Cre to produce K-RasG12D, B-RafΔ/Δ; K-RasG12D and C-RafΔ/Δ; K-RasG12D cells or with empty adenovirus (Ad-Mock). Ablation of B-Raf expression in otherwise wild-type BMK cells had no effect on proliferation (Figure 2B), or pERK and cyclin D1 expression (Suppl. Figure 3A). Similar to K-RasG12D BMK cells activation of K-RasG12D in the absence of B-Raf elevated proliferation of B-RafΔ/Δ; K-RasG12D BMK cells (Figure 2B). These data indicate that B-Raf is dispensable for proliferation of wild-type and K-Ras mutant epithelial cells in vitro. In contrast, C-RafΔ/Δ BMK cells proliferated less than wild-type control cells (Figure 2C), and accumulated in the G2-M phase of the cell cycle (Suppl. Figure 3A). No changes in pERK and only modest and heterogeneous changes in cyclin D1 levels were observed (Suppl. Figure 3B). Reduced cell numbers could not be attributed to apoptosis in the absence of C-Raf as the levels of cleaved caspase 3 and cleaved PARP did not increase upon C-Raf deletion (Suppl. Figure 3C), implying that C-Raf may have additional functions in BMK cells. Interestingly, C-RafΔ/Δ; K-RasG12D BMK cells did not display an increase in proliferation compared to Ad-Mock infected control cells (Figure 2C), suggesting that oncogenic K-RasG12D could not override the proliferative defect of BMK cells elicited by the absence of C-Raf. In addition, cell cycle analysis of BMK cells revealed that expression of oncogenic K-RasG12D resulted in a larger proportion of cells in G1 and S phase and fewer cells in G2-M phase (Figure 2D), similar to endogenous expression of K-RasG12D in MEFs (14). The absence of B-Raf had no impact on the cell cycle profile of K-RasG12D mutant BMK cells, while the C-RafΔ/Δ; K-RasG12D BMK cells displayed similar cell cycle distribution as wild-type control cells (Figure 2D), thus corroborating the proliferation analysis (Figure 2A-C). Expression of K-RasG12D in MEFs induced the expression of several cell cycle regulatory proteins (14). We determined the expression levels of cell cycle proteins in BMK cells to address if the absence of C-Raf alters the induction of such proteins, thereby rescuing the proliferation defect of K-RasG12D-expressing cells. K-RasG12D modestly increased phosphorylation of ERK and expression of CDK4, which was negated by depletion of either B-Raf or C-Raf (Figure 2E, F and data not shown). In addition, induction of cyclin D1 expression in response to K-RasG12D was diminished in the absence of C-Raf but not B-Raf (Figure 2D, E). Other cell cycle regulators were not induced by K-RasG12D or affected by Raf deficiency (data not shown). In addition, RNAi-mediated depletion of Cyclin D1 in K and BBK BMK cells resulted in a markedly reduced number of cells in S phase, as measured by BrdU incorporation (Suppl. Figure 3D). Thus, impaired upregulation of cyclin D1 in the absence of C-Raf may diminish the proliferative response of BMK cells to expression of K-RasG12D. Taken together, C-Raf is a crucial mediator of proliferation in K-RasG12D expressing epithelial cells, while B-Raf is not required.
Figure 2.
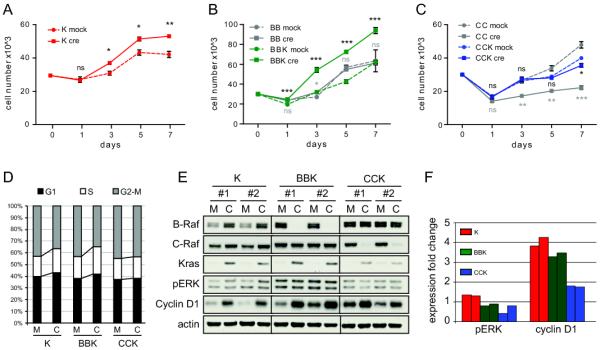
C-Raf is required for the induction of proliferation by K-RasG12D in mutant baby mouse kidney cells. (A) Proliferation of K-RasLSL and K-RasG12D BMKs. (B) Proliferation of BBK and B-Raffl/fl BMKs. (C) Proliferation of CCK and C-Raffl/fl BMKs. Two to three cell lines were analyzed in triplicates and one representative cell line is shown in (A-C). Statistical analysis: gray asterisks refer to BB and CC cells, black asterisks refer to K, BBK and CCK cells. ns, not significant; *, p<0.05; **, p<0.01; ***, p<0.001. (D) Cell cycle analysis of Ad-Cre or Ad-Mock infected K, BBK, and CCK cells. M, Ad-Mock infected cells; C, Ad-Cre infected cells. (E) Western blot for MAPK activation and expression of cyclin D1 in K, BBK, and CCK BMK cells. M, Ad-Mock infected cells; C, Ad-Cre infected cells. (F) Quantification of (E). Expression of pERK and cyclin D1 was normalized to actin.
