

Abstract
Sirtuin 2 (SIRT2) is one of seven known mammalian protein deacetylases homologous to the yeast master lifespan regulator Sir2. In recent years, the sirtuin protein deacetylases have emerged as candidate therapeutic targets for many human diseases, including metabolic and age-dependent neurological disorders. In non-neuronal cells, SIRT2 has been shown to function as a tubulin deacetylase and a key regulator of cell division and differentiation. However, the distribution and function of the SIRT2 microtubule (MT) deacetylase in differentiated, postmitotic neurons remain largely unknown. Here, we show abundant and preferential expression of specific isoforms of SIRT2 in the mammalian central nervous system and find that a previously uncharacterized form, SIRT2.3, exhibits age-dependent accumulation in the mouse brain and spinal cord. Further, our studies reveal that focal areas of endogenous SIRT2 expression correlate with reduced α-tubulin acetylation in primary mouse cortical neurons and suggest that the brain-enriched species of SIRT2 may function as the predominant MT deacetylases in mature neurons. Recent reports have demonstrated an association between impaired tubulin acetyltransferase activity and neurodegenerative disease; viewed in this light, our results showing age-dependent accumulation of the SIRT2 neuronal MT deacetylase in wild-type mice suggest a functional link between tubulin acetylation patterns and the aging brain.
INTRODUCTION
Sirtuin 2 (SIRT2) is one of seven known mammalian sirtuins (SIRT1-7), which comprise the non-canonical, NAD+-dependent class III family of histone deacetylases (HDACs) homologous to the yeast master lifespan regulator Sir2 (1). The founding member of this family, SIRT1, has been shown to modulate energy metabolism and longevity in a wide variety of species (1–3), and extension of lifespan in response to calorie restriction in yeast (4) and in Drosophila (5) requires sirtuin activity. The documented influence of sirtuin proteins on energy metabolism and lifespan extension in yeast and in invertebrate animals has prompted intense interest in this family of proteins as potential molecular targets for the treatment of metabolic and age-related diseases, including age-dependent neurological disorders, in humans (6–8).
Although the mammalian sirtuins all function as NAD+-dependent protein deacetylases (NDACs), SIRTs1-7 appear to act on different sets of substrates and thus to mediate distinct biological processes (6). Several lines of evidence suggest that enhanced expression and/or activity of SIRT1, the best characterized of the mammalian sirtuins, promotes survival of both neuronal and non-neuronal cells (6,9). Although less is known about the biological functions of the other sirtuins, mounting evidence indicates that—in contrast to SIRT1—excess SIRT2 might be deleterious to neurons (9,10) and suggests inhibition of SIRT2 as a therapeutic strategy for neurological disease. Our previous studies have shown that pharmacologic inhibition of SIRT2 exerts neuroprotective effects in diverse models of neurodegenerative disease, including Parkinson's disease (PD) models of α-synuclein toxicity (11) and invertebrate and primary neuronal models of Huntington's disease (HD) (12). In addition, reduced SIRT2 activity has been implicated in the resistance to axonal degeneration exhibited by slow Wallerian degeneration (WldS) mice, and enhanced expression of SIRT2 abolishes this resistance (10).
One possible mechanism by which inhibiting SIRT2 might mediate neuroprotective effects is via modification of the microtubule (MT) cytoskeleton. In non-neuronal cells, SIRT2 has been shown to function as an α-tubulin deacetylase and as a key regulator of cell division and differentiation (13–16). In neurons, recent studies have shown that MT acetylation is essential for normal neuronal development and function (17,18). Moreover, several pathologic features common to diverse neurodegenerative diseases, such as accumulation of misfolded or aggregated proteins, abnormal mitochondrial trafficking, and decreased synaptic connectivity, are MT-dependent cellular processes (19–21). Thus, in the central nervous system (CNS), modulation of neuronal SIRT2 levels and activity could potentially exert profound effects on fundamental cellular pathways that are directed by acetylated MTs, including trafficking, neurite outgrowth and/or synaptic remodeling (19,20,22,23). Previous studies of neuronal MT acetylation (24–26) have focused mainly on the activity of the other known α-tubulin deacetylase, histone deacetylase 6 (HDAC6) (27–29); however, SIRT2 is abundantly expressed in the CNS (16) and among the 18 known HDACs and sirtuins, it exhibits the highest RNA levels in postnatal brain tissue (30). Nonetheless, at present the distribution and function of the SIRT2 MT deacetylase in differentiated, postmitotic neurons remain largely unexplored.
