

Abstract
Cinnamaldehyde derivatives were synthesized in good to excellent yields in one step by a mild and selective, base-free palladium(II)-catalyzed oxidative Heck reaction starting from acrolein and various arylboronic acids. Prepared α,β-unsaturated aldehydes were used for synthesis of novel α-aryl substituted fosmidomycin analogues, which were evaluated for their inhibition of Mycobacterium tuberculosis 1-deoxy-d-xylulose 5-phosphate reductoisomerase. IC50 values between 0.8 and 27.3 μM were measured. The best compound showed activity comparable to that of the most potent previously reported α-aryl substituted fosmidomycin-class inhibitor.
Introduction
The palladium(II)-mediated oxidative Heck reaction with an organoborane substrate was first reported by Dieck and Heck in 1975,(1) but it was not until the development of a catalytic protocol(2) that this reaction began to receive more attention.(3) Initially the Cu(OAc)2 reoxidant(4) was used to regenerate Pd(II) from Pd(0) but could in 2003 be replaced by molecular oxygen,(5) avoiding the generation of stoichiometric amounts of heavy metal salts. In 2004 the ligand-modulated oxidative Heck reaction with arylboronic acids was introduced, in which the 2,9-dimethyl-1,10-phenanthroline (dmphen) ligand facilitated palladium reoxidation, catalytic stability, and control of the regioselectivity with electron-rich olefins.6,7 The reaction conditions became even milder when the base-free reaction using boronic acids was discovered.(8) Some recent developments involve oxygen and base-free reactions without external oxidant(9) and the identification of new nonphenanthroline type ligands.(10)
α,β-Unsaturated aldehydes are important starting materials in various synthetic applications.11,12 Cinnamaldehydes are commonly synthesized in one or more steps by the Wittig reaction(13) or crossed aldol condensation,(13) but various additional methods can be employed, such as Horner–Wadsworth–Emmons reaction,14,15 Peterson reaction,(16) oxidation of primary allylic alcohols,(13) and reduction of carboxylic acid derivatives.(13) The use of a palladium-catalyzed reaction with aryl halides and acetal protected acrolein, with subsequent acetal deprotection under acidic conditions, is another convenient possibility to obtain cinnamaldehyde derivatives.(17) A major drawback of many of the methods mentioned is the harsh reaction conditions. In contrast, the oxidative Heck reaction employs very mild, base-free conditions at room temperature.8,18 The readily available, low toxicity, and easily handled starting materials in the form of boronic acids,(19) used together with various olefins, provides an excellent framework for the synthesis of α,β-unsaturated aromatic derivatives. The use of acrolein as the olefin has been troublesome in the base-requiring palladium(0)-catalyzed Heck–Mizoroki reaction at elevated temperatures, providing low yields due to competing polymerization processes.20,21 Thus only a limited number of palladium(0)-catalyzed Heck–Mizoroki reactions with acrolein22−30 have been reported. The use of acrolein in an oxidative Heck is limited,(18) but the palladium(II)-catalyzed Heck coupling of the related methyl vinyl ketone with various boronic acids has been reported with yields between 50% and 88%.4,8,10,31
Tuberculosis (TB) is still one of the most serious infectious diseases, with 9.4 million new cases and almost 1.7 million deaths in the year 2009.(32) The lengthy and complicated treatment and the emergence of multidrug-resistant strains make the need for new drugs acting on new targets urgent. DXR (EC 1.1.1.267) is the second enzyme in the nonmevalonate pathway that is present in most eubacteria, including Mycobacterium tuberculosis (Mt), many parasites, and the plastids of plants.(33) It catalyzes the conversion of 1-deoxy-d-xylulose 5-phosphate (DOXP) to 2-C-methyl-d-erythrose 4-phosphate (MEP), which is used for the biosynthesis of isopentyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP), two basic precursors for the essential isoprenoids. All enzymes in the non-mevalonate pathway are attractive drug targets, since humans make use of the mevalonate pathway for the synthesis of isoprenoids instead.(34) DXR has been established as a drug target in the malaria parasite in clinical trials35−37 with the DXR inhibitor fosmidomycin (1, Figure 1).38,39 Together with its acetyl derivative FR-900098(40) (2, Figure 1), fosmidomycin acts as a potent inhibitor of Plasmodium falciparum DXR in vitro and in vivo in P. vinckei infected mice.(41) Despite the good inhibition of DXR, fosmidomycin suffers from poor pharmacokinetic properties42,43 and is inactive on Mt at the whole cell level due to poor uptake.44,45 Many attempts to improve in vitro and in vivo activity of fosmidomycin have been made,12,46−55 and some of the most successful DXR inhibitors have been compounds with an aryl substituent in the α-position relative to the phosphonate group (3, Figure 1).(12) Recently, three X-ray structures of Escherichia coli DXR in complex with inhibitors comprising only the phosphonate group and the aryl substituent were published.(56)
Figure 1.

Structures of known MtDXR inhibitors.
Herein the development and scope of the oxidative Heck reaction with acrolein or methyl vinyl ketone as olefins and arylboronic acids as arylating agents are reported. Five of the resulting cinnamaldehyde derivatives were used for the synthesis of 11 fosmidomycin analogues to further explore α-aryl substitutions, which were evaluated for inhibition of MtDXR.
Results and Discussion
In the preparation of novel α-aryl substituted fosmidomycin analogues we aimed to explore the effect of introducing biaryl substituents and other aromatic rings, as compared to the phenyl and thiophene groups investigated previously.12,52 It has recently been shown that biaryl and bicyclic aromatic groups in this position can be accommodated by the E. coli enzyme.56,57 It is reasonable to assume that MtDXR can also accommodate such substituents. Recently, the complex structure of MtDXR with 3 (2YIG, 3RAS)55,57 and its formyl analogue (2Y1D)(55) were published, showing that the 3,4-dichlorophenyl ring binds in a large solvent-exposed site. To investigate this area further a series of α-aryl analogues incorporating heteroraomatic and aliphatic rings that varied in lipophilicity were prepared. Different fused heterocycles such as naphthalene and 2-benzofuran or biaryl rings were considered. The Clog P value of the selected substituents varied between 1.5 and 4.5. The synthesis of α-aryl substituted fosmidomycin analogues starts from the appropriately substituted cinnamaldehyde.(12)
An initial optimization study was conducted to identify suitable reaction conditions for the synthesis of the functionalized cinnamaldehydes from acrolein and p-tolylboronic acid (Table 1). Two different palladium(II) catalysts, with a phenanthroline (8a) or a phosphine ligand (8b), p-benzoquinone (p-bzq, 9) versus air as reoxidant, and different solvents were investigated, as well as a microwave protocol. The response was measured by GC–MS as the peak area of product divided by the internal standard peak, multiplied by the internal standard concentration divided by the concentration of the yield-determining reactant according to eq 1.
 |
Table 1. Optimization of the Reaction Conditions with Acroleina.

| entry | catalytic mixb | solvent | time | temp | GC–MS responsec | ratio 4a:5j |
|---|---|---|---|---|---|---|
| 1 | 7a/8a/9 | DMF | 24 h | rt | 80 | 1:2 |
| 2 | 7a/8a/9 | MeCN | 24 h | rt | 85 | 1:2 |
| 3 | 7a/8a/9 | ethanol | 24 h | rt | <1 | 1:2 |
| 4 | 7b/8a/9 | MeCN | 24 h | rt | 77 | 1:2 |
| 5 | 7a/8b/9 | MeCN | 24 h | rt | <1 | 1:2 |
| 6 | 7a/8a/air | MeCN | 24 h | rt | 81 | 1:2 |
| 7 | 7a/8a/9 | MeCN | 0.5 h | 100 °C | 65 | 1:2 |
| 8 | 7a/8a/9 | MeCN | 24 h | rt | 66 | 2:1 |
| 9 | 7a/8a/9 | MeCN | 24 h | rt | 60 | 5:1 |
All reactions were carried out with acrolein (4a) and p-tolylboronic acid (5j), on a 1 mmol scale with Pd(II) catalyst (0.02 mmol), ligand (0.024 mmol), reoxidant (p-bzq, 1 mmol or air), and solvent (2 mL) for the time and temperature indicated.
Combination of Pd(II) catalyst, ligand, and reoxidant, consisting of either Pd(OAc)2 (7a) or Pd(OCOCF3)2 (7b), dmphen (8a) or dppp (8b), and p-bzq (9) or air as indicated.
Naphthalene was added to each reaction and the GC–MS response was calculated according to (peak area of 6j/naphthalene peak area) × (cnaphthalene/c4a or 5j) × 100. The amount of homocoupled product was <5%. No double arylated Heck product was detected.
Either air or p-bzq could be used as reoxidant, and DMF could be replaced by acetonitrile; all provided equally successful reaction conditions (Table 1, entries 1, 2, and 6). Pd(OCOCF3)2 was not superior to Pd(OAc)2, and the microwave protocol indicated a lower yield in agreement with previous results for the reaction at room temperature or with microwave heating (Table 1, entries 4 and 7).(18) Use of 1,3-bis(diphenylphosphino)propane (dppp, Table 1, entry 5) or ethanol (Table 1, entry 3) was associated with poor yields. Due to the volatile nature of the olefin employed and the long reaction time (24 h), p-bzq was generally used as a reoxidant instead of air, and thus the reaction vessel could be capped. The use of p-bzq also decreased phenol formation resulting from the oxidation of the boronic acid by hydrogen peroxide(58) that occurs in the catalytic process under air.(59) Phenol formation was especially prominent in the case of 2-benzofuranyl boronic acid. Further, traces of the expected homocoupled bitolyl byproduct could be observed in all seven test reactions.8,60 Acetonitrile was selected over DMF due to its lower toxicity. Attempts to invert the reaction stoichiometry did not improve the outcome (Table 1, entries 8 and 9). In summary, the best reaction conditions were essentially the same as those previously used (Table 1, entry 2).(18)
With the selected standard conditions, an investigation of the scope and limitations of different boronic acids was performed (Table 2). As expected, terminal arylation with E-selectivity was evident for all the cinnamaldehyde derivatives synthesized.5,9 In the set of boronic acids examined, variations in the electronic properties of the aromatic system did not have a significant effect on the yields. Further, the sterically hindered ortho-substituted 5g gave cinnamaldehyde 6g in high yield (92%, Table 2, entry 7).
Table 2. Oxidative Heck Reactions with Acrolein (4a) or Methyl Vinyl Ketone (4b) and Different Boronic Acidsa.


Reaction conditions: closed vessel charged with olefin (1.0 mmol), arylboronic acid (2.0 mmol), p-bzq (1.0 mmol), Pd(OAc)2 (0.02 mmol), dmphen (0.024 mmol), and acetonitrile (7.5 mL) stirred at room temperature for 24–48 h.
Isolated yield with purity ≥95% (GC–MS). Yields were calculated according to 100% acrolein.
Reaction conditions: sealed microwave vessel charged with olefin (8.58 mmol), arylboronic acid (0.613 mmol), p-bzq (0.333 mmol), Pd(OAc)2 (0.0062 mmol), dmphen (0.0077 mmol), and acetonitrile (2 mL) with microwave heating at 100 °C for 30 min.
DMF as a solvent instead of acetonitrile.
Good chemoselectivity was also found, since the halogenated boronic acids (5d and 5k, Table 2, entries 4 and 11) afforded the corresponding cinnamaldehydes (6d and 6k) without side-product formation from a competing Pd(0)-catalyzed Heck reaction or dehalogenation. Heteroaromaticboronic acids (5h and 5m) furnished products in 52% and 92% yield, respectively (Table 2, entries 8 and 13), and the sulfur-containing 5m did not disturb the efficiency of the catalytic system. The acid-labile Boc group of 5i remained unaffected (Table 2, entry 9). For the boronic acids with larger aromatic groups (5f, 5h, and 5l, Table 2, entries 6, 8 and 12), DMF was used as a solvent due to poor solubility in acetonitrile. Attempts with several six-membered nitrogen-containing heterocyclicboronic acids were unsuccessful.(61)
Heterocyclic 5e did not provide a useful yield under the standard reaction conditions, and a large amount of homocoupled product was detected (Table 2, entry 5). Since we were interested in a fosmidomycin analogue with 2-benzofuran as an aryl substituent, the reaction conditions were altered to produce 6e in an acceptable yield of 43%. To achieve this outcome, the olefin:boronic acid ratio was inverted to provide acrolein in a 14-fold excess, and the reaction was performed with microwave heating at 100 °C for 30 min.
For the final fosmidomycin analogues (13a–k) various aryl substituents were selected to span a range in lipophilicity, considering the size of the biaryl and fused ring substituents employed by Deng et al.56,57 and the solvent exposed area from the published X-ray structures.55,57 The fosmidomycin analogues 13a–k (Table 3) were prepared, essentially as previously described,(12) from the cinnamaldehydes (6a–e) synthesized in the oxidative Heck reaction (Scheme 1). The phosphonate functionality was introduced by a 1,4-addition of triethylphosphite in the presence of phenol, which gave the acetal intermediate. After acetal deprotection, the aldehyde (10a–e) was isolated in 26–76% yield. The benzyloxyamine was introduced by a reductive amination, and the intermediates were further reacted with acetyl chloride to give 11a–e in 39–99% yield over three steps.
Table 3. Activity of α-Aryl Substituted Fosmidomycin Analogues as MtDXR Inhibitors.


Scheme 1.

The linear synthetic scheme could be shortened by using (E)-3-(4-bromophenyl)acrylaldehyde (6d) as a starting material since the bromo substituent was unaffected by the Pd(II)-catalyzed reaction. Diversification could then be smoothly conducted by microwave-assisted Pd(0)-catalyzed Suzuki reactions62,63 of 11d employing m-tolylboronic acid and five- or six-membered heterocycles to furnish compounds 11f–j in yields between 61% and 92% (Scheme 2). A Buchwald–Hartwig amination(64) with morpholine was employed in the synthesis of 11k. The basic reaction conditions yielded a mixture of 11k and deacetylated product. The product mixture was reacetylated using the reaction conditions from Scheme 1. Compound 11k was obtained in 33% yield from 11d, with no detected debenzylation (Scheme 3). Benzyl deprotection was carried out by classical catalytic hydrogenation to afford 12a–c, 12e–h, and 12j–k (61–96%). Deprotection of 11d and 11i was done with BCl3,(52) and 12d and 12i could be isolated in 76% and 49% yield, respectively. The phosphonate esters were cleaved with TMSBr to afford the final compounds 13a–k in 54–98% yield after purification on preparative HPLC (Schemes 1, 2, and 3).
Scheme 2.

Scheme 3.

The inhibitory capacity of 13a–k was evaluated in a spectrophotometric assay, in which the MtDXR-catalyzed NADPH-dependent rearrangement and reduction of DXP to form MEP is monitored at 340 nm. All compounds were found to inhibit MtDXR with IC50 values between 0.8 and 27.3 μM (Table 3). The best compound in the series was the 4-(pyridin-3-yl)phenyl functionalized analogue (13f, IC50 = 0.8 μM), which had activity comparable to that of the 3,4-dichloro substituted 3, IC50 = 0.7 μM (Figure 2). Overall there was no correlation between the calculated log P values of the substituents and the IC50 values. For example, the second most potent compound (13a), with a benzo[d][1,3]dioxolyl substituent, had an IC50 = 1.5 μM and the lowest calculated Clog P = 1.5, while the least active compound (13k) had the second lowest Clog P = 2.0.
Figure 2.