Lung tumor development and survival of K-RasLSL mice in the absence of B-Raf or C-Raf
To investigate the functions of Raf proteins in K-RasG12D-mediated transformation in vivo, we generated compound mutant mice harboring conditional B-Raf (15) or C-Raf (16) knock-out alleles (B-Raffl/fl or C-Raffl/fl, respectively) in combination with an endogenous, conditional K-RasLSL allele where oncogenic K-RasG12D is expressed upon Cre mediated excision of a lox-stop-lox (LSL) element (12). Compound mutant B-Raffl/fl; K-RasLSL and C-Raffl/fl; K-RasLSL mice and compound mutant mice bearing heterozygous conditional Raf alleles (B-Raffl/wt; K-RasLSL and C-Raffl/wt; K-RasLSL) as well as K-RasLSL mice on the same genetic background were infected with adenoviral Cre recombinase (Ad-Cre) via intranasal instillation at 4-6 weeks of age to induce lung tumorigenesis. This route of Cre delivery has been demonstrated to successfully recombine floxed DNA sequences, including the K-RasLSL allele (12). Mice were aged for 6, 9, and 12 weeks, and lung tumorigenesis examined. B-Raffl/fl; K-RasLSL (BBK) mice displayed similar overall tumor burden as B-Raffl/+; K-RasLSL (BK) and K-RasLSL (K) mice (Figure 3A). In contrast, C-Raffl/fl; K-RasLSL (CCK) mice demonstrated drastically reduced lung tumor burden compared to C-Raffl/+; K-RasLSL (CK) and K-RasLSL (K) mice (Figure 3A). In addition, C-Raffl/fl; K-RasLSL mice exhibited a significant reduction in bronchiolar hyperplasia (BH), adenomatous alveolar hyperplasia (AAH), and adenomas (Figure 3B) while the size of neoplasms was not affected by the absence of C-Raf (Figure 3C), suggesting that the lower number of neoplasms results in reduced lung tumor burden. Thus, K-RasG12D-induced formation of alveolar and bronchiolar tumors is impaired in the absence of C-Raf (Figure 3D). Interestingly, while B-Raffl/fl; K-RasLSL mice displayed a specific reduction in the number of BHs but not AAHs or adenomas (Figure 3B), the size of all neoplasms was reduced (Figure 3C). Thus, B-Raf plays a role in the biology of K-RasG12D expressing alveolar and bronchiolar neoplasms but its function is less prominent than that of C-Raf.
Figure 3.
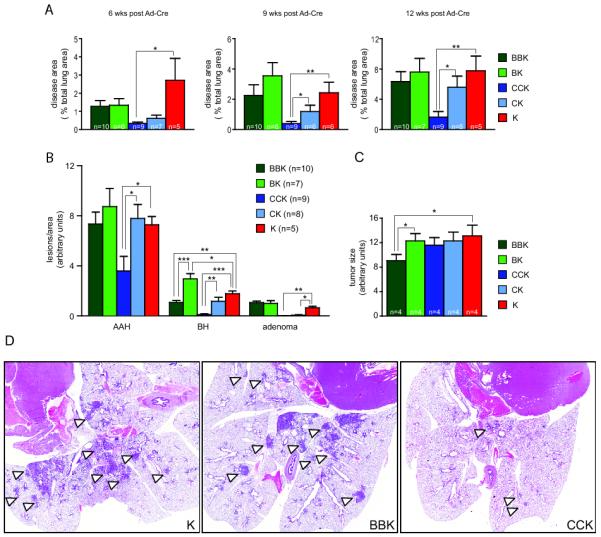
Lung tumor development in the absence of Raf proteins. (A) Tumor burden in B-Raffl/fl; K-RasLSL (BBK), B-Raffl/+; K-RasLSL (BK), C-Raffl/fl; K-RasLSL (CCK), C-Raffl/+; K-RasLSL (CK) and K-RasLSL (K) mice at 6, 9, and 12 weeks post Ad-Cre infection. Burden was determined as percent disease area per total lung area and is significantly decreased in the absence of C-Raf. (B) Characterization of the spectrum of pulmonary preneoplasms in BBK, BK, CCK, CK, and K mice at 12 weeks post Ad-Cre infection. C-Raf loss reduced the number of BH, AAH, and adenomas, while B-Raf deficiency reduces only the number of BH. (C) Size of neoplasms was unchanged in BK, CCK and CK mice but was decreased in BBK mice at 12 weeks post Ad-Cre infection. (D) H&E staining of representative lung sections from BBK, CCK, and K mice 12 weeks post Ad-Cre infection.
