

Abstract
Kinesin is believed to generate force for the movement of organelles in anterograde axonal transport. The identification of genes that encode kinesin-like proteins suggests that other motors may provide anterograde force instead of or in addition to kinesin. To gain insight into the specific functions of kinesin, the effects of mutations in the kinesin heavy chain gene (khc) on the physiology and ultrastructure of Drosophila larval neurons were studied. Mutations in khc impair both action potential propagation in axons and neurotransmitter release at nerve terminals but have no apparent effect on the concentration of synaptic vesicles in nerve terminal cytoplasm. Thus kinesin is required in vivo for normal neuronal function and may be active in the transport of ion channels and components of the synaptic release machinery to their appropriate cellular locations. Kinesin appears not to be required for the anterograde transport of synaptic vesicles or their components.
Kinesin is a ubiquitous mechanochemical adenosine triphosphatase (ATPase) that can move toward the plus ends of microtubules in vitro (1, 2). The native molecule is a heterotetramer consisting of two identical heavy chains and two light chains (2). The kinesin heavy chain, and more specifically the NH2-terminal head of the heavy chain, appears to contain all of the mechanochemical elements necessary for generating microtubule-based movements (3). The light chains and the COOH-terminal tail of the heavy chain are thought to be involved in binding kinesin to vesicles or other cargo destined for microtubule-based transport (2, 4).
On the basis of the polarity of kinesin’s movement and its presence in neural cells, it was originally suggested that kinesin might act as a motor for anterograde axonal transport (1). It since has been shown that vesicle movement in axoplasm (both anterograde and retrograde) can be inhibited in vitro by an antibody that binds to the kinesin heavy chain (5). It also has been shown that kinesin heavy chain (khc) mutations in Drosophila cause behavioral defects that suggest a requirement for kinesin in normal neuromuscular functions (6). Although these data provide a persuasive argument that kinesin acts as a motor for axonal transport, the argument remains indirect.
Kinesin is a member of a superfamily of kinesin-like proteins that share similar mechanochemical domains (7), and multiple members of the kinesin superfamily are co-expressed in neural and other tissues (8). Thus, if kinesin is a motor for anterograde axonal transport, it is likely to function in the context of a complex set of anterograde motors. Consequently, understanding anterograde axonal transport and other microtubule-based transport processes will require critical analyses of the specific functions of different motor proteins.
We have taken advantage of khc mutations to study the effects of impaired kinesin function on specific physiological functions of motor neurons in Drosophila larvae. Action potential propagation by segmental nerves and synaptic transmission at neuromuscular junctions were tested in both the second (A2) and sixth (A6) abdominal segments of control and khc mutant larvae (9) . Two hypomorphic khc alleles (khc5 and khc6) were used to construct both control and mutant genotypes (10) because these alleles are severe enough to cause a strong behavioral phenotype in larvae (6), yet mild enough to permit larval growth to an appropriate size for manipulations. Similar results were obtained with both alleles, and the data were pooled.
Figure 1 shows recordings of compound action potentials and corresponding excitatory junctional potentials (EJPs) evoked by stimulation of segmental nerves in control and khc mutant larvae (9). Recordings from segment A6 indicate that khc mutations caused a fourfold reduction in the mean amplitudes of both compound action potentials and EJPs (Table 1) (11). Similar differences were observed for segment A2, but the short segmental nerve that innervates A2 made the dual recordings and subsequent quantification of compound action potentials difficult. Our best recordings (greatest amplitudes) from A2 are shown in Fig. 1.
Fig. 1.
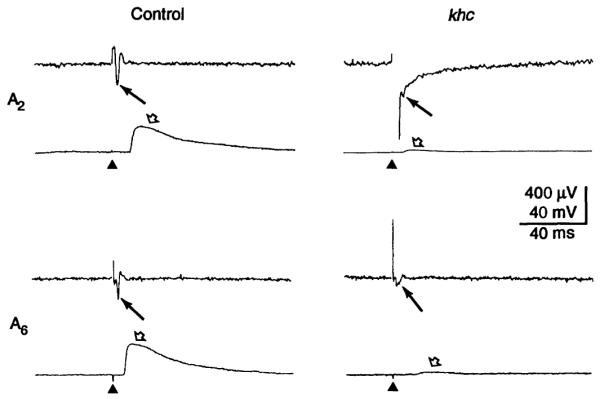
The effects of khc mutations on action potentials and synaptic transmission. Segmental nerves for A2 and A6 were stimulated (triangles) and recordings were made of compound action potential amplitude (filled arrows) and the corresponding EJP amplitudes (open arrows) from muscle 6 in control (left) and khc mutant (right) larvae. The upper panels show responses obtained from segment A2 and the lower panels those obtained from segment A6. The large stimulus artifact that precedes the action potential in mutants was a result of the strong stimulation required to evoke a measurable response. Stimulation with reversed polarity did not evoke any responses. Care was taken to evoke all-or-none EJP responses associated with a measurable delay of at least 2 ms, indicating that the response was elicited by an axonal action potential rather than by direct depolarization of the terminal. Stimulation parameters were 5 V and 0.1 ms for controls and 10 V and 0.1 ms for mutants.