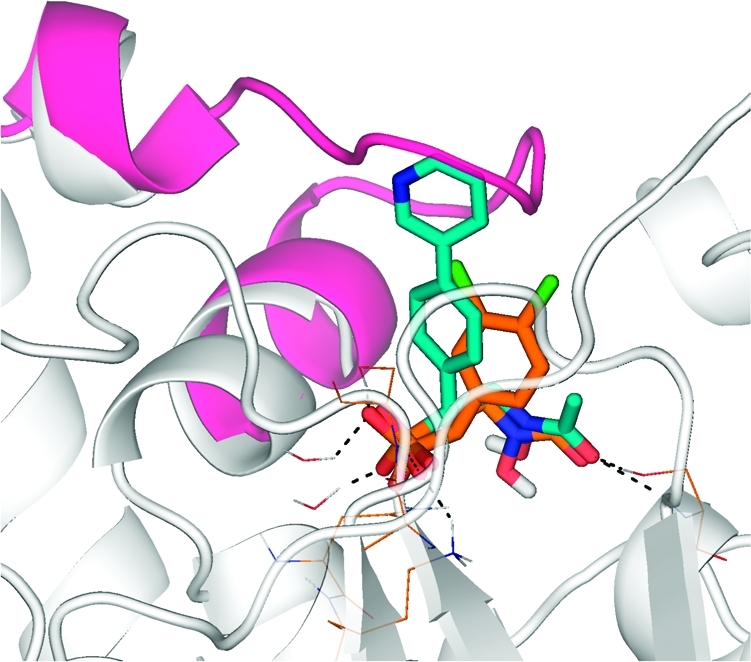
Compound 13f (turquoise carbon atoms) docked in the X-ray structure of MtDXR in complex with 3 (PDB code 2Y1G,(55) orange carbon atoms). Included in pink ribbon is the Gly198–Met208 flap from the 2JVC structure(65) representing MtDXR bound to 1.
The relative insensitivity of the inhibitory activity to the nature of the R1 group probably arises from the limited number of interactions this group makes with the protein once the Gly198–Met208 flap is displaced (Figure 2). However, given both the flexibility of the protein and the different conformation seen for the backbone of fosmidomycin compared to its derivatives,(55) predictions of the precise mode of binding the various compounds are difficult to make.
Conclusions
Acrolein has been employed as the olefin in the oxidative Heck reaction with arylboronic acids for the smooth synthesis of cinnamaldehydes to serve as starting materials for α-aryl substituted fosmidomycin analogues. Despite the volatile nature and the tendency of the olefin used to polymerize, the mild reaction conditions in the oxidative Heck furnished the synthesis of terminally arylated (E)-α,β-unsaturated aldehydes from acrolein and various boronic acids in yields between 43% and 92%. Diversity could thereafter be introduced in the preparation of biaryl fosmidomycin analogues through Suzuki cross-coupling reactions. The activity of the fosmidomycin analogues tested demonstrated that a variety of α-aromatic substituents can be tolerated by the DXR enzyme. The best new compound (13f, α = 4-(pyridin-3-yl)phenyl) had an IC50 value of 0.8 μM, an activity comparable to that of the most potent previously reported α-aryl substituted fosmidomycin derivative (3, α = 3,4-dichlorophenyl, IC50 = 0.7 μM).
Experimental Section
Inhibition Assay
Inhibition of MtDXR activity was measured in a spectrophotometric assay(65) by monitoring the NADPH-dependent rearrangement and reduction of DXP to form MEP, using the absorption of NADPH at 340 nm. Reactions had a final volume of 50 μL and contained 50 mM HEPES-NaOH pH 7.5, 100 mM NaCl, 1.5 mM MnCl2, 0.2 mM NADPH, 0.2 mM DXP, and 0.096 μM MtDXR, as well as inhibitory compound at various concentrations. Initial screening was performed with an inhibitor concentration of 100 μM. IC50 measurements were performed using six inhibitor concentrations ranging between 0.01 and 1000 μM. Reactions were initiated by adding DXP and followed simultaneously in a 96-well plate (UV-Star, Greiner) at 22 °C. Absorbance was measured every 5 s during a 250 s period. The slope of the linear phase of each reaction was used to calculate the initial velocity. This was compared to the velocity of the uninhibited reaction to calculate enzyme activity. Enzyme activities were plotted against the corresponding inhibitor concentration and data points were fitted to eq 2, where Hi is the estimated highest enzyme activity at zero inhibitor concentration, Lo is the estimated lowest enzyme activity at infinite inhibitor concentration, X is the concentration of inhibitor, and Y is the measured enzyme activity. IC50 values presented are the average of three independent experiments.
 |
Molecular Modeling
Docking calculations were done with Glide66,67 in SP mode. The protein (chain A of 2Y1G) was prepared using the protein preparation wizard implemented in Maestro(68) with default settings. All waters but 2262 and 2133, which are close to the phosphonate, were deleted. The gridbox was defined from 3 in the X-ray structure. Poses resembling the X-ray pose of 3 were selected.
General
Nuclear magnetic resonance (NMR) spectra were recorded on two instruments: 1H (at 400 MHz) and 13C (at 101 MHz). NMR chemical shifts were reported as δ (ppm) and referenced using the residual solvent signal (1H, CDCl3 at 7.26 ppm, CD3OD at 3.31 ppm; 13C, CDCl3 at 77.16 ppm, CD3OD at 49.00 ppm). Molecular mass (HR-ESI-MS) was determined on a mass spectrometer equipped with an electrospray ion source. GC–MS analyses were performed with a CP-SIL 8 CB Low Bleed (30 m × 0.25 mm) or a Factor Four VF 5 ms (30 m × 0.25 mm) capillary column using a 70–300 °C temperature gradient and EI ionization at 70 eV.
Analytical HPLC–MS was performed using a C18 column (50 × 4.6 mm) on an HPLC system (detection by UV-DAD) coupled with a quadrupole mass spectrometer (ESI-MS). Analytical UHPLC–MS was performed with an ion trap mass spectrometer and UV-DAD detection using a C18 column (50 × 3 mm). Acetonitrile or methanol in 0.05% aqueous formic acid was used as mobile phase at a flow rate of 4 mL/min. The final compounds (13a–k) were purified on a preparative HPLC, using an SB-C8 (21.2 × 150 mm) or Nucleodur C18 HTec (21.2 × 150 mm) column with UV detection at 220 nm. The compounds were eluted with acetonitrile in 0.1% aqueous trifluoroacetic acid at a flow rate of 5–15 mL/min and isolated after freeze-drying. Purity analysis of the final fosmidomycin analogues was done on an HPLC system equipped with two different columns, biphenyl (4.6 × 50 mm) and C18 (4.6 × 50 mm), with UV detection at 220 and 254 nm. Compounds were eluted with acetonitrile in 0.1% aqueous trifluoroacetic acid at a flow rate of 2 mL/min. Silica gel (Merck 60, 40–63 μm) was used for flash chromatography. Analytical thin layer chromatography was done using aluminum sheets precoated with silica gel (Merck, F254); detection was by UV (254 nm).
The microwave reactions were performed in a single-mode microwave reactor (Initiator, Biotage AB) producing controlled irradiation at 2450 MHz with a power of 0–300 W. The reaction temperature was determined using the built-in online IR-sensor.
All reagents were purchased from commercial suppliers and used without further purification. Dichloromethane (DCM) was freshly distilled from calcium hydride under nitrogen immediately before use. Compounds 6a,(69)6b,17,706c,(17)6d,766e,(71)6j,(17)6k,(17)6m,(72) and 6n(73) are known compounds; 6g, 6h, and 6i are new compounds. Compounds 6f(74) and 6l(75) are known, but there are no NMR data reported in the literature. All final compounds were ≥95% pure as determined by GC–MS, NMR or HPLC–UV. The fosmidomycin analogues (13a–k) decompose upon storage at −20 °C showing a loss of m/z 42 indicating a deacetylation according to ESI+ LC–MS.
Oxidative Heck Reaction at Room Temperature
Pd(OAc)2 (0.05 mmol, 0.020 equiv) and dmphen (0.06 mmol, 0.024 equiv) were dissolved in acetonitrile (2.5 mL), and the mixture was stirred for 30 min at room temperature. A 10 mL round bottomed flask was charged with acrolein (1 mmol, 1 equiv), p-bzq (1 mmol, 1 equiv), and arylboronic acid (2 mmol, 2 equiv). The catalyst–ligand mixture and more acetonitrile (5 mL) were added, the flask was sealed with a stopper and the reaction mixture was stirred at room temperature for 24–48 h. The reaction was evaporated on silica and purified by column chromatography on silica gel.
Oxidative Heck with Microwave Heating
Pd(OAc)2 (0.0062 mmol, 0.010 equiv) and dmphen (0.062 mmol, 0.010 equiv) were dissolved in acetonitrile (1 mL), and the mixture was stirred for 30 min at room temperature. A 2–5 mL microwave vial was charged with acrolein (9.3 mmol, 15 equiv), p-bzq (0.31 mmol, 0.5 equiv), and arylboronic acid (0.62 mmol, 1 equiv). The catalyst–ligand mixture and more acetonitrile (1 mL) were added, the vial was sealed, and the reaction mixture was irradiated to 100 °C for 30 min. The reaction was evaporated on silica and purified by column chromatography on silica gel.
(E)-3-(Benzo[d][1,3]dioxol-5-yl)acrylaldehyde (6a)
Reagents: Pd(OAc)2 (0.0119 g, 0.053 mmol), dmphen (0.0126 g, 0.061 mmol), acrolein (0.126 g, 2.25 mmol), p-bzq (0.270 g, 2.50 mmol), benzo[d][1,3]dioxol-5-ylboronic acid (0.997 g, 6.01 mmol), acetonitrile (7.5 mL). Time: 24 h. Eluent: pentane/diethylether, 5/1. 6a (0.305 g, 1.73 mmol) was isolated as a white solid in 77% yield. 1H NMR (400 MHz, CDCl3) δ 9.58 (d, J = 7.7 Hz, 1H), 7.32 (d, J = 15.8 Hz, 1H), 6.98–7.03 (m, 2H), 6.78–6.82 (m, 1H), 6.49 (dd, J = 7.9, 15.8 Hz, 1H), 5.98–5.99 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 193.5, 152.6, 150.5, 148.5, 128.5, 126.7, 125.3, 108.7, 106.7, 101.8.
(E)-3-(Naphthalen-2-yl)acrylaldehyde (6b)
Reagents: Pd(OAc)2 (0.094 g, 0.042 mmol), dmphen (0.0104 g, 0.050 mmol), acrolein (0.104 g, 1.86 mmol), p-bzq (0.224 g, 2.07 mmol), naphthalene-2-ylboronic acid (0.712 g, 4.14 mmol), acetonitrile (6 mL). Time: 24 h. Eluent: pentane/diethylether, 4/1. 6b (0.290 g, 1.59 mmol) was isolated as a pale yellow solid in 85% yield, mp 125–126 °C. 1H NMR (400 MHz, CDCl3) δ 9.73 (d, J = 7.6 Hz, 1H), 7.93 (s, 1H), 7.80–7.90 (m, 3H), 7.64 (dd, J = 1.5, 8.6 Hz, 1H), 7.49–7.60 (m, 3H), 6.80 (dd, J = 7.8, 16.0 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 193.8, 152.8, 134.7, 133.2, 131.6, 130.8, 129.0, 128.8, 128.7, 127.92, 127.87, 127.0, 123.6; HRMS (ESI+) calcd for C13H11O (M + H+), 183.0810; found, 183.0806.
(E)-3-([1,1′-Biphenyl]-4-yl)acrylaldehyde (6c)
Reagents: Pd(OAc)2 (0.0116 g, 0.052 mmol), dmphen (0.013 g, 0.062 mmol), acrolein (0.151 g, 2.70 mmol), p-bzq (0.276 g, 2.55 mmol), [1,1′-biphenyl]-4-ylboronic acid (1.059 g, 5.35 mmol), acetonitrile (7.5 mL). Time: 24 h. Eluent: pentane/diethylether, 4/1. 6c (0.430 g, 2.07 mmol) was isolated as a pale yellow solid in 77% yield, mp 121–123 °C. 1H NMR (400 MHz, CDCl3) δ 9.73 (d, J = 7.8 Hz, 1H), 7.60–7.72 (m, 6H), 7.45–7.55 (m, 3H), 7.38–7.45 (m, 1H), 6.76 (dd, J = 7.5, 16.0 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 193.8, 152.4, 144.0, 139.8, 132.9, 129.1, 129.0, 128.4, 128.2, 127.7, 127.1; HRMS (ESI+) calcd for C15H13O (M + H+), 209.0966; found, 209.0971.
(E)-3-(4-Bromophenyl)acrylaldehyde (6d)
Reagents: Pd(OAc)2 (0.138 g, 0.061 mmol), dmphen (0.0160 g, 0.077 mmol), acrolein (0.130 g, 2.33 mmol), p-bzq (0.299 g, 2.77 mmol), 4-bromophenylboronic acid (1.06 g, 5.29 mmol), acetonitrile (7.5 mL). Time: 24 h. Eluent: pentane/diethylether, 4/1. 6d (0.378 g, 1.79 mmol) was isolated as a pale yellow solid in 77% yield, mp 81–82 °C. 1H NMR (400 MHz, CDCl3) δ 9.65 (d, J = 7.6 Hz, 1H), 7.47–7.52 (m, 2H), 7.33–7.40 (m, 3H), 6.64 (dd, J = 7.6, 16.0 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 193.3, 151.0, 132.9, 132.3, 129.8, 128.9, 125.6; HRMS (ESI+) calcd for C9H8BrO (M + H+), 210.9759; found, 210.9763.
(E)-3-(Benzofuran-2-yl)acrylaldehyde (6e)
Prepared according to the oxidative Heck protocol with microwave heating. Reagents: Pd(OAc)2 (0.0014 g, 0.0062 mmol), dmphen (0.0016 g, 0.0077 mmol), acrolein (0.481 g, 8.58 mmol), p-bzq (0.0360 g, 0.333 mmol), 2-benzofuranylboronic acid (0.0992 g, 0.613 mmol), acetonitrile (2 mL). Eluent: isohexane/etylacetate, 4/1. 6e (0.045 g, 0.26 mmol) was isolated as a pale yellow solid in 43% yield, mp 64–65 °C. 1H NMR (400 MHz, CDCl3) δ 9.69 (d, J = 7.7 Hz, 1H), 7.60 (ddd, J = 0.7, 1.3, 7.9 Hz, 1H), 7.49 (qd, J = 0.9, 8.4 Hz, 1H), 7.36–7.42 (m, 1H), 7.31 (d, J = 15.9 Hz, 1H), 7.23–7.28 (m, 1H), 7.04–7.07 (m, 1H), 6.79 (ddd, J = 0.5, 7.8, 15.7 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 192.8, 156.0, 151.9, 137.9, 128.4, 128.3, 127.3, 123.7, 122.1, 113.0, 111.7; HRMS (ESI+) calcd for C11H9O2 (M + H+), 173.0603; found, 173.0600.
(E)-3-(4-Phenoxyphenyl)acrylaldehyde (6f)
Reagents: Pd(OAc)2 (0.0114 g, 0.051 mmol), dmphen (0.0128 g, 0.061 mmol), acrolein (0.144 g, 2.57 mmol), p-bzq (0.272 g, 2.52 mmol), 4-phenoxyphenylboronic acid (1.07 g, 4.98 mmol), DMF (7.5 mL). Time: 48 h. Eluent: pentane/diethylether, 4/1. 6f (0.426 g, 1.90 mmol) was isolated as a white solid in 74% yield, mp 72–73 °C. 1H NMR (400 MHz, CDCl3) δ 9.67 (d, J = 7.4 Hz, 1H), 7.50–7.57 (m, 2H), 7.35–7.47 (m, 3H), 7.15–7.22 (m, 1H), 7.04–7.10 (m, 2H), 6.99–7.04 (m, 2H), 6.64 (dd, J = 7.6, 15.8 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 193.7, 160.6, 155.8, 152.2, 130.4, 130.1, 128.7, 127.5, 124.5, 120.0, 118.4; HRMS (ESI+) calcd for C15H13O2 (M + H+), 225.0916; found, 225.0913.