To assess whether loss of either C-Raf or B-Raf affects survival of K-RasG12D mutant mice, cohorts of K-RasLSL mice harboring floxed Raf alleles were aged. Heterozygous genetic deletion of B-Raf in K-RasG12D mutant lung epithelia resulted in a short, albeit statistically insignificant increase in survival compared to K-RasLSL control mice (200.5 vs. 167 days median survival, Figure 4A). However, the lifespan of B-Raffl/fl; K-RasLSL mice was extended compared to K-RasLSL or B-Raffl/wt; K-RasLSL mice (249 vs. 167 or 200.5 days, respectively, Figure 4A). Heterozygous loss of C-Raf in C-Raffl/wt; K-RasLSL mice resulted in a slight prolongation of survival compared to K-RasLSL mice (211 vs. 167 days, Figure 4B). Moreover, homozygous deletion of C-Raf extended survival even further (268 days, Figure 4B). Hence, lower tumor burden in K-RasLSL mutant mice deficient for C-Raf correlates with extended survival. The increased lifespan of C-Raffl/wt; K-RasLSL mice was unexpected as these mice displayed a similar tumor burden compared to K-RasLSL mice at early time points (data not shown). It suggests, however, that loss of only one copy of C-Raf is disadvantageous to K-RasG12D mutant tumors. Similarly, C-Raf heterozygosity strongly impairs the development of SOS-driven epidermal tumors (17).
Figure 4.
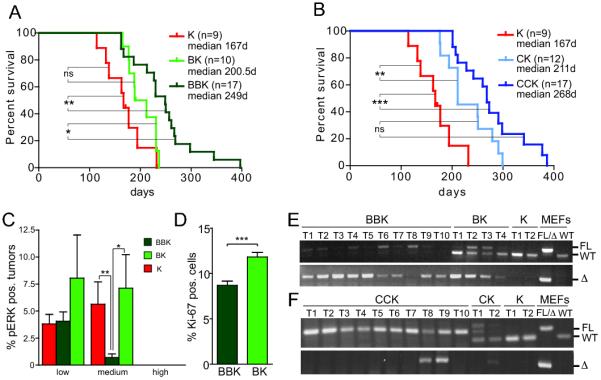
Effect of Raf loss on survival, MAPK activation and proliferation. (A,B) Survival is extended by homozygous loss of B-Raf (A) or C-Raf (B) in the context of K-RasG12D. (C) Reduced MAPK activation in BBK tumors at endpoint. (D) Reduced proliferation of BBK tumors at 12 weeks post Ad-Cre infection. (E, F) Evaluation of recombination of floxed Raf alleles by genotyping PCR of laser capture micro-dissected tumors.