In the studies described here, we investigated distinct protein isoforms of SIRT2 in the CNS and found that specific SIRT2 species are abundantly and preferentially expressed in the mammalian brain and spinal cord. We further observed age-dependent accumulation of this MT deacetylase in the CNS of wild-type mice. These results imply a role for SIRT2 in regulating MT acetylation patterns in neurons and suggest a novel mechanism by which neuronal function might become impaired in the aging brain.
RESULTS
Cohesive interpretation of published findings describing SIRT2 distribution and function is complicated by the existence of multiple isoforms of the protein. Three splice variants are predicted for mouse SIRT2 (Fig. 1A and B); these encode polypeptides of differing lengths with distinct amino-termini, and the two larger species have been documented in previous reports (10,31). To identify and characterize the SIRT2 isoforms expressed in the brain, we performed 5′ RACE experiments and cloned three full-length SIRT2 cDNAs from mouse cortex RNA. The sequences of these cloned cDNAs, referred to here as SIRT2.1 (sirtuin 2, isoform 1) (calculated MW 43 kDa), SIRT2.2 (sirtuin 2, isoform 2) (39.5 kDa) and SIRT2.3 (sirtuin 2, isoform 3) (35.6 kDa), are identical to those predicted for the three alternatively spliced isoforms of mouse SIRT2, suggesting that all three proteins are expressed in the brain. Transfection of the cloned cDNAs into mouse neuroblastoma (Neuro-2a—N2a) cells demonstrated co-migration of SIRT2.1 and SIRT2.2 with the two endogenous SIRT2 species present in these cells (Fig. 1C); no endogenous protein species corresponding to SIRT2.3 was detectable in these studies.
Figure 1.

Multiple protein isoforms are produced from the SIRT2 gene. (A) Diagram showing structural organization of the mouse SIRT2 gene (Entrez GeneID 64383). The locations of key features in the predicted coding sequence are indicated; patterned boxes denote exons encoding the conserved NDAC domain. The human SIRT2 gene (not shown, Entrez GeneID 22933) shares the same overall structure. (B) Schematics of the three cDNAs cloned from the mouse cortex, showing alignment with genomic sequence in (A). Sequences of cloned cDNAs are identical to the splice variants predicted by in silico algorithms (NCBI Accessions NM_022432.4, NM_001122765.1 and NM_001122766.1). Isoform 1 (SIRT2.1) contains all 16 exons. In isoform 2 (SIRT2.2), skipping of exon 2 results in premature termination due to a frameshift in exon 3; translation of the NDAC-containing polypeptide then initiates at a downstream AUG located in exon 4. This downstream translation initiation site is also present in full-length SIRT2.1 mRNA. Isoform 3 (SIRT2.3) results from splicing of exons 1 to 5 (skipping exons 2, 3 and 4); the exon 1–5 junction yields in-frame expression of the NDAC domain from the initiator in exon 1. (C) Immunoblot showing SDS–PAGE migration of SIRT2 isoforms in extracts of N2a cells transfected with the indicated SIRT2 cDNA clones. As the GAPDH signal illustrates (lower panel), samples from mock-transfected and SIRT2.3-transfected cells were deliberately overloaded for this blot, to permit comparison of electrophoretic mobilities of the cDNA-encoded SIRT2 proteins with endogenous SIRT2 in N2a cells. Doublet bands visible for each isoform represent phosphorlyated (upper) and unphosphorylated (lower) forms (30,31). (D) All three cloned SIRT2 isoforms promote deacetylation of α-tubulin in transfected N2a cells. Proteins were harvested 48 h following transfection with expression constructs containing SIRT2 or control (GFP) cDNAs; acetylated α-tubulin levels in the NP40-insoluble (i.e. MT-containing) fraction are shown. The apparent increase of SIRT2.2 expression in the SIRT2.1-transfected sample is likely due to the fact that, as illustrated in (A) and (B), the SIRT2.1 and SIRT2.2 isoforms can both be produced from wild-type SIRT2.1 mRNA depending on whether translation initiates at the upstream (SIRT2.1) or downstream (SIRT2.2) start codon.