(E)-3-(5-(tert-Butyl)-2-methoxyphenyl)acrylaldehyde (6g)
Reagents: Pd(OAc)2 (0.0117 g, 0.050 mmol), dmphen (0.0127 g, 0.061 mmol), acrolein (0.126 g, 2.25 mmol), p-bzq (0.301 g, 2.78 mmol), (5-(tert-butyl)-2-methoxyphenyl)boronic acid (0.997 g, 4.79 mmol), acetonitrile (7.5 mL). Time: 24 h. Eluent: pentane/diethylether, 4/1. 6g (0.451 g, 2.06 mmol) was isolated as a dark red oil in 92% yield. 1H NMR (400 MHz, CDCl3) δ 9.67 (d, J = 7.9 Hz, 1H), 7.82 (d, J = 16.0 Hz, 1H), 7.54 (d, J = 2.5 Hz, 1H), 7.43 (dd, J = 2.5, 8.7 Hz, 1H), 6.88 (d, J = 8.7 Hz, 1H), 6.82 (dd, J = 7.9, 16.0 Hz, 1H), 3.87 (s, 3H), 1.30 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 194.6, 156.3, 148.9, 143.5, 129.9, 128.9, 125.8, 122.2, 111.0, 55.6, 34.13, 31.37; HRMS (ESI+) calcd for C14H19O2 (M + H+), 219.1385; found, 219.1389.
(E)-3-(Dibenzo[b,d]furan-4-yl)acrylaldehyde (6h)
Reagents: Pd(OAc)2 (0.0127 g, 0.057 mmol), dmphen (0.0140 g, 0.067 mmol), acrolein (0.127 g, 2.26 mmol), p-bzq (0.266 g, 2.46 mmol), dibenzo[b,d]furan-3-ylboronic acid (1.489 g, 7.02 mmol), acetonitrile (2.5 mL), DMF (5 mL). Time: 48 h. Eluent: pentane/diethylether, 4/1. 6h (0.262 g, 1.18 mmol) was isolated as a pale yellow solid in 52% yield, mp 87–88 °C. 1H NMR (400 MHz, CDCl3) δ 9.74 (d, J = 7.8 Hz, 1H), 7.85–7.92 (m, 2H), 7.64 (d, J = 16.1 Hz, 1H), 7.53–7.57 (m, 1H), 7.43–7.49 (m, 2H), 7.32–7.37 (m, 1H), 7.27–7.32 (m, 1H), 7.21 (dd, J = 7.8, 16.1 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 194.3, 156.1, 154.2, 146.9, 131.3, 127.9, 127.8, 125.2, 123.4, 123.4 (as confirmed by HMBC and HSQC), 123.3, 123.1, 120.8, 119.2, 111.9; HRMS (ESI+) calcd for C15H11O2 (M + H+), 223.0759; found, 223.0763.
(E)-tert-Butyl (4-(3-Oxoprop-1-en-1-yl)phenyl)carbamate (6i)
Reagents: Pd(OAc)2 (0.0041, 0.018 mmol), dmphen (0.0051 g, 0.024 mmol), acrolein (0.049 g, 0.88 mmol), p-bzq (0.093 g, 0.86 mmol), 4-aminophenylboronic acid Boc protected (0.412 g, 1.74 mmol), acetonitrile (3 mL). Time: 25 h. Eluent: pentane/diethylether, 7/3. 6i (0.143 g, 0.576 mmol) was isolated as an orange solid in 66% yield, mp 180–181 °C. 1H NMR (400 MHz, CDCl3:CD3OD, 2:1) δ 9.55 (d, J = 7.9 Hz, 1H), 8.36 (br. s., 1H), 7.39–7.51 (m, 5H), 6.58 (dd, J = 7.9, 15.8 Hz, 1H), 1.48 (s, 9H); 13C NMR (101 MHz, CDCl3) δ 193.8, 152.6, 152.4, 141.5, 129.8, 128.7, 127.2, 118.5, 81.4, 28.4; HRMS (ESI+) calcd for C14H18NO3 (M + H+), 248.1287; found, 248.1290.
(E)-3-(p-Tolyl)acrylaldehyde (6j)
Reagents: Pd(OAc)2 (0.047 g, 0.021 mmol), dmphen (0.0054 g, 0.026 mmol), acrolein (0.052 g, 0.923 mmol), p-bzq (0.107 g, 0.990 mmol), p-tolylboronic acid (0.272 g, 2.00 mmol), acetonitrile (2 mL). Time: 17 h. Eluent: isohexane/ethyl acetate, 9/1. 6j (0.0786 g, 0.538 mmol) was isolated as a pale yellow solid in 58% yield, mp 39–41 °C. 1H NMR (400 MHz, CDCl3) δ 9.66 (d, J = 7.8 Hz, 1H), 7.38–7.47 (m, 3H), 7.22 (d, J = 8.2 Hz, 2H), 6.66 (dd, J = 7.8, 16.0 Hz, 1H), 2.38 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 193.9, 153.0, 142.1, 131.4, 130.0, 128.6, 127.8, 21.7; HRMS (ESI+) calcd for C10H11O (M + H+), 147.0810; found, 147.0806.
(E)-3-(4-Chlorophenyl)acrylaldehyde (6k)
Reagents: Pd(OAc)2 (0.0115 g, 0.051 mmol), dmphen (0.0138 g, 0.066 mmol), acrolein (0.125 g, 2.23 mmol), p-bzq (0.275 g, 2.54 mmol), 4-chlorophenylboronic acid (0.790 g, 5.05 mmol), acetonitrile (7.5 mL). Time: 24 h. Eluent: pentane/diethylether, 4/1. 6k (0.292 g, 1.75 mmol) was isolated as a pale yellow solid in 78% yield, mp 58–59 °C. 1H NMR (400 MHz, CDCl3) δ 9.66 (d, J = 7.6 Hz, 1H), 7.43–7.49 (m, 2H), 7.33–7.42 (m, 3H), 6.64 (dd, J = 7.6, 16.0 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 193.3, 151.0, 137.2, 132.5, 129.6, 129.4, 128.9; HRMS (ESI+) calcd for C9H8ClO (M + H+), 167.0264; found, 167.0266.
(E)-3-(4-Benzoylphenyl)acrylaldehyde (6l)
Reagents: Pd(OAc)2 (0.0117 g, 0.052 mmol), dmphen (0.0130 g, 0.062 mmol), acrolein (0.194 g, 3.46 mmol), p-bzq (0.278 g, 2.57 mmol), 4-benzoylphenylboronic acid (1.15 g, 5.08 mmol), DMF (7.5 mL). Time: 48 h. Eluent: pentane/diethylether, 4/1. 6l (0.610 g, 2.58 mmol) was isolated as a white solid in 75% yield, mp 175–176 °C. 1H NMR (400 MHz, CDCl3) δ 9.76 (d, J = 7.6 Hz, 1H), 7.82–7.88 (m, 2H), 7.77–7.82 (m, 2H), 7.65–7.70 (m, 2H), 7.58–7.64 (m, 1H), 7.46–7.56 (m, 3H), 6.79 (dd, J = 7.6, 16.0 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 195.8, 193.4, 150.9, 139.6, 137.6, 137.2, 132.9, 130.7, 130.4, 130.1, 128.5, 128.3; HRMS (ESI+) calcd for C16H13O2 (M + H+), 237.0916; found, 237.0923.
(E)-3-(Thiophen-3-yl)acrylaldehyde (6m)
Reagents: Pd(OAc)2 (0.124 g, 0.055 mmol), dmphen (0.0147 g, 0.071 mmol), acrolein (0.144 g, 2.57 mmol), p-bzq (0.272 g, 2.52 mmol), thiophen-3-ylboronic acid (0.648 g, 5.06 mmol), acetonitrile (7.5 mL). Time: 24 h. Eluent: pentane/diethylether, 4/1. 6m (0.272 g, 1.97 mmol) was isolated as a yellow oil in 92% yield. 1H NMR (400 MHz, CDCl3) δ 9.62 (d, J = 7.8 Hz, 1H), 7.60 (dd, J = 1.3, 2.9 Hz, 1H), 7.44 (d, J = 15.8 Hz, 1H), 7.35 (ddd, J = 0.5, 2.9, 5.0 Hz, 1H), 7.28–7.31 (m, 1H), 6.50 (dd, J = 7.8, 15.8 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 193.8, 145.8, 137.4, 129.6, 128.4, 127.5, 125.4; HRMS (ESI+) calcd for C7H7OS (M + H+), 139.0218; found, 139.0216.
(E)-4-(Naphthalen-2-yl)but-3-en-2-one (6n)
Reagents: Pd(OAc)2 (0.0045 g, 0.020 mmol), dmphen (0.0054 g, 0.026 mmol), methyl vinyl ketone (0.070 g, 1.00 mmol), p-bzq (0.109 g, 1.00 mmol), naphthalene-2-ylboronic acid (0.344 g, 2.00 mmol), acetonitrile (3 mL). Time: 24 h. Eluent: pentane/diethylether, 4/1. 6n (0.167 g, 0.85 mmol) was isolated as a white solid in 85% yield, mp 94–95 °C. 1H NMR (400 MHz, CDCl3) δ 7.87 (s, 1H), 7.75–7.83 (m, 3H), 7.58–7.65 (m, 2H), 7.45–7.53 (m, 2H), 6.78 (d, J = 16.3 Hz, 1H), 2.38 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 198.3, 143.4, 134.3, 133.2, 131.8, 130.3, 128.7, 128.5, 127.8, 127.4, 127.1, 126.7, 123.4, 27.5; HRMS (ESI+) calcd for C14H13O (M + H+), 197.0966; found, 197.0964.
1,4-Addition(12)
A round-bottom flask equipped with a condenser was charged with α,β-unsaturated aldehyde 6 (1 equiv), phenol (2.6 equiv), and triethylphosphite (1.2 equiv). The reaction mixture was stirred at 100 °C for the time indicated below. The reaction mixture was concentrated. H2O, HCl (2 M), and acetone were added, and the mixture was refluxed for 24 h. The reaction mixture was extracted with diethylether and dried over MgSO4. The product was purified by column chromatography on silica gel with ethyl acetate as eluent.
Diethyl (1-(Benzo[d][1,3]dioxol-5-yl)-3-oxopropyl)phosphonate (10a)
Reagents: 6a (0.30 g, 1.7 mmol), phenol (0.417 g, 4.43 mmol) and triethylphosphite (0.334 g, 2.01 mmol). Time: 4.5 h. Reagents: H2O (2.4 mL), HCl (2M, 6.3 mL), acetone (14 mL). Time: 24 h. 10a (0.342 g, 1.09 mmol) was isolated as an oil in 64% yield. 1H NMR (400 MHz, CDCl3) δ 9.57 (s, 1H), 6.58–6.86 (m, 3H), 5.76–5.92 (m, 2H), 3.81–4.08 (m, 3H), 3.68–3.81 (m, 1H), 3.49–3.65 (m, 1H), 2.87–3.12 (m, 2H), 1.21 (t, J = 7.4 Hz, 3H), 1.08 (t, J = 7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 198.7 (d, JC–P = 16.2 Hz), 147.7 (d, JC–P = 2.2 Hz), 146.8 (d, JC–P = 3.0 Hz), 128.5 (d, JC–P = 7.4 Hz), 122.3 (d, JC–P = 7.4 Hz), 109.2 (d, JC–P = 5.9 Hz), 108.2 (d, JC–P = 3.0 Hz), 101.0, 62.8 (d, JC–P = 7.4 Hz), 62.0 (d, JC–P = 7.4 Hz), 43.9 (d, JC–P = 2.2 Hz), 37.4 (d, JC–P = 143.0 Hz), 16.2 (d, JC–P = 5.9 Hz), 16.1 (d, JC–P = 5.9 Hz); HRMS (ESI+) calcd for C14H20O6P (M + H+), 315.0998; found, 315.0991.
Diethyl (1-(nNaphthalen-2-yl)-3-oxopropyl)phosphonate (10b)
Reagents: 6b (0.290 g, 1.60 mmol), phenol (0.391 g, 4.15 mmol) and triethylphosphite (0.692 g, 4.16 mmol). Time: 3.5 h. Reagents: H2O (2.2 mL), HCl (2M, 5.9 mL), acetone (13 mL). Time: 24 h. 10b (0.286 g, 892 mmol) was isolated as an oil in 56% yield. 1H NMR (400 MHz, CDCl3) δ 9.63 (s, 1H), 7.72–7.81 (m, 4H), 7.36–7.51 (m, 3H), 3.95–4.12 (m, 2H), 3.81–3.94 (m, 2H), 3.64–3.78 (m, 1H), 3.12–3.29 (m, 2H), 1.24 (t, J = 6.9 Hz, 3H), 1.05 (t, J = 6.9 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 198.7 (d, JC–P = 15.5 Hz), 133.2 (d, JC–P = 3.0 Hz), 132.7 (d, JC–P = 7.4 Hz), 132.6 (d, JC–P = 2.2 Hz), 128.3 (d, JC–P = 2.2 Hz), 127.9 (d, JC–P = 8.1 Hz), 127.7 (d, JC–P = 1.5 Hz), 127.5 (d, JC–P = 1.5 Hz), 126.9 (d, JC–P = 5.2 Hz), 126.2, 126.0 (d, JC–P = 1.5 Hz), 62.9 (d, JC–P = 7.4 Hz), 62.2 (d, JC–P = 7.4 Hz), 44.0 (d, JC–P = 2.2 Hz), 38.0 (d, JC–P = 140.8 Hz), 16.3 (d, JC–P = 5.9 Hz), 16.2 (d, JC–P = 5.9 Hz); HRMS (ESI+) calcd for C17H22O4P (M + H+), 321.1256; found, 321.1258.
Diethyl (1-([1,1′-Biphenyl]-4-yl)-3-oxopropyl)phosphonate (10c)
Reagents: 6c (0.430 g, 2.07 mmol), phenol (0.510 g, 5.42 mmol) and triethylphosphite (0.860 g, 5.18 mmol). Time: 3 h. Reagents: H2O (2.9 mL), HCl (2M, 7.7 mL), acetone (16 mL). Time: 24 h. 10c (0.544 g, 1.57 mmol) was isolated as an oil in 76% yield. 1H NMR (400 MHz, CDCl3) δ 9.58 (s, 1H), 7.43–7.54 (m, 4H), 7.29–7.40 (m, 4H), 7.20–7.27 (m, 1H), 3.94–4.07 (m, 2H), 3.82–3.93 (m, 1H), 3.64–3.81 (m, 2H), 2.99–3.18 (m, 2H), 1.21 (t, J = 7.0 Hz, 1H), 1.06 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 198.6 (d, JC–P = 15.5 Hz), 140.2 (d, JC–P = 1.5 Hz), 140.0 (d, JC–P = 3.0 Hz), 134.1 (d, JC–P = 7.4 Hz), 129.3 (d, JC–P = 6.6 Hz), 128.6, 127.2, 127.0 (d, JC–P = 3.0 Hz), 126.7, 62.7 (d, JC–P = 7.4 Hz), 62.0 (d, JC–P = 6.6 Hz), 43.7 (d, JC–P = 2.2 Hz), 36.6 (d, JC–P = 141.5 Hz), 16.2 (d, JC–P = 5.9 Hz), 16.0 (d, JC–P = 5.9 Hz); HRMS (ESI+) calcd for C19H24O4P (M + H+), 347.1412; found, 347.1415.