MAPK activity and proliferation of lung neoplasms
To evaluate Raf/MEK/ERK pathway activation, immunohistochemistry was employed to measure phosphorylated ERK in lung tumors of K-RasLSL mice harboring conditional Raf knock-out alleles. Premalignant lesions of the lung have been previously reported to lack increased levels of pERK, while advanced tumors harboring concomitant loss of the tumor suppressor gene p53 demonstrated augmented activation of the MAPK pathway (18-20). Similarly, AAH, BH and adenomas at 12 weeks post Ad-Cre infection showed no elevation of pERK levels compared to surrounding normal lung epithelia in all genotypes (data not shown). Thus, we examined pERK expression in lung tumors of the survival cohorts, as these tumors presumably had progressed to more advanced stages. While the majority of tumors at the time of death of these animals displayed no increase in pERK staining (~80%, Figure 4C, Suppl. Figure 4A) a fraction of tumors exhibited elevated levels of pERK. We devised a grading system to assess the intensity of ERK phosphorylation in lung tumors. Weak pERK staining in less than 25% of cells in a tumor was considered low intensity staining. Weak to moderate staining in more than 25% of cells or strong pERK positivity in less than half of the tumor was counted as medium intensity staining. Tumors with high intensity pERK staining displayed strong staining for pERK in more than 50% of cells. Intriguingly, while the lack of C-Raf had no effect on the percentage of tumors with increased ERK phosphorylation (Suppl. Figure 4A), B-Raffl/fl; K-RasLSL mice displayed fewer lung tumors with pERK positivity (Figure 2C). Thus, K-RasG12D may activate the MAPK pathway in advanced lung tumors through B-Raf signaling.
Since the MAPK pathway is a major regulatory pathway of cell proliferation, we examined proliferation of lung neoplasms at 12 weeks post Ad-Cre infection by Ki-67 immunohistochemistry. The percentage of Ki-67 positive, neoplastic cells in C-Raffl/fl; K-RasLSL mice was unchanged (Suppl. Figure 4B). In contrast, proliferation of AAH and early grade adenomas was reduced in B-Raffl/fl; K-RasLSL mice (Figure 4D), suggesting that reduced MAPK pathway activation in these tumors may result in impaired proliferation. Thus, while B-Raf is dispensable for initiation of alveolar neoplasms, B-Raf deficiency impairs proliferation of K-RasG12D mutant lung tumors, correlating with an extended survival of B-Raffl/fl; K-RasLSL mice. These data support the notion that both B-Raf and C-Raf are important for K-RasG12D-driven lung tumorigenesis, albeit at different stages.
Recombination of Raf alleles in vivo
To confirm that decreased MAPK activation and proliferation in conditional B-Raf knock-out tumors is due to B-Raf deficiency, tumor tissue was isolated from mice at endpoint by laser capture microdissection and genotyping PCRs were performed on the DNA isolated from these tissues. Notably, in 50% of tumors (5 out of 10) a recombined floxed B-Raf product could be amplified, while the intact flox allele was absent (Figure 4E), indicating that these tumors had recombined both floxed B-Raf alleles and are deficient for B-Raf. Examination of the remaining five tumors revealed that four (T1, T2, T4, and T6) had deleted at least one B-Raf flox allele and retained a variable but usually reduced content of unrecombined B-Raffl allele, potentially representing the contribution of non-neoplastic cells. Only one tumor retained both wildtype copies of B-Raf (T8; Figure 4E). Thus, B-Raf deficiency does not prevent tumor formation but correlates with impaired MAPK activation and proliferation. Tumor tissue was similarly isolated from C-Raffl/fl; K-RasLSL mice and recombination of floxed C-Raf alleles evaluated. Strikingly, any presence of a recombined C-Raf allele could only be detected in two of the ten isolated tumors (T8, T9; Figure 4F). However, these tumors displayed an incomplete loss of C-Raf, supporting the notion that K-RasG12D-induced tumor formation is severely compromised in the absence of C-Raf. Interestingly, even heterozygous loss of C-Raf in C-Raffl/wt; K-RasLSL mice was minimally detected, further supporting the importance of C-Raf in K-RasG12D mediated oncogenesis (Figure 4B). The fact that tumors that develop in C-Raffl/fl; K-RasLSL mice are indistinguishable from control tumors in terms of proliferation and MAPK activation (Suppl. Figure 1A, B) substantiates the notion that these lesions are ‘escaper’ tumors with normal C-Raf expression. Similar results have been obtained in Ras-driven epidermal tumors (17). While excision of the conditional B-Raf and C-Raf alleles is easily accomplished in cell culture, there may be differential recombination efficiencies in vivo, or simply the outgrowth of Raffl/fl; K-RasG12D lung epithelial “escaper” cells that have a fitness advantage in comparison to C-RaflΔ/Δ; K-RasG12D cells but not in comparison to B-RaflΔ/Δ; K-RasG12D cells. Nonetheless, this experiment highlights the unique importance of intact C-Raf expression for the genesis of lung adenocarcinoma.