Table 1.
Mean amplitudes of compound action potentials (CAP) and excitatory junctional potentials (EJP) in segment A6.
| Larvae | CAP (μV) | EJP (mV) |
|---|---|---|
| Control (n = 9) | 366 ± 130 | 23 ± 9 |
| khc mutant (n = 10) | 86 ± 46 | 5±3 |
The voltage required to evoke responses in khc mutant larvae was twice that for control larvae (legend to Fig. 1). Even with the increased voltage, only about one out of four nerve stimuli evoked a response. Control larvae never failed to respond under the same conditions. Because it usually was possible to elicit compound action potentials and corresponding EJPs, functional motor neurons were present in khc mutants. However, the frequent failures and the reduced amplitudes of compound action potentials in mutant nerves indicate that axonal function was impaired. A compound action potential is the sum of many simple action potentials in the individual sensory and motor axons within the segmental nerve. Thus, reduced compound action potential amplitude could reflect a reduction in the proportion of axons that fire in response to a given stimulation or a reduction in the action potential amplitudes produced by individual axons. In either case, khc mutations cause impaired action potential propagation. Electron micrographs of cross sections of larval nerves (not shown) indicate that khc mutations do not alter the number of axons in a nerve bundle.
The observation that EJP amplitudes in khc mutants are reduced relative to controls suggest that wild-type kinesin is required for proper synaptic transmission. To analyze whether the reduction in synaptic transmission resulted from defective release of neurotransmitter or in the postsynaptic response to neurotransmitter, we studied spontaneous miniature excitatory junctional currents (mEJCs) in control and khc mutant muscles (Fig. 2). Such currents are thought to be evoked in a muscle cell by the random spontaneous release of single quanta of a neurotransmitter from the presynaptic terminal, probably due to the exocytosis of individual synaptic vesicles (12). The mEJC amplitude serves as a measure of postsynaptic response to quantal stimulation by neurotransmitter. Under voltage-clamp conditions (13), the mean amplitude of mEJCs was not appreciably different in control and khc mutant muscles (Fig. 2B). Furthermore, comparable results were obtained at different muscle membrane potentials (VH = −60, −80, −100, and −120 mV) indicating by extrapolation that the reversal potential of the postsynaptic currents was not modified by the khc mutations. These results show that postsynaptic response to neurotransmitter was normal in khc mutants. Therefore, the effect of khc mutations on synaptic transmission must be due to impaired neuronal function.
Fig. 2.

Spontaneous miniature excitatory junctional currents recorded from control and khc mutant muscles. (A) Representative traces of mEJCs obtained from muscle 6 in the second (A2) and sixth (A6) abdominal segments. Histograms show mean and standard deviation values for the amplitude (B) and frequency (C) of mEJCs in control and khc mutant muscles. In (C) the values for segment A6 are significantly different as determined by the Student’s test (P < 0.01). The number of animals tested was five controls and five mutants for segment A2 and six controls and six mutants for segment A6. Data were collected for at least 30 s from each muscle. To obtain good resolution in mEJC recordings, the holding potential was ~100 mV. The external calcium concentration was 0.15 mM.
Although the amplitude of mEJCs was not affected by khc mutations, in segment A6 the frequency of mEJCs was depressed fourfold relative to that of controls in both hypertonic (Fig. 2C) (9) and normal saline solutions (not shown). These results suggest that khc mutations affect the function of the axon terminal itself, at least in the more posterior segment. An occurrence versus amplitude histogram was prepared for mEJCs recorded from segment A6 of mutant and wild-type larvae to address the possibility that the reduction in mEJC frequency resulted from the absence of a distinct subpopulation of synaptic vesicles. The amplitude histograms from khc mutants and controls were not distinguishable (not shown). Thus it is unlikely that kinesin is responsible for the preferential transport of a particular class of synaptic vesicles to motor terminals.