Diethyl (1-(4-Bromophenyl)-3-oxopropyl)phosphonate (10d)
Reagents: 6d (1.13 g, 5.37 mmol), phenol (1.34 g, 14.3 mmol) and triethylphosphite (1.07 g, 6.45 mmol). Time: 3 h. Reagents: H2O (7.5 mL), HCl (2M, 20 mL), acetone (30 mL). Time: 26 h. 10d (0.915 g, 2.62 mmol) was isolated as an oil in 49% yield. 1H NMR (400 MHz, CDCl3) δ 9.56 (td, J = 1.09, 2.17 Hz, 1H), 7.32–7.37 (m, 2H), 7.12–7.17 (m, 2H), 3.88–4.03 (m, 2H), 3.78–3.88 (m, 1H), 3.66–3.78 (m, 1H), 3.60 (ddd, J = 4.7, 9.4, 22.7 Hz, 1H), 2.92–3.13 (m, 2H), 1.18 (t, J = 7.0 Hz, 3H), 1.05 (dt, J = 0.4, 7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 198.2 (d, JC–P = 15.3 Hz), 134.4 (d, JC–P = 6.9 Hz), 131.61, 131.58, 130.7 (d, JC–P = 6.9 Hz), 121.3 (d, JC–P = 3.8 Hz), 62.9 (d, JC–P = 6.9 Hz), 62.2 (d, JC–P = 7.7 Hz), 43.7 (d, JC–P = 2.3 Hz), 37.2 (d, JC–P = 141.9 Hz), 16.2 (d, JC–P = 5.4 Hz), 16.1 (d, JC–P = 5.4 Hz); HRMS (ESI+) calcd for C13H19BrO4P (M + H+), 349.0204; found, 349.0197.
Diethyl (1-(Benzofuran-2-yl)-3-oxopropyl)phosphonate (10e)
Reagents: 6e (0.300 g, 1.74 mmol), phenol (0.430 g, 4.57 mmol) and triethylphosphite (0.341 g, 2.05 mmol). Time: 2.5 h. Reagents: H2O (2.4 mL), HCl (2M, 6.5 mL), acetone (15 mL). Time: 25 h. 10e (0.141 g, 0.454 mmol) was isolated as an oil in 26% yield. 1H NMR (400 MHz, CDCl3) δ 9.68 (td, J = 1.1, 2.0 Hz, 1H), 7.42–7.48 (m, 1H), 7.36–7.42 (m, 1H), 7.11–7.22 (m, 2H), 6.62 (td, J = 0.7, 4.0 Hz, 1H), 3.88–4.11 (m, 5H), 3.06–3.26 (m, 2H), 1.25 (dt, J = 0.4, 7.0 Hz, 3H), 1.17 (dt, J = 0.5, 7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 198.1 (d, JC–P = 13.8 Hz), 154.6 (d, JC–P = 1.5 Hz), 151.6 (d, JC–P = 9.2 Hz), 128.2 (d, JC–P = 3.1 Hz), 124.0 (d, JC–P = 1.5 Hz), 122.8, 120.7, 110.9, 105.2 (d, JC–P = 8.4 Hz), 63.0 (d, JC–P = 7.7 Hz), 62.8 (d, JC–P = 6.9 Hz), 41.7 (d, JC–P = 2.3 Hz), 32.3 (d, JC–P = 144.2 Hz), 16.2 (d, JC–P = 4.6 Hz), 16.2 (d, JC–P = 3.8 Hz); HRMS (ESI+) calcd for C15H20O5P (M + H+), 311.1048; found, 311.1053.
Imine Formation(12)
The (3-oxopropyl)phosphonate derivative (1 equiv) was dissolved in ethanol. O-Benzylhydroxylamine hydrochloride (1.5 equiv) and pyridine were added, and the reaction was stirred for the time indicated below. The reaction mixture was coevaporated with toluene three times.
Reduction of the Imine(12)
The crude imine product was dissolved in methanol, and NaBH3CN (3 equiv) was added. After the reaction mixture was stirred at room temperature for 45 min, it was cooled in an ice bath (0 °C), and aqueous HCl (37%) was added dropwise over 45 min. The reaction mixture was stirred for an additional 2 h. The reaction mixture was basified with aqueous NaOH (10% w/w) and extracted with DCM. The combined organic layers were dried with MgSO4 and evaporated. The crude material was used without further purification.
Acetylation(12)
The crude product from reductive amination was dissolved in DCM. Triethylamine (3 equiv) and acetyl chloride (2 equiv) were added, and the reaction mixture was stirred for the time indicated below. After the reaction was finished, H2O was added, and the reaction mixture was extracted with DCM. The combined organic layers were dried with MgSO4 and evaporated. The product was purified by column chromatography on silica gel (ethyl acetate).
Diethyl (1-(Benzo[d][1,3]dioxol-5-yl)-3-(N-(benzyloxy)acetamido)propyl)phosphonate (11a)
Reagents: 10a (0.34 g, 1.08 mmol) was dissolved in ethanol (6 mL). O-Benzylhydroxylamine hydrochloride (0.262 g, 1.64 mmol) and pyridine (4.5 mL). Time: 2.75 h. Reagents: NaBH3CN (0.208 g, 3.31 mmol), methanol (14 mL), HCl (37%, 1.6 mL). Time: 2 h. Reagents: Triethylamine (0.327 g, 3.23 mmol), acetyl chloride (0.180 g, 2.29 mmol) and DCM (6 mL). Time: 2.5 h. 11a (0.145 g, 0.312 mmol) was isolated as an oil in 39% yield. 1H NMR (400 MHz, CDCl3) δ 7.29–7.35 (m, 3H), 7.22–7.28 (m, 2H), 6.80 (d, J = 0.9 Hz, 1H), 6.69 (s, 2H), 5.89 (s, 2H), 4.62–4.71 (m, 2H), 3.92–4.06 (m, 2H), 3.82–3.91 (m, 1H), 3.67–3.79 (m, 1H), 3.48–3.61 (m, 1H), 3.34–3.46 (m, 1H), 2.93 (ddd, J = 3.5, 11.6, 22.9 Hz, 1H), 2.29–2.43 (m, 1H), 2.05–2.19 (m, 1H), 1.99 (s, 3H), 1.21 (t, J = 7.0 Hz, 3H), 1.07 (t, J = 7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 172.2, 147.8 (d, JC–P = 2.2 Hz), 146.8 (d, JC–P = 3.7 Hz), 134.3, 129.1, 128.9, 128.7 (d, JC–P = 7.4 Hz), 128.6, 122.7 (d, JC–P = 8.1 Hz), 109.3 (d, JC–P = 5.9 Hz), 108.2 (d, JC–P = 2.2 Hz), 101.0, 76.3, 62.5 (d, JC–P = 6.6 Hz), 61.9 (d, JC–P = 7.4 Hz), 43.7, 41.7 (d, JC–P = 140.1 Hz), 27.1, 20.4, 16.4 (d, JC–P = 5.9 Hz), 16.3 (d, JC–P = 5.9 Hz); HRMS (ESI+) calcd for C23H31NO7P (M + H+), 464.1838; found, 464.1836.
Diethyl (3-(N-(Benzyloxy)acetamido)-1-(naphthalen-2-yl)propyl)phosphonate (11b)
Reagents: 10b (0.28 g, 0.87 mmol) was dissolved in ethanol (5 mL). O-Benzylhydroxylamine hydrochloride (0.213 g, 1.33 mmol) and pyridine (3 mL). Time: 3.5 h. Reagents: NaBH3CN (0.165 g, 2.63 mmol), methanol (11 mL), HCl (37%, 1.2 mL). Time: 2 h. Reagents: Triethylamine (0.265 g, 2.62 mmol), acetyl chloride (0.157 g, 2.00 mmol) and DCM (5 mL). Time: 3 h. 11b (0.41 g, 0.86 mmol) was isolated as an oil in 99% yield. 1H NMR (400 MHz, CDCl3) δ 7.72–7.87 (m, 4H), 7.41–7.53 (m, 3H), 7.25–7.35 (m, 3H), 7.17–7.24 (m, 2H), 4.56–4.73 (m, 2H), 3.94–4.13 (m, 2H), 3.79–3.93 (m, 1H), 3.54–3.75 (m, 2H), 3.39–3.54 (m, 1H), 3.24 (ddd, J = 3.9, 11.3, 22.8 Hz, 1H), 2.47–2.59 (m, 1H), 2.31–2.47 (m, 1H), 1.99 (s, 3H), 1.26 (t, J = 7.1 Hz, 3H), 1.04 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 171.6, 133.9, 132.9, 132.4 (d, JC–P = 7.4 Hz), 132.2, 128.7, 128.4, 128.1, 127.9 (d, JC–P = 5.2 Hz), 127.8, 127.3, 127.2, 126.7 (d, JC–P = 5.2 Hz), 125.7, 125.5, 75.8, 62.1 (d, JC–P = 6.6 Hz), 61.5 (d, JC–P = 6.6 Hz), 42.5 (d, JC–P = 137.9 Hz), 26.5, 20.0 (d, JC–P = 3.0 Hz), 16.0 (d, JC–P = 5.9 Hz), 15.8 (d, JC–P = 5.9 Hz); HRMS (ESI+) calcd for C26H33NO5P (M + H+), 470.2096; found, 470.2092.
Diethyl (1-([1,1′-Biphenyl]-4-yl)-3-(N-(benzyloxy)acetamido)propyl)phosphonate (11c)
Reagents: 10c (0.54 g, 1.56 mmol) was dissolved in ethanol (9 mL). O-Benzylhydroxylamine hydrochloride (0.374 g, 2.34 mmol) and pyridine (6 mL). Time: 3 h. Reagents: NaBH3CN (0.294 g, 4.68 mmol), methanol (19 mL), HCl (37%, 2.1 mL). Time: 2 h. Reagents: Triethylamine (0.230 g, 2.27 mmol), acetyl chloride (0.148 g, 1.89 mmol) and DCM (9 mL). Time: 4.5 h. 11c (0.408 g, 0.823 mmol) was isolated as an oil in 53% yield. 1H NMR (400 MHz, CDCl3) δ 7.51–7.63 (m, 4H), 7.24–7.48 (m, 10H), 4.64–4.77 (m, 2H), 3.97–4.14 (m, 2H), 3.85–3.97 (m, 1H), 3.71–3.82 (m, 1H), 3.56–3.70 (m, 1H), 3.45–3.56 (m, 1H), 3.05–3.20 (m, 1H), 2.42–2.56 (m, 1H), 2.24–2.40 (m, 1H), 1.94–2.09 (s, 3H), 1.28 (t, J = 7.0 Hz, 3H), 1.11 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 170.6, 140.0, 139.5 (d, JC–P = 3.7 Hz), 133.9, 133.8, 129.2 (d, JC–P = 6.6 Hz), 128.7, 128.4, 128.3, 128.1, 126.8, 126.6 (d, JC–P = 2.2 Hz), 126.4, 75.8, 62.1 (d, JC–P = 6.6 Hz), 61.4 (d, JC–P = 7.4 Hz), 43.4, 41.3 (d, JC–P = 138.6 Hz), 26.4, 19.9, 15.9 (d, JC–P = 5.9 Hz), 15.8 (d, JC–P = 5.9 Hz); HRMS (ESI+) calcd for C28H35NO5P (M + H+), 496.2253; found, 496.2257.
Diethyl (3-(N-(Benzyloxy)acetamido)-1-(4-bromophenyl)propyl)phosphonate (11d)
Reagents: 10d (0.915 g, 2.62 mmol) was dissolved in ethanol (14 mL). O-Benzylhydroxylamine hydrochloride (0.627 g, 3.93 mmol) and pyridine (10 mL). Time: 5 h. Reagents: NaBH3CN (0.495 g, 7.88 mmol), methanol (35 mL), HCl (37%, 3.5 mL). Time: 2 h. Reagents: Triethylamine (0.80 g, 7.9 mmol), acetyl chloride (0.41 g, 5.2 mmol) and DCM (15 mL). Time: 1.5 h. 11d (0.922 g, 1.85 mmol) was isolated as an oil in 71% yield. 1H NMR (400 MHz, CDCl3) δ 7.35–7.41 (m, 2H), 7.29–7.34 (m, 3H), 7.20–7.26 (m, 2H), 7.10–7.17 (m, 2H), 4.60–4.70 (m, 2H), 3.92–4.06 (m, 2H), 3.79–3.92 (m, 1H), 3.65–3.77 (m, 1H), 3.45–3.57 (m, 1H), 3.34–3.45 (m, 1H), 2.98 (ddd, J = 3.7, 11.3, 22.8 Hz, 1H), 2.34–2.47 (m, 1H), 2.08–2.22 (m, 1H), 1.97 (s, 3H), 1.21 (t, J = 7.1 Hz, 3H), 1.06 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 172.1, 134.4 (d, JC–P = 7.7 Hz), 134.2, 131.6 (d, JC–P = 2.3 Hz), 130.9 (d, JC–P = 6.1 Hz), 129.1, 128.9, 128.6, 121.2 (d, JC–P = 3.8 Hz), 76.4, 62.6 (d, JC–P = 6.9 Hz), 62.0 (d, JC–P = 7.7 Hz), 43.6, 41.6 (d, JC–P = 139.6 Hz), 26.8 (d, JC–P = 2.3 Hz), 20.4, 16.3 (d, JC–P = 6.1 Hz), 16.2 (d, JC–P = 6.1 Hz); HRMS (ESI+) calcd for C22H30BrNO5P (M + H+), 498.1045; found, 498.1044.
Diethyl (1-(Benzofuran-2-yl)-3-(N-(benzyloxy)acetamido)propyl)phosphonate (11e)
Reagents: 10e (0.141 g, 0.454 mmol) was dissolved in ethanol (2.5 mL). O-Benzylhydroxylamine hydrochloride hydrochloride (0.111 g, 0.695 mmol) and pyridine (1.8 mL). Time: 2.5 h. Reagents: NaBH3CN (0.091 g, 1.45 mmol), methanol (6 mL), HCl (37%, 0.6 mL). Time: 2 h. Reagents: Triethylamine (0.13 g, 1.3 mmol), acetyl chloride (0.07 g, 0,9 mmol) and DCM (2.5 mL). Time: 3 h. 11e (0.135 g, 0.294 mmol) was isolated as an oil in 65% yield. 1H NMR (400 MHz, CDCl3) δ 7.46–7.52 (m, 1H), 7.37–7.44 (m, 1H), 7.13–7.34 (m, 7H), 6.63 (d, J = 4.0 Hz, 1H), 4.70 (s, 2H), 3.85–4.15 (m, 4H), 3.54–3.74 (m, 2H), 3.40 (ddd, J = 4.4, 10.7, 22.8 Hz, 1H), 2.26–2.52 (m, 2H), 2.02 (s, 3H), 1.26 (t, J = 7.4 Hz, 3H), 1.17 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 172.2, 154.7 (d, JC–P = 1.5 Hz), 152.4 (d, JC–P = 10.0 Hz), 134.2, 129.1, 128.9, 128.6, 128.4 (d, JC–P = 2.3 Hz), 123.9, 122.8, 120.7 (d, JC–P = 1.5 Hz), 111.0, 105.4 (d, JC–P = 8.4 Hz), 76.4, 62.7 (d, JC–P = 6.9 Hz), 62.5 (d, JC–P = 6.9 Hz), 43.8, 36.6 (d, JC–P = 141.1 Hz), 25.7, 20.4, 16.4 (d, JC–P = 6.1 Hz), 16.3 (d, JC–P = 6.1 Hz); HRMS (ESI+) calcd for C24H31NO6P (M + H+), 460.1889; found, 460.1893.