DISCUSSION
In this study we describe the role of B-Raf and C-Raf in K-RasG12D-mediated transformation. In primary mouse embryonic fibroblasts, a cell system commonly utilized to assess the oncogenicity of mutant Ras, simultaneous ablation of B-Raf and C-Raf is required to prevent the proliferative effects of K-RasG12D. In contrast, K-RasG12D promotes proliferation of cultured epithelial cells through C-Raf but not B-Raf. Furthermore, lung tumor initiation by K-RasG12D is completely prevented by the absence of C-Raf.
Oncogenic K-RasG12D is one of the most common cancer-associated genetic lesions; however, devising therapeutic approaches to target K-RasG12D has been unsuccessful thus far and inhibition of Ras effector pathways may yield better results. In this study we sought to determine the contribution of the Raf-MEK-ERK pathway to K-RasG12D-mediated transformation in vitro and in vivo. Conditional A-Raf knock-out mice have not been generated yet and, therefore, we had to limit our analysis to B-Raf and C-Raf, the two best characterized Raf family members. K-RasG12D has been demonstrated to partially transform primary MEFs (14) and we assessed the contribution of B-Raf and C-Raf to this phenotype. Individual ablation of either B-Raf or C-Raf did not obviously alter the cellular effects conferred by K-RasG12D. Curiously, the levels of steady state ERK phosphorylation were influenced by the absence of both B-Raf and C-Raf. Recently, a negative feedback loop was described in which ERK-dependent phosphorylation of C-Raf results in inactivation of the latter (21), and ERK-dependent phosphorylation of B-Raf reduces its binding to activated Ras and disrupts heterodimerization with C-Raf (22). Thus, feedback regulation of the MAPK pathway may be perturbed in the absence of B-Raf or C-Raf, leading to alterations in steady state levels of pERK. However, the absence of either B-Raf or C-Raf does not impinge on the kinetics of MAPK activation upon serum stimulation and steady state pERK levels do not correlate with proliferation rates of these MEFs. Decreased pERK levels upon B-Raf ablation and elevated pERK upon C-Raf ablation has also been observed in fibroblasts expressing wild-type K-Ras as well as in other cell types (reviewed in 23). Whether these differences in ERK activation have cell biological consequences may depend upon the cell type and/or the mutational status of MAPK-activating genes. In K-RasG12D-mutant MEFs, B-Raf and C-Raf collectively relay the oncogenic signals from K-RasG12D. Simultaneous ablation of B-Raf and C-Raf was required to negate the hyperproliferative effect of K-RasG12D, supporting the notion of overlapping functions of B-Raf and C-Raf in this context.
To determine whether B-Raf and C-Raf are similarly redundant in epithelial cells, the cell type most commonly affected by K-Ras mutations in humans (9), we employed baby mouse kidney cells. Kissil et al. have previously shown that BMK cells are hyperproliferative following expression of K-RasG12D (7). Indeed, we found that levels of pERK and the cell cycle regulator cyclin D1 are elevated upon K-RasG12D expression, resulting in higher proliferation rates (Figure 2). Ablation of C-Raf impaired induction of cyclin D1, which correlated with a complete abrogation of the K-RasG12D-mediated elevation of proliferation. These data show that C-Raf and B-Raf have unique functions in BMK cells and suggest that induction of at least cyclin D1 may be critical for the higher proliferative rates of epithelial cells upon K-RasG12D expression.
In a lung cancer mouse model of oncogenic K-RasG12D we show that C-Raf is crucial for lung tumorigenesis. Unexpectedly, none of the tumors analyzed displayed homozygous deletion of C-Raf. In fact, the vast majority of tumors retained both alleles of C-Raf, indicating that loss of only one allele is detrimental to tumor development. This is further supported by the observed extension of survival in C-Raffl/wt; K-RasG12D mice. C-Raf appears to be important at the early stages of tumor development, potentially tumor initiation, as suggested by the reduction in tumor burden and tumor number at early time points. We were unable to detect any increase in apoptosis immediately following Ad-Cre delivery or at later time points, thus the fate of such cells remains unclear. Homozygous deletion of B-Raf was tolerated by lung epithelial cells; however, it resulted in reduced MAPK activation and decreased proliferation of B-Raf-deficient low grade tumors and correlated with an extended lifespan of B-Raffl/fl; K-RasG12D mice. Due to the viral titer used to deliver Ad-Cre, mice in this study succumbed to the overwhelming lung tumor burden and respiratory distress. Whether B-Raf affects proliferation of or progression to high-grade adenocarcinoma could not be addressed in this study and remains to be determined.