The analysis of EJP amplitudes after nerve stimulation (Fig. 1) revealed that khc mutations reduce the amount of evoked neurotransmitter release from motor neurons. This result could be either a direct consequence of impaired presynaptic terminal function or an indirect consequence of impaired action potential propagation in motor axons. To evaluate the function of presynaptic terminals in the absence of axonal conduction, excitatory junctional currents (EJCs) evoked by direct electrotonic stimulation of the presynaptic terminal were measured (Fig. 3) (14). The mean amplitude of EJCs evoked by direct stimulation in segment A2 of controls was 159 ± 35 nA (n = 3 larvae). Under similar conditions the mean amplitude in khc mutants was 61 ± 20 nA (n = 4 larvae). The reduction in amplitude in khc mutants was more extreme in segment A6: 118 ± 27 nA for controls (n = 7 larvae) and 25 ± 16 nA for khc mutants (n = 6 larvae). These observations show that khc mutations inhibit the function of presynaptic terminals in both anterior and posterior segments.
Fig. 3.
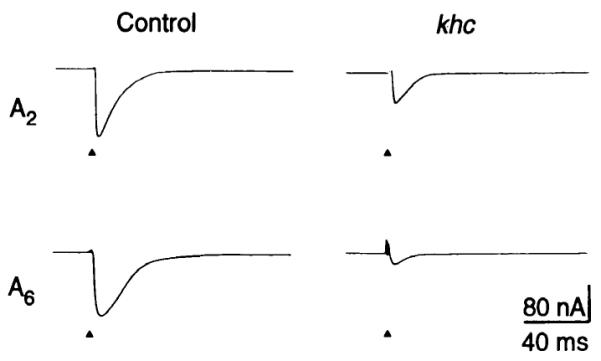
The effect of khc mutations on excitatory junctional currents evoked by direct electrotonic stimulation of the presynaptic terminal. Terminals were stimulated (triangles) and recordings of EJCs were taken from muscle 6 of segments A2 and A6 of control and khc mutant larvae. The bathing solution contained 1 μM tetrodotoxin. Stimulation parameters were 5 V and 1 ms.
The preceding observations provide direct evidence that kinesin is required for proper neuronal function. In addition, comparison of results from segments A2 and A6 suggest that the effect of impaired kinesin function on motor terminals is more severe in posterior segments. Motor terminals in posterior segments are served by relatively long axons, and thus there is a correlation between phenotype severity and axon length. This correlation supports the hypothesis that kinesin functions in vivo as a motor for axonal transport.
The axonal components transported by kinesin have not yet been identified. Hall and Hedgecock (15) have reported a severe reduction in synaptic vesicle concentration in nerve terminals of nematodes carrying mutations in unc-104, a Caenorhabditis elegans gene that encodes a kinesin-like protein (16). This suggests that UNC-104 protein is responsible for the anterograde transport of synaptic vesicle components (15). A similar role for kinesin might explain the impaired transmitter release reported here for khc mutants.
To determine if the defects in transmitter release in khc mutants were due to reduced transport of synaptic vesicle components, we examined nerve terminals by electron microscopy (17). In khc mutant nerve terminals corresponding to those assayed by electrophysiology, synaptic vesicles appeared to be abundant (Fig. 4). Morphometric analysis confirmed that the concentrations of synaptic vesicles in the terminals of khc and control larvae were not different (Table 2). These results, bolstered by our observations that the frequency of spontaneous transmitter release in khc mutants was normal in segment A2 (Fig. 2) while the evoked transmitter release was substantially reduced (Fig. 3), suggests defects in the mechanism of transmitter release. Kinesin must be responsible for the transport of material other than synaptic vesicle components. Presumably the components of synaptic vesicles in Drosophila are transported by a kinesin-like protein that is similar to UNC-104.
Fig. 4.

A comparison of the ultrastructure of nerve terminals from wild-type and khc mutant larvae. Synaptic vesicles (arrows), mitochondria (m), and dense bodies (arrowheads) are noted in sections of boutons from junction of muscles 6 and 7 in segment A6 of wild-type (A) and khc mutant (B) larvae. Scale bar, 0.2 μm.
Table 2.