Synthesis of Biaryl Compounds by Suzuki Reaction
Method A:(62) a 2–5 mL microwave vial was charged with 11d (0.050 g, 0.10 mmol), boronic acid (0.50 mmol), Pd(PPh3)2Cl2 (7.0 mg, 0.010 mmol), Na2CO3 (2 M, 0.15 mL), DME (2.4 mL), and ethanol (95%, 0.6 mL). The vial was sealed, and the reaction mixture was irradiated for 30 min at 120 °C. After the reaction was finished the solvent was evaporated, and the compound was purified on silica gel (100% EtOAc).
Method B:(63) a 2–5 mL microwave vial was charged with 11d (0.05 g, 0.10 mmol), boronic acid (0.60 mmol), Pd(OAc)2 (0.02 mmol), [HP(t-Bu)3]BF4 (0.04 mmol), K2CO3 (0.60 mmol), DME (2 mL), and H2O (0.6 mL). The vial was sealed, and the reaction mixture was irradiated for 15 min at 100 °C. After the reaction was finished, it was filtered through a Celite plug, and the compound was purified on silica gel (100% EtOAc).
Diethyl (3-(N-(Benzyloxy)acetamido)-1-(4-(pyridin-3-yl)phenyl)propyl)phosphonate (11f)
Compound 11f was prepared according to method A, and the synthesis was repeated three times. Reagents: 11d (0.048 g, 0.096 mmol), 3-pyridinylboronic acid (0.068 g, 0.55 mmol), Pd(PPh3)2Cl2 (7.4 mg, 0.011 mmol). 11f (0.128 g, 0.259 mmol) was isolated as an oil in an average yield of 90%. 1H NMR (400 MHz, CDCl3) δ 9.06 (s, 1H), 8.75 (d, J = 4.9 Hz, 1H), 8.39 (d, J = 8.2 Hz, 1H), 7.83 (dd, J = 5.4, 8.0 Hz, 1H), 7.54–7.61 (m, 2H), 7.45–7.53 (m, 2H), 7.35–7.40 (m, 3H), 7.28–7.34 (m, 2H), 4.74 (s, 2H), 4.00–4.15 (m, 2H), 3.90–4.00 (m, 1H), 3.77–3.90 (m, 1H), 3.46–3.66 (m, 2H), 3.09–3.23 (m, 1H), 2.42–2.59 (m, 1H), 2.20–2.37 (m, 1H), 2.02 (s, 3H), 1.42 (d, J = 12.9 Hz, 1H), 1.28 (t, J = 7.1 Hz, 6H), 1.15 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 172.6, 161.7 (d, JC–P = 38.3 Hz), 141.7 (d, JC–P = 38.3 Hz), 140.7, 139.6, 137.7 (d, JC–P = 6.9 Hz), 134.3, 133.5 (d, JC–P = 3.1 Hz), 130.8 (d, JC–P = 6.9 Hz), 129.3, 129.2, 128.9, 127.4 (d, JC–P = 2.3 Hz), 126.3, 76.6, 63.1 (d, JC–P = 7.7 Hz), 62.7 (d, JC–P = 6.9 Hz), 43.7, 42.0 (d, JC–P = 138.8 Hz), 29.0, 26.9, 20.5, 16.5 (d, JC–P = 6.1 Hz), 16.4 (d, JC–P = 6.1 Hz); HRMS (ESI+) calcd for C27H34N2O5P (M + H+), 497.2205; found, 497.2201.
Diethyl (3-(N-(Benzyloxy)acetamido)-1-(3′-methyl-[1,1′-biphenyl]-4-yl)propyl)phosphonate (11g)
Compound 11g was prepared according to method B, and the synthesis was repeated three times. Reagents: 11d (0.050 g, 0.10 mmol), m-tolylboronic acid (0.084 g, 0.62 mmol), Pd(OAc)2 (4.2 mg, 0.019 mmol), [HP(t-Bu)3]BF4 (0.012, 0.040 mmol), K2CO3 (0.088 g, 0.64 mmol). 11g (0.133 g, 0.262 mmol) was isolated as an oil in an average yield of 87%. 1H NMR (400 MHz, CDCl3) δ 7.55 (d, J = 7.7 Hz, 2H), 7.27–7.43 (m, 10H), 7.13–7.18 (m, 1H), 4.70 (s, 2H), 3.97–4.13 (m, 2H), 3.85–3.97 (m, 1H), 3.70–3.81 (m, 1H), 3.56–3.69 (m, 1H), 3.45–3.56 (m, 1H), 3.11 (ddd, J = 4.0, 11.3, 23.4 Hz, 1H), 2.43–2.55 (m, 1H), 2.39–2.43 (m, 3H), 2.24–2.38 (m, 1H), 2.02 (s, 3H), 1.27 (t, J = 7.3 Hz, 3H), 1.11 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 172.3, 140.6, 140.3 (d, JC–P = 3.1 Hz), 138.4, 134.4, 134.2 (d, JC–P = 6.9 Hz), 129.7 (d, JC–P = 6.1 Hz), 129.2, 129.0, 128.7 (d, JC–P = 3.8 Hz), 128.0 (d, JC–P = 29.9 Hz), 127.3 (d, JC–P = 3.1 Hz), 124.1, 76.4, 62.7 (d, JC–P = 6.9 Hz), 62.1 (d, JC–P = 6.9 Hz), 43.9, 41.9 (d, JC–P = 138.0 Hz), 26.9, 21.6, 20.5, 16.5 (d, JC–P = 6.1 Hz), 16.3 (d, JC–P = 6.1 Hz); HRMS (ESI+) calcd for C29H37NO5P (M + H+), 510.2409; found, 510.2410.
Diethyl (3-(N-(Benzyloxy)acetamido)-1-(4-(pyridin-4-yl)phenyl)propyl)phosphonate (11h)
Compound 11h was prepared according to method A, and the synthesis was repeated two times. Reagents: 11d (0.048 g, 0.097 mmol), 4-pyridinylboronic acid (0.071 g, 0.58 mmol), Pd(PPh3)2Cl2 (10 mg, 0.014 mmol). 11h (0.0731 g, 0.147 mmol) was isolated as an oil in an average yield of 76%. 1H NMR (400 MHz, CDCl3) δ 8.59–8.66 (m, 2H), 7.54–7.62 (m, 2H), 7.46–7.50 (m, 2H), 7.38–7.45 (m, 2H), 7.31–7.36 (m, 3H), 7.24–7.31 (m, 2H), 4.64–4.75 (m, 2H), 3.97–4.11 (m, 2H), 3.84–3.97 (m, 1H), 3.70–3.83 (m, 1H), 3.41–3.66 (m, 2H), 3.11 (ddd, J = 3.6, 11.0, 22.5 Hz, 1H), 2.40–2.55 (m, 1H), 2.19–2.38 (m, 1H), 1.99 (s, 3H), 1.25 (t, J = 7.1 Hz, 3H), 1.10 (t, J = 7.3 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 172.1, 150.3, 147.7, 137.0 (d, JC–P = 3.8 Hz), 136.5 (d, JC–P = 6.9 Hz), 134.3, 130.0 (d, JC–P = 6.1 Hz), 129.1, 128.9, 128.6, 127.0 (d, JC–P = 3.1 Hz), 121.4, 76.4, 62.6 (d, JC–P = 6.9 Hz), 62.1 (d, JC–P = 7.7 Hz), 43.8, 41.9 (d, JC–P = 138.8 Hz), 26.8 (d, JC–P = 2.3 Hz), 20.4, 16.4 (d, JC–P = 6.1 Hz), 16.2 (d, JC–P = 5.4 Hz); HRMS (ESI+) calcd for C27H34N2O5P (M + H+), 497.2205; found, 497.2202.
Diethyl (3-(N-(Benzyloxy)acetamido)-1-(4-(thiophen-3-yl)phenyl)propyl)phosphonate (11i)
Compound 11i was prepared according to method A, and the synthesis was repeated two times. Reagents: 11d (0.048 g, 0.097 mmol), thiophen-3-ylboronic acid (0.068 g, 0.53 mmol), Pd(PPh3)2Cl2 (9.2 mg, 0.013 mmol). 11i (0.0594 g, 0.118 mmol) was isolated as an oil in an average yield of 61%. 1H NMR (400 MHz, CDCl3) δ 7.55 (d, J = 7.6 Hz, 2H), 7.42–7.46 (m, 1H), 7.37–7.40 (m, 2H), 7.31–7.37 (m, 5H), 7.25–7.31 (m, 2H), 4.69 (s, 2H), 3.96–4.11 (m, 2H), 3.84–3.95 (m, 1H), 3.68–3.79 (m, 1H), 3.54–3.67 (m, 1H), 3.43–3.54 (m, 1H), 3.08 (ddd, J = 3.9, 11.4, 22.9 Hz, 1H), 2.40–2.53 (m, 1H), 2.21–2.36 (m, 1H), 2.01 (m, 3H), 1.26 (t, J = 7.0 Hz, 3H), 1.10 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 172.3, 141.9 (d, JC–P = 1.5 Hz), 134.9 (d, JC–P = 3.8 Hz), 134.4, 134.1 (d, JC–P = 6.9 Hz), 129.8 (d, JC–P = 6.9 Hz), 129.2, 129.0, 128.8, 126.6 (d, JC–P = 2.3 Hz), 126.4, 126.3, 120.3 (d, JC–P = 1.5 Hz), 76.4, 62.8 (d, JC–P = 6.9 Hz), 62.1 (d, JC–P = 6.9 Hz), 44.0, 42.0 (d, JC–P = 138.8 Hz), 26.9, 20.6, 16.5 (d, JC–P = 6.1 Hz), 16.4 (d, JC–P = 6.1 Hz); HRMS (ESI+) calcd for C26H33NO5PS (M + H+), 502.1817; found, 502.1819.
Diethyl (3-(N-(Benzyloxy)acetamido)-1-(4-(3,5-dimethylisoxazol-4-yl)phenyl)propyl)phosphonate (11j)
Compound 11j was prepared according to method A, and the synthesis was repeated two times. Reagents: 11d (0.048 g, 0.097 mmol), (3,5-dimethylisoxazol-4-yl)boronic acid (0.074 g, 0.52 mmol), Pd(PPh3)2Cl2 (11 mg, 0.016 mmol). 11j (0.0920 g, 0.179 mmol) was isolated as an oil in an average yield of 92%. 1H NMR (400 MHz, CDCl3) δ 7.31–7.40 (m, 5H), 7.26–7.31 (m, 2H), 7.18 (d, J = 7.8 Hz, 2H), 4.65–4.76 (m, 2H), 3.96–4.11 (m, 2H), 3.83–3.95 (m, 1H), 3.70–3.82 (m, 1H), 3.45–3.66 (m, 2H), 3.08 (ddd, J = 3.9, 11.1, 23.1 Hz, 1H), 2.40–2.54 (m, 1H), 2.36 (s, 3H), 2.24–2.33 (m, 1H), 2.23 (s, 3H), 1.99 (s, 3H), 1.25 (t, J = 7.0 Hz, 3H), 1.07 (t, J = 7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 172.7, 165.2, 158.6, 134.7 (d, JC–P = 6.9 Hz), 134.4, 129.8 (d, JC–P = 6.1 Hz), 129.5 (d, JC–P = 3.8 Hz), 129.2, 129.1, 129.0, 128.7, 116.2 (d, JC–P = 1.5 Hz), 76.4, 62.7 (d, JC–P = 6.9 Hz), 62.1 (d, JC–P = 7.7 Hz), 43.8, 42.0 (d, JC–P = 138.8 Hz), 26.9 (d, JC–P = 2.4 Hz), 20.5, 16.4 (d, JC–P = 5.4 Hz), 16.2 (d, JC–P = 5.4 Hz), 11.7, 10.9; HRMS (ESI+) calcd for C27H36N2O6P (M + H+), 515.2311; found, 515.2309.
Diethyl (3-(N-(Benzyloxy)acetamido)-1-(4-morpholinophenyl)propyl)phosphonate (11k).(64)
A 2–5 mL microwave vial was charged with 11d (0.097 g, 0.19 mmol), morpholine (0.20 g, 2.3 mmol), Pd(OAc)2 (1.3 mg, 5.8 μmol), [HP(t-Bu)3]BF4 (1.3 mg, 4.5 μmol), NaO-t-Bu (0.028 g, 0.29 mmol), and anhydrous toluene (2 mL). The vial was sealed and purged with N2(g), and the reaction mixture was irradiated for 30 min at 100 °C. The reaction was repeated three times. After the reaction was finished, the mixture was filtered through a Celite plug, and the compound was purified on silica gel (DCM/methanol, 95/5). A mixture of the product and the deacetylated product was obtained, so the crude product mixture was further reacted with acetyl chloride (0.07 mL, 0.08 mmol), triethylamine (0.21 mL, 0.15 mmol) in DCM (5 mL) for 2 h at room temperature. H2O (10 mL) was added, and the reaction mixture was extracted with DCM (2 × 10 mL) dried with MgSO4 and purified on silica gel (100% EtOAc). 11k (0.098 g, 0.19 mmol) was obtained as an oil in 33% yield. 1H NMR (400 MHz, CDCl3) δ 7.30–7.36 (m, 3H), 7.23–7.29 (m, 2H), 7.13–7.21 (m, 2H), 6.82 (d, J = 8.5 Hz, 2H), 4.67 (s, 2H), 3.90–4.05 (m, 2H), 3.76–3.90 (m, 5H), 3.61–3.74 (m, 1H), 3.49–3.61 (m, 1H), 3.36–3.48 (m, 1H), 3.05–3.15 (m, 4H), 2.95 (ddd, J = 3.8, 11.2, 23.1 Hz, 1H), 2.31–2.46 (m, 1H), 2.09–2.23 (m, 1H), 1.98 (s, 3H), 1.22 (t, J = 7.7 Hz, 3H), 1.06 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 173.0, 150.4 (d, JC–P = 3.1 Hz), 134.4, 130.0 (d, JC–P = 6.9 Hz), 129.1, 128.9, 128.6, 126.0 (d, JC–P = 6.9 Hz), 115.7 (d, JC–P = 2.3 Hz), 76.3, 66.8, 62.6 (d, JC–P = 7.7 Hz), 61.9 (d, JC–P = 7.7 Hz), 49.1, 43.8, 41.1 (d, JC–P = 140.3 Hz), 26.6, 20.5, 16.4 (d, JC–P = 6.1 Hz), 16.3 (d, JC–P = 5.4 Hz); HRMS (ESI+) calcd for C26H38N2O6P (M + H+), 505.2468; found, 505.2460.
Deprotection of the Benzyl Group
Method C:(12) The acetylated product was dissolved in methanol, and Pd/C (10%) was added. The reaction mixture was stirred under H2 (g) at atmospheric pressure while the reaction was monitored by TLC (ethyl acetate 100% and DCM/methanol, 95/5). After the reaction was finished, the mixture was filtered through a Celite plug, and the solvent was removed by evaporation. The product was purified by column chromatography on silica gel (DCM/methanol, 95/5).
Method D:(52) The acetylated product was stirred in dry DCM at −50 °C under N2. BCl3 (4 equiv) was added. The reaction was stirred for the time indicated. After the reaction was finished NaHCO3 (satd) was added, and the mixture was extracted with DCM, dried with MgSO4, filtered, and purified by column chromatography on silica gel (DCM/methanol, 95/5).