Our findings support the model that K-RasG12D signals through C-Raf in both bronchiolar hyperplasia and adenomatous alveolar hyperplasia cells or a common progenitor cell type and that the loss of C-Raf impinges on the initiation of both of these preneoplasms. B-Raf, on the other hand, may participate once a cell fate decision has been made. Indeed, germline and conditional B-Raf and C-Raf knock-out mice revealed that these Raf proteins serve different functions in distinct cell types (24). Alternatively, the absence of B-Raf could influence this cell fate decision, leading to fewer bronchiolar hyperplasias. Interestingly, it has been noted that the importance of individual Raf proteins changes depending on the transformation status of a cell (25). Thus, it is plausible that B-Raf is dispensable for tumor initiation but becomes more important once an epithelial cell has been transformed by K-RasG12D.
Recently, three studies reported that ATP-competitive Raf inhibitors have unexpected adverse effects in Ras mutant cancer cells, where they promote Raf dimerization and MAPK hyperactivation (26-28). These pharmacological results argue that treatment of patients with Raf inhibitors could be detrimental if the Ras mutation status is unknown. However, our genetic analysis supports Raf proteins, and especially C-Raf, as potential therapeutic targets in K-Ras mutant lung cancer. As genetic ablation is not equivalent to pharmacological inhibition, future experiments are required to determine whether our genetic data are accurately predictive of a pharmacological response to complete Raf inhibition. In addition, once the genetic tools become available, the effect of Raf ablation in established tumors can be addressed. Patients treated with currently available Raf inhibitors often develop therapy-induced squamous cell carcinoma, potentially due to increased MAPK activation as mentioned above. Thus, our finding of the critical role of C-Raf in K-RasG12D-mediated lung cancer development should stimulate further research to validate C-Raf as a therapeutic target and prompt the development of Raf inhibitors without detrimental side effects. Given the importance of C-Raf during tumor initiation, such agents may be useful not only for the treatment of advanced cancer but also as chemopreventative agents.
Previously, B-Raf was considered the main activator of the MAPK pathway as deletion of B-Raf but not A-Raf or C-Raf abrogated ERK phosphorylation in several cell types (23). However, our data show that in the context of K-RasG12D expressing mutant epithelial cells, C-Raf plays a more prominent role than B-Raf. Interestingly, two other reports implicated C-Raf as the major Raf member downstream of oncogenic Ras in different cancer types. In melanoma cells, oncogenic N-Ras signals to MEK/ERK via C-Raf (25). Moreover, C-Raf was required for the development and maintenance of squamous cell carcinomas induced by either an activated SOS transgene or DMBA/TPA carcinogen treatment (17). Thus, in several oncogenic contexts C-Raf appears to be the most critical Raf family member, providing an attractive target to inhibit during chemoprevention and intervention studies in patients.
EXPERIMENTAL PROCEDURES
Mouse strains and intranasal Ad-Cre instillation
Mice were maintained according to CRI, CRUK, and UK Home Office regulations. Conditional knock-out strains of B-Raf and C-Raf and LSL-K-RasG12D mice were described previously (12, 15, 16). Mice were interbred and all mice analyzed were on the same mixed 129/SvHsd × 129/SvJae genetic background. Infection of lungs with ~5×107 PFU Ad-Cre (University of Iowa) was performed as described (12).
Histopathology and Immunohistochemistry
Mice were sacrificed at indicated time points, lungs inflated with and fixed in 4% formalin and embedded in paraffin. Sectioning, H&E stainings and immunohistochemistry for Ki-67, cleaved Caspase 3, phospho-ERK and GFP were performed by the CRI Histopathology core. For disease quantification H&E stained lungs were assessed using Image J software.
Laser capture microdissection and PCR analysis
Tumors were dissected from H&E stained lungs using a P.A.L.M Laser Capture System (Zeiss). DNA was extracted with a QIAamp DNA Micro Kit (Qiagen) and genotyping PCR performed as published (15, 16).