The number of synaptic vesicles (SVs) per cubic micrometer of cytoplasm estimated from electron micrographs of sections of boutons on muscles 6 and 7.
| Animal | Segment | Sections | SVs/μm3 |
|---|---|---|---|
| Mutant | |||
| 1 | A2 | 13 | 1440 |
| A6 | 19 | 1750 | |
| 2 | A6 | 12 | 2280 |
| 3 | A6 | 23 | 2390 |
| Control | |||
| 1 | A2 | 32 | 2100 |
| A6 | 12 | 1320 | |
| 2 | A6 | 9 | 2260 |
| 3 | A6 | 26 | 1790 |
Both of the defects of khc mutant neurons—faulty action potential propagation and impaired neurotransmitter release—could be explained by reduced anterograde transport of ion channels. Voltage-gated sodium and potassium channels are likely to be carried in the membranes of vesicles that are transported along microtubules to points of insertion into the axonal plasma membrane (18). Kinesin could be responsible for transporting such channel-bearing vesicles. Impaired kinesin function would then lead to reduced sodium and potassium channel density in the axonal membrane and consequently to a reduced capacity to propagate action potentials. Similarly, impaired kinesin function could lead to reduced calcium channel density in terminal membranes, causing impaired neurotransmitter release. However, reduced anterograde transport of other terminal components could also cause impaired transmitter release. These include presynaptic membrane components responsible for forming release sites and cytoplasmic proteins that are active in preparing synaptic vesicles for productive association with release sites.
Acknowledgments
We thank B. Diaz, E. Raff, and S. Strome for critical reading of this manuscript. Supported by grants from the National Institutes of Health (W.M.S. and B.G.) the Markey Charitable Trust, and by an award from the McKnight Foundation (B.G.). M.G. was partially supported by the Foundation pour la Recherche Medicale. K.M. was supported by a grant from the National Institutes of Health to the Boulder Laboratory for Three Dimensional Fine Structure (J. R. Mcintosh).
Contributor Information
M. Gho, Laboratory of Genetics, University of Wisconsin, Madison, WI 53706..
K. McDonald, Department of Molecular, Cellular, and Developmental Biology, University of Colorado, Boulder, CO 80309.
B. Ganetzky, Laboratory of Genetics, University of Wisconsin, Madison, WI 53706.
W. M. Saxton, Department of Biology, Indiana University, Bloomington, IN 47405.
REFERENCES AND NOTES
- 1.Vale RD, Reese TS, Sheetz MP. Cell. 1985;42:39. doi: 10.1016/s0092-8674(85)80099-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; Vale RD, et al. ibid. 1985;43:623. [Google Scholar]
- 2.Vallee RB, Shpetner HS. Annu. Rev. Biochem. 1990;59:909. doi: 10.1146/annurev.bi.59.070190.004401. [DOI] [PubMed] [Google Scholar]; Brady ST. Neuron. 1991;7:521. doi: 10.1016/0896-6273(91)90365-7. [DOI] [PubMed] [Google Scholar]
- 3.Yang JT, Saxton WM, Stewart RJ, Raff EC, Goldstein LSB. Science. 1990;249:42. doi: 10.1126/science.2142332. [DOI] [PubMed] [Google Scholar]
- 4.Hollenbeck PJ, Swanson JA. Nature. 1990;346:864. doi: 10.1038/346864a0. [DOI] [PubMed] [Google Scholar]; Gyoeva FK, Gelfand VI. ibid. 1991;353:445. doi: 10.1038/353445a0. [DOI] [PubMed] [Google Scholar]
- 5.Brady ST, Pfister KK, Bloom GS. Proc. Natl. Acad. Sci. U.S.A. 1990;87:1061. doi: 10.1073/pnas.87.3.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saxton WM, Hicks J, Goldstein LSB, Raff EC. Cell. 1991;64:1093. doi: 10.1016/0092-8674(91)90264-y. [DOI] [PubMed] [Google Scholar]
- 7.Vale RD, Goldstein LSB. ibid. 1990;60:883. doi: 10.1016/0092-8674(90)90334-b. [DOI] [PubMed] [Google Scholar]; Goldstein LSB. Trends Cell Bioi. 1991;1:93. doi: 10.1016/0962-8924(91)90036-9. [DOI] [PubMed] [Google Scholar]
- 8.Stewart RJ, Pesavento PA, Woerpel DN, Goldstein LSB. Proc. Natl. Acad. Sci. U.S.A. 1991;88:8470. doi: 10.1073/pnas.88.19.8470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conventional intracellular voltage and two-microelectrode voltage-clamp recordings were made from body wall muscle 6 [for nomenclature see Johansen J, Halpern ME, Johansen KM, Keshishian H. J. Neurosci. 1989;9:710. doi: 10.1523/JNEUROSCI.09-02-00710.1989.] of third instar larvae that were dissected as described by Jan LY, Jan YN. J. Physiol. (London) 1976;262:189. doi: 10.1113/jphysiol.1976.sp011592.] and maintained in hypertonic saline to prevent muscle contraction (129 mM NaCl, 2 mM KCl, 4 mM MgCl2, 5.4 mM CaCl2, 5 mM Hepes, pH 7.1, and 353 nM sucrose). Normal saline (36 mM sucrose) was also used in some experiments. Microelectrodes were filed with 3 M KCl. During conventional voltage recordings, membrane potential was held at −60 mV by using a bridge circuit unless otherwise indicated. The bridge was continually checked by hyperpolarizing current pulses. Segmental nerves were stimulated via a suction electrode at 0.3 Hz. Compound action potentials were recorded extracellularly by a second suction electrode placed midway between the central nervous system and the appropriate segment.