We verified activity of all three isoforms cloned from the mouse brain by testing their ability to promote deacetylation of α-tubulin, a known protein substrate for SIRT2 (13,16,31) in transfected cells (Fig. 1D). Our results show that overexpression of each cDNA isoform results in a decrease in the amount of acetylated α-tubulin present in the NP40-insoluble (i.e. MT-containing) fraction in transfected cells. As shown in Figure 1D, SIRT2.1-transfected cells also express a significant amount of SIRT2.2, presumably due to initiation of translation at the downstream start codon present in wild-type SIRT2.1 mRNA (Fig. 1A and B). Thus, it is possible that modulation of acetylated α-tubulin in SIRT2.1-transfected cells could be mediated by both SIRT2.1 and SIRT2.2, or by the SIRT2.2 deacetylase alone. To ensure that the observed decrease in acetylated α-tubulin in these studies does not result from increased HDAC6 expression, we also assessed HDAC6 levels in SIRT2-tranfected cells (Supplementary Material, Fig. S1). HDAC6 levels were not increased in these studies; on the contrary, HDAC6 levels appear to be reduced in N2a cells in response to ectopic overexpression of SIRT2. Lastly, in our studies, the SIRT2.3 cDNA consistently yields lower expression levels than SIRT2.1 and SIRT2.2 constructs, yet the effects on α-tubulin acetylation are comparable for all three constructs. Although our experiments did not address the relative potencies of the different isoforms as MT deacetylases, these results suggest that even a modest increase in one or more SIRT2 proteins yields a marked reduction of acetylated α-tubulin in N2a cells.
Next, we used RNA interference-validated antibodies that detect endogenous SIRT2 (see Supplementary Material, Fig. S2, and Materials and Methods) to determine the expression patterns of SIRT2 isoforms across multiple mouse tissues. As shown in Figure 2A, SIRT2 exhibits strikingly abundant and preferential expression in the CNS but is present at much lower levels in peripheral tissues (Fig. 2A) and in cultured cell lines (Fig. 2 and data not shown). We further determined that SIRT2.1 (∼43 kDa) and SIRT2.2 (∼39 kDa) are readily detected in tissue extracts and that these isoforms exhibit distinct tissue- and cell type-specific expression patterns. The shorter SIRT2.2 isoform is expressed at extremely high levels in the CNS, whereas the larger SIRT2.1 species predominates in the skeletal muscle (Fig. 2A) and in immortalized cells in culture. Given that neurons contain significantly more acetylated α-tubulin than other cell types, we assessed whether preferential expression in the CNS is a general feature of tubulin deacetylases (Fig. 1A, lower panels). Our results show that the tissue distribution of HDAC6 is distinct from that of SIRT2 and confirm that, as has been reported previously (32), HDAC6 expression is not specifically enriched in the brain.
Figure 2.
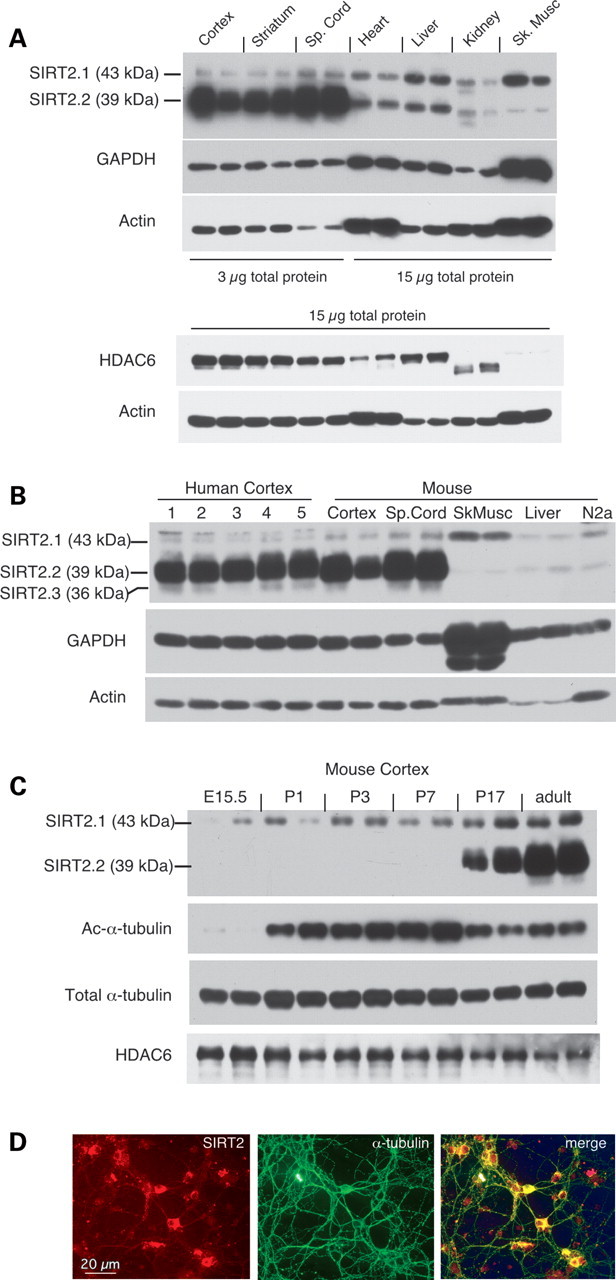