Cell lines
MEFs were derived from 13.5-14.5dpc embryos and propagated according to standard procedures. All experiments with MEFs were carried out between passages 3-6. Baby mouse kidney cells were derived from 4 day old mice as previously described (7) and propagated in DMEM/F12 media containing 5% Nu-serum IV, 5mg/ml glucose, 1.22mg/ml nicotinamide, 100ng/ml cholera toxin, 5% ITS+, 20ng/ml EGF, 25μg/ml bovine pituitary extract, 50nM T3, and 1μM Dexamethasone. To delete floxed Raf alleles and induce expression of conditional oncogenic K-Ras, cells were infected with Ad-Cre (~0.5-1.5×108 PFU, 150 MOI) or empty adenoviral control (Ad-Mock). Cells were incubated for three days post-infection to allow recombination to occur. For siRNA transfections, ON-TARGETplus SMARTpool siRNA and Dharmafect transfection reagent (both Dharmacon) were used according to the manufacturer’s instructions.
Proliferation and focus formation assays
For proliferation assays, 2.0×104 (MEFs) or 3.0×104 (BMKs) cells were plated in triplicates in 12 well plates and counted every other day for 5-7 days. For focus formation, 5×103 MEFs were plated onto a confluent layer of growth arrested MEFs in 6 well plates, grown for 21 days and stained with Giemsa.
Western blotting
Western blots were performed as previously described (29). The following primary antibodies were used: phospho-ERK1/2, ERK1/2(Cell Signaling), C-Raf, B-Raf, K-Ras, cyclin D1, actin (Santa Cruz Biotechnologies), Membranes were incubated with secondary HRP-antibodies (Jackson ImmunoResearch).
Statistical analysis
All experiments were repeated at least three times and done in triplicates. Statistical analysis was performed with the Mantel-Cox test or Student’s t-test, P<0.05 was accepted as significant. GraphPad Prism software was used for both analyses. Data are shown as mean, error bars represent the standard error. *, P<0.05; **, P<0.01; ***, P<0.001.
Supplementary Material
SIGNIFICANCE.
Ras is one of the most prevalent oncogenes in human cancer; however, it is considered ‘undruggable’ and thus increasing our understanding of the importance of Ras effectors, including the Raf-MEK-ERK pathway, will create novel avenues for therapeutic intervention.
ACKNOWLEDGEMENTS
We thank Leisa Johnson and Mariano Barbacid for sharing their results prior to publication and members of the Tuveson lab for helpful discussion. F.A.K. was supported by a Boehringer Ingelheim Fonds PhD student fellowship. D.A.T. is a group leader of Cancer Research UK, and this work was funded by the University of Cambridge and Cancer Research UK, The Li Ka Shing Foundation, and Hutchison Whampoa.
REFERENCES
- 1.Bos JL. ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–9. [PubMed] [Google Scholar]
- 2.Mitin N, Rossman KL, Der CJ. Signaling interplay in Ras superfamily function. Curr Biol. 2005;15:R563–74. doi: 10.1016/j.cub.2005.07.010. [DOI] [PubMed] [Google Scholar]
- 3.Basso AD, Kirschmeier P, Bishop WR. Lipid posttranslational modifications. Farnesyl transferase inhibitors. J Lipid Res. 2006;47:15–31. doi: 10.1194/jlr.R500012-JLR200. [DOI] [PubMed] [Google Scholar]
- 4.Lim KH, Baines AT, Fiordalisi JJ, Shipitsin M, Feig LA, Cox AD, et al. Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer Cell. 2005;7:533–45. doi: 10.1016/j.ccr.2005.04.030. [DOI] [PubMed] [Google Scholar]
- 5.Lim KH, Counter CM. Reduction in the requirement of oncogenic Ras signaling to activation of PI3K/AKT pathway during tumor maintenance. Cancer Cell. 2005;8:381–92. doi: 10.1016/j.ccr.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 6.Rangarajan A, Hong SJ, Gifford A, Weinberg RA. Species- and cell type-specific requirements for cellular transformation. Cancer Cell. 2004;6:171–83. doi: 10.1016/j.ccr.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 7.Kissil JL, Walmsley MJ, Hanlon L, Haigis KM, Kim CF Bender, Sweet-Cordero A, et al. Requirement for Rac1 in a K-ras induced lung cancer in the mouse. Cancer Res. 2007;67:8089–94. doi: 10.1158/0008-5472.CAN-07-2300. [DOI] [PubMed] [Google Scholar]
- 8.Gupta S, Ramjaun AR, Haiko P, Wang Y, Warne PH, Nicke B, et al. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell. 2007;129:957–68. doi: 10.1016/j.cell.2007.03.051. [DOI] [PubMed] [Google Scholar]