Diethyl (1-(Benzo[d][1,3]dioxol-5-yl)-3-(N-hydroxyacetamido)propyl)phosphonate (12a)
Compound 12a was prepared by method C. Reagents: 11a (0.14 g, 0.30 mmol), Pd/C (10%, 0.035 g), and methanol (15 mL). Time: 4 h. 12a (0.087 g, 0.23 mmol) was isolated as an oil in 77% yield. 1H NMR (400 MHz, CDCl3) δ 9.21–9.94 (m, 1H), 6.65–6.85 (m, 3H), 5.85–5.99 (m, 2H), 3.70–4.10 (m, 5H), 3.30–3.42 (m, 1H), 2.92–3.09 (m, 1H), 2.40 (d, J = 4.0 Hz, 1H), 2.02–2.13 (m, 4H), 1.20–1.32 (m, 3H), 1.15 (t, J = 6.7 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 172.2, 147.9, 147.0, 129.3 (d, JC–P = 7.4 Hz), 122.7 (d, JC–P = 8.1 Hz), 109.3 (d, JC–P = 5.9 Hz), 108.4, 101.2, 63.1 (d, JC–P = 6.6 Hz), 62.6 (d, JC–P = 5.9 Hz), 46.3 (d, JC–P = 14.7 Hz), 41.6 (d, JC–P = 139.3 Hz), 27.8, 20.6, 16.4, 16.3; HRMS (ESI+) calcd for C16H25NO7P (M + H+), 374.1369; found, 374.1359.
Diethyl (3-(N-Hydroxyacetamido)-1-(naphthalen-2-yl)propyl)phosphonate (12b)
Compound 12b was prepared by method C. Reagents: 11b (0.4 g, 0.85 mmol), Pd/C (10%, 0.102 g), and methanol (25 mL). Time: 7 h. 12b (0.198 g, 0.52 mmol) was isolated as an oil in 61% yield. 1H NMR (400 MHz, CDCl3) δ 9.68 (br. s., 1H), 7.69–7.83 (m, 4H), 7.37–7.48 (m, 3H), 3.90–4.04 (m, 2H), 3.60–3.88 (m, 3H), 3.19–3.43 (m, 2H), 2.44–2.57 (m, 1H), 2.26–2.42 (m, 1H), 2.03 (s, 3H), 1.16–1.24 (m, 3H), 1.04 (t, J = 7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 172.1, 133.3 (d, JC–P = 2.2 Hz), 133.0 (d, JC–P = 8.1 Hz), 132.6, 128.3, 128.2, 127.8, 127.6, 126.8 (d, JC–P = 4.4 Hz), 126.2, 126.0, 63.0 (d, JC–P = 7.4 Hz), 62.4 (d, JC–P = 7.4 Hz), 46.3 (d, JC–P = 15.5 Hz), 42.1 (d, JC–P = 137.9 Hz), 27.4, 20.4, 16.3 (d, JC–P = 5.2 Hz), 16.2 (d, JC–P = 5.2 Hz); HRMS (ESI+) calcd for C19H27NO5P (M + H+), 380.1627; found, 380.1624.
Diethyl (1-([1,1′-Biphenyl]-4-yl)-3-(N-(benzyloxy)acetamido)propyl)phosphonate (12c)
Compound 12c was prepared by method C. Reagents: 11c (0.35 g, 0.71 mmol), Pd/C (10%, 0.074 g), and methanol (20 mL). Time: 2.25 h. 12c (0.231 g, 0.569 mmol) was isolated as an oil in 80% yield. 1H NMR (400 MHz, CDCl3) δ 9.71 (br. s., 1H), 7.46–7.63 (m, 4H), 7.15–7.45 (m, 5H), 3.92–4.10 (m, 2H), 3.81–3.92 (m, 1H), 3.67–3.81 (m, 2H), 3.32–3.48 (m, 1H), 3.03–3.24 (m, 1H), 2.36–2.54 (m, 1H), 2.17–2.35 (m, 1H), 2.05 (s, 2H), 1.17–1.33 (m, 3H), 1.08 (t, J = 7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 172.0, 140.3, 140.1, 134.3 (d, JC–P = 7.4 Hz), 129.5 (d, JC–P = 6.6 Hz), 128.7, 127.3, 127.1, 126.8, 63.0, 62.4 (d, JC–P = 7.4 Hz), 46.2 (d, JC–P = 16.2 Hz), 41.6 (d, JC–P = 139.3 Hz), 27.2, 20.4, 16.3 (d, JC–P = 5.2 Hz), 16.1 (d, JC–P = 5.2 Hz); HRMS (ESI+) calcd for C21H29NO5P (M + H+), 406.1783; found, 406.1774.
Diethyl (1-(4-Bromophenyl)-3-(N-hydroxyacetamido)propyl)phosphonate (12d)
Compound 12d was prepared by method D. Reagents: 11d (0.0975 g, 0.196 mmol), BCl3 (0.8 mL, 1 M). Time: 2 h. 12d (0.061 g, 0.149 mmol) was isolated as an oil in 76% yield. 1H NMR (400 MHz, CDCl3) δ 9.55 (br. s., 1H), 7.42 (d, J = 8.1 Hz, 2H), 7.08–7.21 (m, 2H), 3.66–4.10 (m, 5H), 3.23–3.61 (m, 1H), 2.96–3.16 (m, 1H), 2.33–2.88 (m, 2H), 1.98–2.19 (s, 3H), 1.22 (t, J = 6.9 Hz, 3H), 1.15 (t, J = 7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 172.4, 135.0 (d, JC–P = 7.7 Hz), 131.8, 130.9 (d, JC–P = 6.9 Hz), 121.5 (d, JC–P = 3.8 Hz), 63.2 (d, JC–P = 6.9 Hz), 62.7 (d, JC–P = 7.7 Hz), 46.1 (d, JC–P = 13.8 Hz), 41.4 (d, JC–P = 138.0 Hz), 27.5, 20.6, 16.4 (d, JC–P = 16.4 Hz), 16.3 (d, JC–P = 16.3 Hz); HRMS (ESI+) calcd for C15H24BrNO5P (M + H+), 408.0575; found, 408.0580.
Diethyl (1-(Benzofuran-2-yl)-3-(N-hydroxyacetamido)propyl)phosphonate (12e)
Compound 12e was prepared by method C. Reagents: 11e (0.135 g, 0.294 mmol), Pd/C (10%, 0.037 g), and methanol (5 mL). Time: 1.5 h. 12e (0.101 g, 0.274 mmol) was isolated as an oil in 93% yield. 1H NMR (400 MHz, CDCl3) δ 9.60 (br. s., 1H), 7.37–7.53 (m, 2H), 7.14–7.25 (m, 2H), 6.65 (d, J = 3.7 Hz, 1H), 3.81–4.13 (m, 5H), 3.39–3.54 (m, 2H), 2.24–2.51 (m, 2H), 2.07 (s, 3H), 1.26 (t, J = 7.0 Hz, 3H), 1.19 (t, J = 7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 172.4, 154.8, 152.2 (d, JC–P = 9.2 Hz), 128.4 (d, JC–P = 2.3 Hz), 124.1, 122.9, 120.9, 111.0, 105.5 (d, JC–P = 7.7 Hz), 63.4 (d, JC–P = 6.9 Hz), 63.0 (d, JC–P = 6.9 Hz), 46.3 (d, JC–P = 13.0 Hz), 36.3 (d, JC–P = 141.1 Hz), 25.9, 20.5, 16.3 (d, JC–P = 5.4 Hz, 2C, confirmed by HSQC); HRMS (ESI+) calcd for C17H25NO6P (M + H+), 370.1420; found, 370.1408.
Diethyl (3-(N-Hydroxyacetamido)-1-(4-(pyridin-3-yl)phenyl)propyl)phosphonate (12f)
Compound 12f was prepared by method C. Reagents: 11f (0.149 g, 0.300 mmol), Pd/C (10%, 0.039 g), and methanol (10 mL). Time: 20 h. 12f (0.0842 g, 0.207 mmol) was isolated as an oil in 69% yield. 1H NMR (400 MHz, CDCl3) δ 10.06 (br. s., 1H), 8.75 (s, 1H), 8.51 (d, J = 4.1 Hz, 1H), 7.81–7.89 (m, 1H), 7.47–7.56 (m, 2H), 7.40 (dd, J = 1.9, 8.2 Hz, 2H), 7.34 (dd, J = 4.9, 7.8 Hz, 1H), 3.74–4.11 (m, 5H), 3.38–3.50 (m, 1H), 3.10–3.24 (m, 1H), 2.43–2.59 (m, 1H), 2.16–2.34 (m, 1H), 2.05–2.12 (m, 3H), 1.23 (t, J = 7.0 Hz, 3H), 1.15 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 172.3, 148.2, 147.8, 136.6, 136.3, 136.2 (d, JC–P = 6.9 Hz), 134.6, 130.0 (d, JC–P = 6.9 Hz), 127.3, 123.8, 63.0 (d, JC–P = 6.9 Hz), 62.6 (d, JC–P = 6.9 Hz), 46.3 (d, JC–P = 14.6 Hz), 41.7 (d, JC–P = 138.0 Hz), 27.6, 20.6, 16.4, 16.3; HRMS (ESI+) calcd for C20H28N2O5P (M + H+), 407.1736; found, 407.1733.
Diethyl (3-(N-Hydroxyacetamido)-1-(3′-methyl-[1,1′-biphenyl]-4-yl)propyl)phosphonate (12g)
Compound 12g was prepared by method C. Reagents: 11g (0.166 g, 0.326 mmol), Pd/C (10%, 0.043 g) and methanol (5 mL). Time: 1.5 h. 12g (0.114 g, 0.272 mmol) was isolated as an oil in 82% yield. 1H NMR (400 MHz, CDCl3) δ 9.60 (br. s., 1H), 7.48–7.62 (m, 2H), 7.27–7.43 (m, 5H), 7.15 (d, J = 7.2 Hz, 1H), 3.70–4.14 (m, 5H), 3.35–3.65 (m, 1H), 3.08–3.23 (m, 1H), 2.45–2.68 (m, 1H), 2.41 (s, 3H), 2.19–2.35 (m, 1H), 2.12 (s, 3H), 1.23–1.34 (m, 3H), 1.15 (t, J = 7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 172.3, 140.5, 140.5 (d, JC–P = 3.1 Hz), 138.5, 134.9 (d, JC–P = 7.7 Hz), 129.6 (d, JC–P = 6.9 Hz), 128.8, 128.2, 127.8, 127.4, 124.1, 63.2 (d, JC–P = 6.9 Hz), 62.7 (d, JC–P = 7.7 Hz), 46.4 (d, JC–P = 13.0 Hz), 41.8 (d, JC–P = 137.3 Hz), 27.8, 21.6, 20.7, 16.4, 16.3; HRMS (ESI+) calcd for C22H31NO5P (M + H+), 420.1940; found, 420.1943.
Diethyl (3-(N-Hydroxyacetamido)-1-(4-(pyridin-4-yl)phenyl)propyl)phosphonate (12h)
Compound 12h was prepared by method C. Reagents: 11h (0.073 g, 0.15 mmol), Pd/C (10%, 0.028 g), and methanol (5 mL). Time: 3 h. 12h (0.040 g, 0.098 mmol) was isolated as an oil in 67% yield. 1H NMR (400 MHz, CDCl3) δ 9.90 (br. s., 1H), 8.43–8.75 (m, 2H), 7.54–7.66 (m, 2H), 7.35–7.52 (m, 4H), 3.79–4.12 (m, 5H), 3.35–3.66 (m, 1H), 3.11–3.25 (m, 1H), 2.46–2.67 (m, 1H), 2.18–2.39 (m, 1H), 2.07–2.17 (m, 3H), 1.22–1.28 (m, 3H), 1.19 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 172.4, 150.0, 147.9, 137.6 (d, JC–P = 7.7 Hz), 136.8, 130.0 (d, JC–P = 6.1 Hz), 127.1, 121.5, 63.0 (d, JC–P = 8.4 Hz), 62.6 (d, JC–P = 6.9 Hz), 46.2 (d, JC–P = 13.0 Hz), 41.7 (d, JC–P = 137.3 Hz), 29.2, 20.6, 16.31, 16.26; HRMS (ESI+) calcd for C20H28N2O5P (M + H+), 407.1736; found, 407.1735.
Diethyl (3-(N-Hydroxyacetamido)-1-(4-(thiophen-3-yl)phenyl)propyl)phosphonate (12i)
Compound 12i was prepared by method D. Reagents: 11i (0.059 g, 0.12 mmol), BCl3 (0.5 mL, 1 M). Time: 1 h. 12i (0.024 g, 0.058 mmol) was isolated as an oil in 49% yield. 1H NMR (400 MHz, CDCl3) δ 9.52 (br. s., 1H), 7.51–7.63 (m, 2H), 7.27–7.49 (m, 5H), 3.71–4.14 (m, 5H), 3.33–3.65 (m, 1H), 3.06–3.22 (m, 1H), 2.44–2.68 (m, 1H), 2.19–2.39 (m, 1H), 2.07–2.18 (m, 3H), 1.23–1.34 (m, 3H), 1.08–1.21 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 13C NMR (101 MHz, CDCl3) δ = 172.5, 141.8, 135.2, 135.0 (d, JC–P = 8.4 Hz), 129.7, 126.7 (d, JC–P = 6.1 Hz), 126.5, 126.3, 120.5, 63.2 (d, JC–P = 6.9 Hz), 62.8 (d, JC–P = 6.9 Hz), 46.4 (d, JC–P = 11.5 Hz), 41.8 (d, JC–P = 138.0 Hz), 28.1, 20.7, 16.4, 16.3; HRMS (ESI+) calcd for C19H27NO5PS (M + H+), 412.1348; found, 412.1344.
Diethyl (1-(4-(3,5-Dimethylisoxazol-4-yl)phenyl)-3-(N-hydroxyacetamido)propyl)phosphonate (12j)
Compound 12j was prepared by method C. Reagents: 11j (0.104 g, 0.202 mmol), Pd/C (10%, 0.025 g), and methanol (5 mL). Time: 3 h. 12j (0.0822 g, 0.194 mmol) was isolated as an oil in 96% yield. 1H NMR (400 MHz, CDCl3) δ 9.58 (br. s., 1H), 7.31–7.40 (m, 2H), 7.15–7.23 (m, 2H), 3.75–4.12 (m, 5H), 3.32–3.47 (m, 1H), 3.05–3.21 (m, 1H), 2.44–2.66 (m, 2H), 2.37 (s, 3H), 2.20–2.26 (m, 3H), 2.10 (s, 3H), 1.20–1.29 (m, 3H), 1.15 (t, J = 7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 172.5, 165.4, 158.6, 135.4 (d, JC–P = 7.7 Hz), 134.4, 129.7 (d, JC–P = 6.9 Hz), 129.2, 116.2, 63.1 (d, JC–P = 7.7 Hz), 62.7 (d, JC–P = 6.9 Hz), 46.2 (d, JC–P = 13.8 Hz), 41.7 (d, JC–P = 138.0 Hz), 27.8, 20.6, 16.4, 16.3, 11.7, 10.9; HRMS (ESI+) calcd for C20H30N2O6P (M + H+), 425.1842; found, 425.1846.
Diethyl (3-(N-Hydroxyacetamido)-1-(4-morpholinophenyl)propyl)phosphonate (12k)
Compound 12k was prepared by method C. Reagents: 11k (0.098 g, 0.19 mmol), Pd/C (10%, 0.023 g), and methanol (5 mL). Time: 3 h. 12k (0.057 g, 0.14 mmol) was isolated as an oil in 71% yield. 1H NMR (400 MHz, CDCl3) δ 9.62 (br. s., 1H), 7.16 (dd, J = 2.2, 8.8 Hz, 2H), 6.82 (d, J = 8.3 Hz, 2H), 3.65–4.07 (m, 9H), 3.24–3.57 (m, 1H), 3.07–3.15 (m, 4H), 2.91–3.06 (m, 1H), 2.62–2.76 (m, 1H), 2.30–2.49 (m, 1H), 2.01–2.18 (m, 3H), 1.18–1.28 (m, 3H), 1.12 (t, J = 7.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 172.2, 150.5, 129.9 (d, JC–P = 6.9 Hz), 126.5 (d, JC–P = 7.7 Hz), 115.7, 66.9, 63.0 (d, JC–P = 6.9 Hz), 62.5 (d, JC–P = 6.9 Hz), 49.2, 46.2 (d, JC–P = 14.6 Hz), 41.0 (d, JC–P = 137.3 Hz), 27.7, 20.6, 16.4, 16.3; HRMS (ESI+) calcd for C19H32N2O6P (M + H+), 415.1998; found, 415.2005.