- 10.Hemizygous mutant genotypes (khcm/Df(2R)Jp6) were constructed by using either khc5 and khc6 and a deletion that removes the khc gene (Df2R)Jp6). Control larvae were of two types: (i) those with the second chromosome genotype khcm/Df(2R)Jp6 but carrying a transposon that includes the wild-type khc gene (P-khc+) on the third chromosome or (ii) those with the second chromosome genotype khcm/+. Results from these two different types of control animals were indistinguishable and were pooled.
- 11.The amplitude of a compound action potential was measured by drawing a line between the two positive peaks that flanked the negative one and measuring the vertical distance between that line and the negative peak.
- 12.Heuser JE, et al. J. Cell Biol. 1979;81:275. doi: 10.1083/jcb.81.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.The measurement of synaptic currents with the voltage-clamp technique avoided the nonlinear summation that occurs when synaptic potential is used as a measurement of synaptic activity [Augustine GJ, Charlton MP, Smith SJ. J. Physiol. (London) 1985;367:163. doi: 10.1113/jphysiol.1985.sp015819.]. Also, the voltage clamp permitted comparison of data from different cells independent of input membrane resistance.
- 14.Direct (electrotonic) stimulation of the presynaptic terminal was accomplished by adding tetrodotoxin (1 μM) to the bathing solution to block axonal action potential propagation. The segmental nerve was then cut, drawn into the suction electrode and stimulated as close to the terminal as possible [Wu C-F, et al. Proc. Natl. Acad. Sci. U.S.A. 1978;75:4047. doi: 10.1073/pnas.75.8.4047.]. A series of stimuli of increasing voltage was used to determine the plateau level for transmitter release, and suprathreshold stimulation was then used for each test.
- 15.Hall DH, Hedgecock EM. Cell. 1991;65:837. doi: 10.1016/0092-8674(91)90391-b. [DOI] [PubMed] [Google Scholar]
- 16.Otsuka AJ, et al. Neuron. 1991;6:113. doi: 10.1016/0896-6273(91)90126-k. [DOI] [PubMed] [Google Scholar]
- 17.Larvae were dissected in hypertonic saline as described in (9), fixed in 2% glutaraldehyde, 100 mM Na cacodylate (pH 7.2) for 1 hour at 25°C, and post-fixed, stained, and embedded as described [McDonald K. J. Ultrastruct. Res. 1984;86:107. doi: 10.1016/s0022-5320(84)80051-9.]. Transverse serial 100-nm sections were cut from appropriate regions, post-stained with uranium and lead, and photographed at x21,000 on a Phillips CM10 electron microscope operating at 80 or 100 kV (K. McDonald, ibid.). Micrographs of sections were analyzed by eye for the number of small (30 to 100 nm in diameter) electron lucent vesicles present in bouton profiles. The area of each bouton profile was then determined with computer digitization of the circumference, and volume was calculated by multiplication by the section thickness. Microscopy and analysis were performed by different people. Samples and micrographs were coded to avoid bias until analysis was complete.
- 18.Brismar T, Gilly WF. Proc. Natl. Acad. Sci. U.S.A. 1987;84:1459. doi: 10.1073/pnas.84.5.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wonderlin WF, French RJ. ibid. 1991;88:4391. doi: 10.1073/pnas.88.10.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]