- 9.Karreth FA, Tuveson DA. Modelling oncogenic Ras/Raf signalling in the mouse. Curr Opin Genet Dev. 2009;19:4–11. doi: 10.1016/j.gde.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vicent S, Lopez-Picazo JM, Toledo G, Lozano MD, Torre W, Garcia-Corchon C, et al. ERK1/2 is activated in non-small-cell lung cancer and associated with advanced tumours. Br J Cancer. 2004;90:1047–52. doi: 10.1038/sj.bjc.6601644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–6. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jackson EL, Willis N, Mercer K, Bronson RT, Crowley D, Montoya R, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15:3243–8. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. http://www.cancer.gov/
- 14.Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, et al. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–87. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- 15.Chen AP, Ohno M, Giese KP, Kuhn R, Chen RL, Silva AJ. Forebrain-specific knockout of B-raf kinase leads to deficits in hippocampal long-term potentiation, learning, and memory. J Neurosci Res. 2006;83:28–38. doi: 10.1002/jnr.20703. [DOI] [PubMed] [Google Scholar]
- 16.Jesenberger V, Procyk KJ, Ruth J, Schreiber M, Theussl HC, Wagner EF, et al. Protective role of Raf-1 in Salmonella-induced macrophage apoptosis. J Exp Med. 2001;193:353–64. doi: 10.1084/jem.193.3.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ehrenreiter K, Kern F, Velamoor V, Meissl K, Galabova-Kovacs G, Sibilia M, et al. Raf-1 addiction in Ras-induced skin carcinogenesis. Cancer Cell. 2009;16:149–60. doi: 10.1016/j.ccr.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 18.Jackson EL, Olive KP, Tuveson DA, Bronson, Crowley D, Brown M, et al. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005;65:10280–8. doi: 10.1158/0008-5472.CAN-05-2193. [DOI] [PubMed] [Google Scholar]
- 19.Feldser DM, Kostova KK, Winslow MM, Taylor SE, Cashman C, Whittaker CA, et al. Stage-specific sensitivity to p53 restoration during lung cancer progression. Nature. 2010;468:572–5. doi: 10.1038/nature09535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Junttila MR, Karnezis AN, Garcia D, Madriles F, Kortlever RM, Rostker F, et al. Selective activation of p53-mediated tumour suppression in high-grade tumours. Nature. 2010;468:567–71. doi: 10.1038/nature09526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dougherty MK, Muller J, Ritt DA, Zhou M, Zhou XZ, Copeland TD, et al. Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell. 2005;17:215–24. doi: 10.1016/j.molcel.2004.11.055. [DOI] [PubMed] [Google Scholar]
- 22.Ritt DA, Monson DM, Specht SI, Morrison DK. Impact of feedback phosphorylation and Raf heterodimerization on normal and mutant B-Raf signaling. Mol Cell Biol. 2010;30:806–19. doi: 10.1128/MCB.00569-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wimmer R, Baccarini M. Partner exchange: protein-protein interactions in the Raf pathway. Trends Biochem Sci. 2010;35:660–8. doi: 10.1016/j.tibs.2010.06.001. [DOI] [PubMed] [Google Scholar]
- 24.Galabova-Kovacs G, Kolbus A, Matzen D, Meissl K, Piazzolla D, Rubiolo C, et al. ERK and beyond: insights from B-Raf and Raf-1 conditional knockouts. Cell Cycle. 2006;5:1514–8. doi: 10.4161/cc.5.14.2981. [DOI] [PubMed] [Google Scholar]
- 25.Dumaz N, Hayward R, Martin J, Ogilvie L, Hedley D, Curtin JA, et al. In melanoma, RAS mutations are accompanied by switching signaling from BRAF to CRAF and disrupted cyclic AMP signaling. Cancer Res. 2006;66:9483–91. doi: 10.1158/0008-5472.CAN-05-4227. [DOI] [PubMed] [Google Scholar]
- 26.Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R, et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–5. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- 27.Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–21. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–30. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karreth FA, DeNicola GM, Winter SP, Tuveson DA. C-Raf inhibits MAPK activation and transformation by B-Raf(V600E) Mol Cell. 2009;36:477–86. doi: 10.1016/j.molcel.2009.10.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.