Deprotection of the Phosphate Ethyl Ester(12)
TMSBr was added to a stirred solution of the phosphonate diethyl ester in dry DCM under N2 at room temperature. After the indicated time, the volatiles were removed in vacuo to give the phosphonic acid derivative. The product was purified by preparative HPLC.
(1-(Benzo[d][1,3]dioxol-5-yl)-3-(N-hydroxyacetamido)propyl)phosphonic Acid (13a)
Reagents: 12a (0.041 g, 0.11 mmol), TMSBr (0.10 mL, 0.76 mmol), and DCM (5 mL). Time: 3 h. Purification: gradient 0–30%, 60 min, 5 mL/min. 13a (33 mg, 0.10 mmol) was isolated as a white lyophilized material in 95% yield. 1H NMR (400 MHz, CD3OD) δ 6.86 (dd, J = 1.7, 1.7 Hz, 1H), 6.72–6.81 (m, 2H), 5.91 (s, 2H), 3.52–3.63 (m, 1H), 3.35–3.46 (m, 1H), 2.96 (ddd, J = 3.4, 11.5, 22.8 Hz, 1H), 2.34–2.46 (m, 1H), 2.07–2.19 (m, 1H), 2.05 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 173.7, 149.2, 148.0, 131.5 (d, JC–P = 7.7 Hz), 123.9 (d, JC–P = 7.7 Hz), 110.4 (d, JC–P = 6.1 Hz), 109.0 (d, JC–P = 2.3 Hz), 102.3, 47.4 (d, JC–P = 18.4 Hz), 43.8 (d, JC–P = 137.3 Hz), 28.5 (d, JC–P = 1.5 Hz), 20.2; HRMS (ESI+) calcd for C12H17NO7P (M + H+), 318.0743; found, 318.0742.
(3-(N-Hydroxyacetamido)-1-(naphthalen-2-yl)propyl)phosphonic Acid (13b)
Reagents: 12b (0.072 g, 0.19 mmol), TMSBr (0.10 mL, 0.76 mmol), and DCM (5 mL). Time: 4 h. Purification: gradient 5–40%, 70 min, 5 mL/min. 13b (42 mg, 0.19 mmol) was isolated as a white lyophilized material in 68% yield. 1H NMR (400 MHz, CD3OD) δ 7.79–7.85 (m, 4H), 7.52 (ddd, J = 1.5, 1.5, 8.7 Hz, 1H), 7.40–7.49 (m, 2H), 3.54–3.68 (m, 1H), 3.45 (s, 1H), 3.24 (ddd, J = 3.5, 11.4, 22.8 Hz, 1H), 2.46–2.61 (m, 1H), 2.27–2.44 (m, 1H), 1.95–2.09 (m, 3H); 13C NMR (101 MHz, CD3OD) δ 173.6, 135.5 (d, JC–P = 7.7 Hz), 134.9 (d, JC–P = 2.3 Hz), 134.1 (d, JC–P = 2.3 Hz), 129.4 (d, JC–P = 8.4 Hz), 128.9, 128.6 (d, JC–P = 22.2 Hz), 128.4 (d, JC–P = 4.6 Hz), 126.9 (d, JC–P = 29.9 Hz), 47.5 (d, JC–P = 18.4 Hz), 44.4 (d, JC–P = 136.5 Hz), 28.2, 20.1; HRMS (ESI+) calcd for C15H19NO5P (M + H+), 324.1001; found, 324.1003.
(1-([1,1′-Biphenyl]-4-yl)-3-(N-hydroxyacetamido)propyl)phosphonic Acid (13c)
Reagents: 12c (0.08 g, 0.2 mmol), TMSBr (0.10 mL, 0.76 mmol), and DCM (5 mL). Time: 5 h. Purification: gradient 5–45%, 70 min, 5 mL/min. 13c (38 mg, 0.11 mmol) was isolated as a white lyophilized material in 56% yield. 1H NMR (400 MHz, CD3OD) δ 7.54–7.63 (m, 4H), 7.38–7.47 (m, 4H), 7.28–7.35 (m, 1H), 3.56–3.67 (m, 1H), 3.42–3.51 (m, 1H), 3.11 (ddd, J = 3.1, 11.5, 23.1 Hz, 1H), 2.41–2.55 (m, 1H), 2.19–2.35 (m, 1H), 2.04 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 173.6, 142.1, 141.2 (d, JC–P = 3.1 Hz), 137.1 (d, JC–P = 6.9 Hz), 131.0 (d, JC–P = 6.9 Hz), 129.8, 128.3, 128.0 (d, JC–P = 2.3 Hz), 127.9, 47.5 (d, JC–P = 17.6 Hz), 44.0 (d, JC–P = 136.5 Hz), 28.2 (d, JC–P = 2.3 Hz), 20.2; HRMS (ESI+) calcd for C17H21NO5P (M + H+), 350.1157; found, 350.1151.
(1-(4-Bromophenyl)-3-(N-hydroxyacetamido)propyl)phosphonic Acid (13d)
Reagents: 12d (0.0608 g, 0.149 mmol), TMSBr (0.10 mL, 0.76 mmol), and DCM (5 mL). Time: 9 h. Purification: gradient 5–45%, 60 min, 5 mL/min. 13d (50 mg, 0.14 mmol) was isolated as a white lyophilized material in 96% yield. 1H NMR (400 MHz, CD3OD) δ 7.47 (d, J = 8.2 Hz, 2H), 7.27 (dd, J = 2.3, 8.5 Hz, 2H), 3.51–3.62 (m, 1H), 3.37–3.49 (m, 1H), 3.04 (ddd, J = 3.4, 11.4, 23.1 Hz, 1H), 2.37–2.52 (m, 1H), 2.11–2.27 (m, 1H), 2.00–2.06 (m, 3H); 13C NMR (101 MHz, CD3OD) δ 173.6, 137.4 (d, JC–P = 7.7 Hz), 132.4, 132.4, 121.8 (d, JC–P = 3.8 Hz), 47.3 (d, JC–P = 17.6 Hz), 43.7 (d, JC–P = 136.5 Hz), 28.1, 20.1; HRMS (ESI+) calcd for C11H16BrNO5P (M + H+), 351.9949; found, 351.9961.
(1-(Benzofuran-2-yl)-3-(N-hydroxyacetamido)propyl)phosphonic Acid (13e)
Reagents: 12e (0.10 g, 0.27 mmol), TMSBr (0.30 mL, 2.3 mmol), and DCM (5 mL). Time: 19 h. Purification: gradient 0–40%, 80 min, 5 mL/min. 13e (72 mg, 0.23 mmol) was isolated as a white lyophilized material in 85% yield. 1H NMR (400 MHz, CD3OD) δ 7.52 (d, J = 7.4 Hz, 1H), 7.44 (d, J = 8.0 Hz, 1H), 7.13–7.28 (m, 2H), 6.70 (d, J = 3.6 Hz, 1H), 3.65–3.78 (m, 1H), 3.50–3.63 (m, 1H), 3.39 (ddd, J = 3.5, 11.1, 23.3 Hz, 1H), 2.24–2.53 (m, 2H), 1.96–2.07 (m, 3H); 13C NMR (101 MHz, CD3OD) δ 173.8, 156.3, 155.1 (d, JC–P = 9.2 Hz), 130.1 (d, JC–P = 3.1 Hz), 124.7, 123.7, 121.6, 111.8, 106.0 (d, JC–P = 8.4 Hz), 47.4 (d, JC–P = 16.1 Hz), 38.7 (d, JC–P = 138.8 Hz), 26.9, 20.1; HRMS (ESI+) calcd for C13H17NO6P (M + H+), 314.0794; found, 314.0790.
(3-(N-Hydroxyacetamido)-1-(4-(pyridin-3-yl)phenyl)propyl)phosphonic Acid (13f)
Reagents: 12f (0.0842 g, 0.207 mmol), TMSBr (0.12 mL, 0.91 mmol), and DCM (5 mL). Time: 4 h. Purification: gradient 0–30%, 60 min, 5 mL/min. 13f (69 mg, 0.20 mmol) was isolated as a white lyophilized material in 95% yield. 1H NMR (400 MHz, CD3OD) δ 8.53–8.69 (m, 3H), 7.95 (dd, J = 5.6, 7.9 Hz, 1H), 7.50–7.63 (m, 4H), 3.55–3.66 (m, 1H), 3.40–3.51 (m, 1H), 3.17 (ddd, J = 3.0, 11.4, 22.7 Hz, 1H), 2.48–2.64 (m, 1H), 2.22–2.37 (m, 1H), 2.04 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 173.6, 143.1, 141.7, 141.6, 141.0, 140.3, 133.4 (d, JC–P = 3.1 Hz), 131.9 (d, JC–P = 6.1 Hz), 128.1, 127.8, 47.4 (d, JC–P = 16.9 Hz), 44.6 (d, JC–P = 133.4 Hz), 28.3, 20.2; HRMS (ESI+) calcd for C16H20N2O5P (M + H+), 351.1110; found, 351.1112.
(3-(N-Hydroxyacetamido)-1-(3′-methyl-[1,1′-biphenyl]-4-yl)propyl)phosphonic Acid (13g)
Reagents: 12g (0.114 g, 0.300 mmol), TMSBr (0.15 mL, 1.14 mmol), and DCM (5 mL). Time: 5 h. Purification: gradient 5–45%, 70 min, 5 mL/min. 13g (53 mg, 0.15 mmol) was isolated as a white lyophilized material in 54% yield. 1H NMR (400 MHz, CD3OD) δ 7.52–7.61 (m, 2H), 7.34–7.46 (m, 4H), 7.29 (t, J = 7.6 Hz, 1H), 7.14 (d, J = 7.5 Hz, 1H), 3.56–3.67 (m, 1H), 3.41–3.52 (m, 1H), 3.10 (ddd, J = 3.2, 11.1, 22.7 Hz, 1H), 2.42–2.56 (m, 1H), 2.39 (s, 3H), 2.19–2.35 (m, 1H), 2.04 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 173.6, 142.1, 141.3 (d, JC–P = 3.1 Hz), 139.5, 136.9 (d, JC–P = 6.9 Hz), 130.9 (d, JC–P = 6.1 Hz), 129.7, 128.9, 128.5, 128.0 (d, JC–P = 2.3 Hz), 125.0, 47.5 (d, JC–P = 18.4 Hz), 44.0 (d, JC–P = 136.5 Hz), 28.2 (d, JC–P = 2.3 Hz), 21.6, 20.2; HRMS (ESI+) calcd for C18H23NO5P (M + H+), 364.1314; found, 364.1319.
(3-(N-Hydroxyacetamido)-1-(4-(pyridin-4-yl)phenyl)propyl)phosphonic Acid (13h)
Reagents: 12h (0.040 g, 0.098 mmol), TMSBr (0.6 mL, 4.9 mmol), and DCM (5 mL). Time: 46 h. Purification: gradient 0–30%, 60 min, 5 mL/min. 13h (34 mg, 0.096 mmol) was isolated as a white lyophilized material in 98% yield. 1H NMR (400 MHz, CD3OD) δ 8.69 (d, J = 6.1 Hz, 2H), 8.27 (d, J = 6.6 Hz, 2H), 7.87 (d, J = 8.2 Hz, 2H), 7.60 (dd, J = 1.9, 8.3 Hz, 2H), 3.55–3.66 (m, 1H), 3.41–3.53 (m, 1H), 3.20 (ddd, J = 3.2, 11.6, 22.8 Hz, 1H), 2.47–2.64 (m, 1H), 2.22–2.38 (m, 1H), 2.03 (s, 3H); 13C NMR (101 MHz, CD3OD) δ 173.7, 158.3, 143.7, 142.9, 134.0, 132.0 (d, JC–P = 6.1 Hz), 128.9, 125.0, 47.3 (d, JC–P = 17.6 Hz), 44.6 (d, JC–P = 133.4 Hz), 28.1, 20.2; HRMS (ESI+) calcd for C16H20N2O5P (M + H+), 351.110; found, 351.1111.
(3-(N-Hydroxyacetamido)-1-(4-(thiophen-3-yl)phenyl)propyl)phosphonic Acid (13i)
Reagents: 12i (0.024 g, 0.058 mmol), TMSBr (0.25 mL, 1.9 mmol), and DCM (5 mL). Time: 19 h. Purification: gradient 5–45%, 20 min, 15 mL/min. 13i (14 mg, 0.041 mmol) was isolated as a white lyophilized material in 69% yield. 1H NMR (400 MHz, CD3OD) δ 7.55–7.68 (m, 3H), 7.42–7.49 (m, 2H), 7.39 (dd, J = 2.0, 8.3 Hz, 2H), 3.54–3.66 (m, 1H), 3.40–3.50 (m, 1H), 3.07 (ddd, J = 3.2, 11.3, 22.7 Hz, 1H), 2.39–2.54 (m, 1H), 2.17–2.32 (m, 1H), 2.01–2.07 (m, 3H); 13C NMR (101 MHz, CD3OD) δ 173.6, 143.3, 136.8 (d, JC–P = 6.9 Hz), 136.0 (d, JC–P = 3.1 Hz), 130.9 (d, JC–P = 6.1 Hz), 127.29, 127.26, 127.1, 121.1, 47.5 (d, JC–P = 18.4 Hz), 44.0 (d, JC–P = 136.5 Hz), 28.2, 20.2; HRMS (ESI+) calcd for C15H19NO5PS (M + H+), 356.0722; found, 356.0725.
(1-(4-(3,5-Dimethylisoxazol-4-yl)phenyl)-3-(N-hydroxyacetamido)propyl)phosphonic Acid (13j)
Reagents: 12j (0.082 g, 0.19 mmol), TMSBr (0.6 mL, 4.9 mmol), and DCM (5 mL). Time: 46 h. Purification: gradient 10–45%, 15 min, 15 mL/min. 13j (56 mg, 0.15 mmol) was isolated as a white lyophilized material in 78% yield. 1H NMR (400 MHz, CD3OD) δ 7.47 (dd, J = 2.0, 8.2 Hz, 2H), 7.30 (d, J = 8.0 Hz, 2H), 3.55–3.67 (m, 1H), 3.41–3.53 (m, 1H), 3.12 (ddd, J = 3.0, 11.3, 22.9 Hz, 1H), 2.43–2.57 (m, 1H), 2.40 (s, 3H), 2.20–2.36 (m, 4H), 2.04 (s, 3H), 13C NMR (101 MHz, CD3OD) δ 173.6, 166.8, 160.0, 137.7 (d, JC–P = 6.9 Hz), 131.0 (d, JC–P = 6.1 Hz), 130.2 (d, JC–P = 2.3 Hz), 130.0 (d, JC–P = 3.1 Hz), 117.6, 47.4 (d, JC–P = 17.6 Hz), 44.0 (d, JC–P = 135.0 Hz), 28.1 (d, JC–P = 1.5 Hz), 20.2, 11.4, 10.7; HRMS (ESI+) calcd for C16H22N2O6P (M + H+), 369.1216; found, 369.1214.
(3-(N-Hydroxyacetamido)-1-(4-morpholinophenyl)propyl)phosphonic Acid (13k)
Reagents: 12k (0.057 g, 0.14 mmol), TMSBr (0.3 mL, 2.3 mmol), and DCM (5 mL). Time: 7 h. Purification: gradient 0–15%, 10 min, 15 mL/min. 13k (48 mg, 0.13 mmol) was isolated as a white lyophilized material in 96% yield. 1H NMR (400 MHz, CD3OD) δ 7.37–7.45 (m, 2H), 7.30 (d, J = 8.5 Hz, 2H), 3.87–3.99 (m, 4H), 3.51–3.61 (m, 1H), 3.35–3.48 (m, 5H), 3.07 (ddd, J = 3.2, 11.5, 22.8 Hz, 1H), 2.40–2.55 (m, 1H), 2.12–2.28 (m, 1H), 1.99–2.07 (m, 3H); 13C NMR (101 MHz, CD3OD) δ 173.6, 146.5, 135.5, 131.9, 119.7, 66.6, 53.7, 47.3 (d, JC–P = 17.6 Hz), 43.6 (d, JC–P = 136.5 Hz), 28.2, 20.2; HRMS (ESI+) calcd for C15H24N2O6P (M + H+), 359.1372; found, 359.1375.
Acknowledgments
We would like to thank the Swedish Foundation for Strategic Research (SSF), the Swedish Research Council (VR), Knut and Alice Wallenberg’s foundation, and the EU Sixth Framework Program NM4TB CT:01892 for financial support.
Supporting Information Available
1H and 13C NMR spectra for compound 6a–6n, 10a–10e, 11a–11k, 12a–12k, 13a–13k as well as GC–MS spectra for 6b–6k, 6j–6n and LC-MS spectra for 6i and 13a–13k. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Dieck H. A.; Heck R. F. J. Org. Chem. 1975, 40, 1083. [Google Scholar]
- Cho C. S.; Uemura S. J. Organomet. Chem. 1994, 465, 85. [Google Scholar]
- Karimi B.; Behzadnia H.; Elhamifar D.; Akhavan P. F.; Esfahani F. K.; Zamani A. Synthesis 2010, 1399. [Google Scholar]
- Du X. L.; Suguro M.; Hirabayashi K.; Mori A.; Nishikata T.; Hagiwara N.; Kawata K.; Okeda T.; Wang H. F.; Fugami K.; Kosugi M. Org. Lett. 2001, 3, 3313. [DOI] [PubMed] [Google Scholar]
- Jung Y. C.; Mishra R. K.; Yoon C. H.; Jung K. W. Org. Lett. 2003, 5, 2231. [DOI] [PubMed] [Google Scholar]
- Andappan M. M.; Nilsson P.; Larhed M. Chem. Commun. 2004, 218. [DOI] [PubMed] [Google Scholar]
- Andappan M. M.; Nilsson P.; von Schenck H.; Larhed M. J. Org. Chem. 2004, 69, 5212. [DOI] [PubMed] [Google Scholar]
- Yoo K. S.; Yoon C. H.; Mishra R. K.; Jung Y. C.; Yi S. W.; Jung K. W. J. Am. Chem. Soc. 2006, 128, 16384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan J. W.; Li X. M.; Saidi O.; Xiao J. L. J. Am. Chem. Soc. 2008, 130, 2424. [DOI] [PubMed] [Google Scholar]
- Gottumukkala A. L.; Teichert J. F.; Heijnen D.; Eisink N.; van Dijk S.; Ferrer C.; van den Hoogenband A.; Minnaard A. J. J. Org. Chem. 2011, 76, 3498. [DOI] [PubMed] [Google Scholar]
- Lee A. W. M.; Martin V. S.; Masamune S.; Sharpless K. B.; Walker F. J. J. Am. Chem. Soc. 1982, 104, 3515. [Google Scholar]
- Haemers T.; Wiesner J.; Van Poecke S.; Goeman J.; Henschker D.; Beck E.; Jomaa H.; Van Calenbergh S. Bioorg. Med. Chem. Lett. 2006, 16, 1888. [DOI] [PubMed] [Google Scholar]
- Mackie P. R.; C.E. F.. Aldehydes: α,β-Unsaturated Aldehydes; Elsevier Pergamon: Amsterdam, London, 2005; Vol. 3. [Google Scholar]
- Blanchette M. A.; Choy Y.; Davis J. T.; Essenfield A. P.; Masamune S.; Roush W. R.; Sasaki T. Tetrahedron Lett. 1984, 25, 2183. [Google Scholar]
- Wadsworth W. S. J.; Emmons W. D. J. Am. Chem. Soc. 1961, 83, 1733. [Google Scholar]
- Ager D. J. Synthesis 1984, 384. [Google Scholar]
- Battistuzzi G.; Cacchi S.; Fabrizi G. Org. Lett. 2003, 5, 777. [DOI] [PubMed] [Google Scholar]
- Lindh J.; Enquist P. A.; Pilotti A.; Nilsson P.; Larhed M. J. Org. Chem. 2007, 72, 7957. [DOI] [PubMed] [Google Scholar]
- Hall D.Boronic Acids: Preparation and Applications in Organic Synthesis and Medicine; Wiley-VCH: Weinheim, 2005. [Google Scholar]
- Zebovitz T. C.; Heck R. F. J. Org. Chem. 1977, 42, 3907. [Google Scholar]
- Berthiol F.; Doucet H.; Santelli M. Catal. Lett. 2005, 102, 281. [Google Scholar]
- Jeffery T. J. Chem. Soc., Chem. Comm. 1984, 1287. [Google Scholar]
- Finkelstein B. L.; Benner E. A.; Hendrixson M. C.; Kranis K. T.; Rauh J. J.; Sethuraman M. R.; McCann S. F. Bioorg. Med. Chem. 2002, 10, 599. [DOI] [PubMed] [Google Scholar]
- Kobayashi S.; Ueda T.; Fukuyama T. Synlett 2000, 883. [Google Scholar]
- Nejjar A.; Pinel C.; Djakovitch L. Adv. Synth. Catal. 2003, 345, 612. [Google Scholar]
- Noel S.; Djakovitch L.; Pinel C. Tetrahedron Lett. 2006, 47, 3839. [Google Scholar]
- Quintiliani M.; Kahnt A.; Wolfle T.; Hieringer W.; Vazquez P.; Gorling A.; Guldi D. M.; Torres T. Chem.—Eur. J. 2008, 14, 3765. [DOI] [PubMed] [Google Scholar]
- Tanaka R.; Rubio A.; Harn N. K.; Gernert D.; Grese T. A.; Eishima J.; Hara M.; Yoda N.; Ohashi R.; Kuwabara T.; Soga S.; Akinaga S.; Nara S.; Kanda Y. Bioorg. Med. Chem. 2007, 15, 1363. [DOI] [PubMed] [Google Scholar]
- Unroe M. R.; Reinhardt B. A. Synthesis 1987, 981. [Google Scholar]
- Gupta A. K.; Song C. H.; Oh C. H. Tetrahedron Lett. 2004, 45, 4113. [Google Scholar]
- Andappan M. M. S.; Nilsson P.; Larhed M. Mol. Diversity 2003, 7, 97. [DOI] [PubMed] [Google Scholar]
- Global Tuberculosis Control 2010; World Health Organization: Geneva, 2010. [Google Scholar]
- Rohmer M.; Knani M.; Simonin P.; Sutter B.; Sahm H. Biochem. J. 1993, 295, 517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beytia E. D.; Porter J. W. Annu. Rev. Biochem. 1976, 45, 113. [DOI] [PubMed] [Google Scholar]
- Missinou M. A.; Borrmann S.; Schindler A.; Issifou S.; Adegnika A. A.; Matsiegui P. B.; Binder R.; Lell B.; Wiesner J.; Baranek T.; Jomaa H.; Kremsner P. G. Lancet 2002, 360, 1941. [DOI] [PubMed] [Google Scholar]
- Borrmann S.; Lundgren I.; Oyakhirome S.; Impouma B.; Matsiegui P. B.; Adegnika A. A.; Issifou S.; Kun J. F. J.; Hutchinson D.; Wiesner J.; Jomaa H.; Kremsner P. G. Antimicrob. Agents Chemother. 2006, 50, 2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrmann S.; Adegnika A. A.; Moussavou F.; Oyakhirome S.; Esser G.; Matsiegui P. B.; Ramharter M.; Lundgren I.; Kombila M.; Issifou S.; Hutchinson D.; Wiesner J.; Jomaa H.; Kremsner P. G. Antimicrob. Agents Chemother. 2005, 49, 3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuhara M.; Kuroda Y.; Goto T.; Okamoto M.; Terano H.; Kohsaka M.; Aoki H.; Imanaka H. J. Antibiot. 1980, 33, 24. [DOI] [PubMed] [Google Scholar]
- Kuzuyama T.; Shimizu T.; Takahashi S.; Seto H. Tetrahedron Lett. 1998, 39, 7913. [Google Scholar]
- Okuhara M.; Kuroda Y.; Goto T.; Okamoto M.; Terano H.; Kohsaka M.; Aoki H.; Imanaka H. J. Antibiot. 1980, 33, 13. [DOI] [PubMed] [Google Scholar]
- Jomaa H.; Wiesner J.; Sanderbrand S.; Altincicek B.; Weidemeyer C.; Hintz M.; Turbachova I.; Eberl M.; Zeidler J.; Lichtenthaler H. K.; Soldati D.; Beck E. Science 1999, 285, 1573. [DOI] [PubMed] [Google Scholar]
- Kuemmerle H. P.; Murakawa T.; Sakamoto H.; Sato N.; Konishi T.; Desantis F. Int. J. Clin. Pharmacol. Ther. 1985, 23, 521. [PubMed] [Google Scholar]
- Tsuchiya T.; Ishibashi K.; Terakawa M.; Nishiyama M.; Itoh N.; Noguchi H. Eur. J. Drug Metab. Pharmacokinet. 1982, 7, 59. [DOI] [PubMed] [Google Scholar]
- Dhiman R. K.; Schaeffer M. L.; Bailey A. M.; Testa C. A.; Scherman H.; Crick D. C. J. Bacteriol. 2005, 187, 8395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A. C.; Parish T. BMC Microbiol. 2008, 8, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuntz L.; Tritsch D.; Grosdemange-Billiard C.; Hemmerlin A.; Willem A.; Bacht T. J.; Rohmer M. Biochem. J. 2005, 386, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo Y.-H.; Fernandes R. P. M.; Proteau P. J. Bioorg. Med. Chem. 2006, 14, 2375. [DOI] [PubMed] [Google Scholar]
- Zingle C.; Kuntz L.; Tritsch D.; Grosdemange-Billiard C.; Rohmer M. J. Org. Chem. 2010, 75, 3203. [DOI] [PubMed] [Google Scholar]
- Devreux V.; Wiesner J.; Goeman J. L.; Van der Eycken J.; Jomaa H.; Van Calenbergh S. J. Med. Chem. 2006, 49, 2656. [DOI] [PubMed] [Google Scholar]
- Kurz T.; Schlüter K.; Kaula U.; Bergmann B.; Walter R. D.; Geffken D. Bioorg. Med. Chem. 2006, 14, 5121. [DOI] [PubMed] [Google Scholar]
- Verbrugghen T.; Cos P.; Maes L.; Van Calenbergh S. J. Med. Chem. 2010, 53, 5342. [DOI] [PubMed] [Google Scholar]
- Devreux V.; Wiesner J.; Jomaa H.; Rozenski J.; Van der Eycken J.; Van Calenbergh S. J. Org. Chem. 2007, 72, 3783. [DOI] [PubMed] [Google Scholar]
- Reichenberg A.; Wiesner J.; Weidemeyer C.; Dreiseidler E.; Sanderbrand S.; Altincicek B.; Beck E.; Schlitzer M.; Jomaa H. Bioorg. Med. Chem. Lett. 2001, 11, 833. [DOI] [PubMed] [Google Scholar]
- Andaloussi M.; Lindh M.; Björkelid C.; Suresh S.; Wieckowska A.; Iyer H.; Karlen A.; Larhed M. Bioorg. Med. Chem. Lett. 2011, 21, 5403. [DOI] [PubMed] [Google Scholar]
- Andaloussi M.; Henriksson L. M.; Wieckowska A.; Lindh M.; Björkelid C.; Larsson A. M.; Surisetti S.; Iyer H.; Srinivasa B. R.; Bergfors T.; Unge T.; Mowbray S. L.; Larhed M.; Jones T. A.; Karlen A. J. Med. Chem. 2011, 54, 4964. [DOI] [PubMed] [Google Scholar]
- Deng L.; Endo K.; Kato M.; Cheng G.; Yajima S.; Song Y. ACS Med. Chem. Lett. 2010, 2, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L.; Diao J.; Chen P.; Pujari V.; Yao Y.; Cheng G.; Crick D. C.; Prasad B. V. V.; Song Y. J. Med. Chem. 2011, (54), 4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawthorne M. F. J. Org. Chem. 1957, 22, 1001. [Google Scholar]
- Enquist P. A.; Nilsson P.; Sjoberg P.; Larhed M. J. Org. Chem. 2006, 71, 8779. [DOI] [PubMed] [Google Scholar]
- Adamo C.; Amatore C.; Ciofini I.; Jutand A.; Lakmini H. J. Am. Chem. Soc. 2006, 128, 6829. [DOI] [PubMed] [Google Scholar]
- 8-Quinolinylboronic acid, 3-quinolinylboronic acid, and 5-bromo-3-pyridinylboronic acid.
- Noteberg D.; Schaal W.; Hamelink E.; Vrang L.; Larhed M. J. Comb. Chem. 2003, 5, 456. [DOI] [PubMed] [Google Scholar]
- Wannberg J.; Sabnis Y. A.; Vrang L.; Samuelsson B.; Karlen A.; Hallberg A.; Larhed M. Bioorg. Med. Chem. 2006, 14, 5303. [DOI] [PubMed] [Google Scholar]
- Hartwig J. F.; Kawatsura M.; Hauck S. I.; Shaughnessy K. H.; Alcazar-Roman L. M. J. Org. Chem. 1999, 64, 5575. [DOI] [PubMed] [Google Scholar]
- Henriksson L. M.; Unge T.; Carlsson J.; Aqvist J.; Mowbray S. L.; Jones T. A. J. Biol. Chem. 2007, 282, 19905. [DOI] [PubMed] [Google Scholar]
- Glide, version 5.7; Schrödinger, LLC: New York, NY, 2011. [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. J. Med. Chem. 2004, 47, 1739. [DOI] [PubMed] [Google Scholar]
- Maestro, version 9.2; Schrödinger, LLC: New York, NY, 2011. [Google Scholar]
- Leung P. S. W.; Teng Y.; Toy P. H. Org. Lett. 2010, 12, 4996. [DOI] [PubMed] [Google Scholar]
- Avery T. D.; Caiazza D.; Culbert J. A.; Taylor D. K.; Tiekink E. R. T. J. Org. Chem. 2005, 70, 8344. [DOI] [PubMed] [Google Scholar]
- Kagawa N.; Sasaki Y.; Kojima H.; Toyota M. Tetrahedron Lett. 2010, 51, 482. [Google Scholar]
- Sagud I.; Faraguna F.; Marinic Z.; Sindler-Kulyk M. J. Org. Chem. 2011, 76, 2904. [DOI] [PubMed] [Google Scholar]
- Zumbansen K.; Dohring A.; List B. Adv. Synth. Catal. 2010, 352, 1135. [Google Scholar]
- Nakayama A.; Iwamura H.; Niwa A.; Nakagawa Y.; Fujita T. J. Agric. Food Chem. 1985, 33, 1034. [Google Scholar]
- Togninelli A.; Gevariya H.; Alongi M.; Botta M. Tetrahedron Lett. 2007, 48, 4801. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.