Specific SIRT2 isoforms are abundantly and preferentially expressed in the CNS. (A). Upper panels: distribution of SIRT2 expression in tissues from adult (5-month-old) C57BL6/J mice. Samples from two animals are shown. To permit detection of SIRT2 in peripheral tissues, protein extracts from these tissues were loaded at 5× higher concentration than those from CNS nonetheless, prolonged exposure of the blot was required to visualize SIRT2 in peripheral samples. GAPDH and actin are shown to indicate sample loading. Lower panels: HDAC6 exhibits a tissue distribution pattern distinct from that of SIRT2. For this experiment, equivalent amounts of total protein extracts were loaded for all tissues; actin signal is shown below. (B). SIRT2 expression and isoform distribution in human cortical extracts is comparable with that in mouse CNS tissues, in contrast to the low levels detected in the mouse skeletal muscle, liver, and in cultured N2a cells. Case information for human samples is presented in Supplementary Material, Table S1. (C). SIRT2 protein expression is a marker of the mature CNS. Immunoblots show SIRT2, acetylated α-tubulin, total α-tubulin and HDAC6 expression in cortical extracts from mice at embryonic day 15.5 (E15.5), at postnatal days 1, 3, 7, and 17, and from adult (5-month-old) animals. Samples from two animals at each stage are shown. (D). Left: SIRT2 labeling, using same antibodies as in (A) and (B), of perikarya and neurites in cultured mouse embryonic cortical neurons at 10 days in vitro (DIV). Middle: α-tubulin; right: merged image showing overlap of SIRT2 (red) and α-tubulin (green) staining of neuronal processes.
To verify that the SIRT2.2 species is a brain-enriched protein in mammals, we analyzed SIRT2 expression in human cortical tissue and found it identical to that observed in the mouse (Fig. 2B). Moreover, although we had not previously appreciated the SIRT2.3 isoform in immunoblots of mouse tissue extracts, the human samples clearly revealed the presence of a third distinct SIRT2 band migrating at the expected size for SIRT2.3. Because of the extremely close migration of SIRT2.2 and SIRT2.3, and the overwhelming abundance of SIRT2.2 in these samples, these species are not easily resolved in mouse extracts; however, the electrophoretic mobilities of human SIRT2.2 and SIRT2.3 isoforms differ slightly from the mouse and thus were more distinctly separated on SDS–PAGE. Lastly, although the SIRT2.3 isoform has not previously been reported for human SIRT2, mining of existing deep-sequencing data sets from human samples (publicly available at http://www.broadinstitute.org/igvdata/BodyMap/IlluminaHiSeq2000_BodySites/) showed that all three splice variants are produced in the human brain (E.T. Wang and D.E. Housman, personal communication).
Our early studies of SIRT2 expression in different cell and tissue types revealed that endogenous SIRT2 is expressed at extremely low levels in immortalized cultured cells, including mouse N2a (Figs 1 and 2) and human SH-SY5Y neuroblastoma and U87-MG glioblastoma lines (not shown). In N2a cells, SIRT2 RNA and protein levels are enhanced upon differentiation to a neuron-like phenotype (Supplementary Material, Fig. S3A and B). In cultured mouse embryonic cortical neurons, SIRT2 expression is low at plating and increases as the cultures mature (Supplementary Material, Fig. S3C and D); these findings are in accordance with published studies showing that the gene is normally expressed postnatally (16,30) and suggested that SIRT2 expression might be a marker of mature, fully developed neurons. To investigate this possibility, we assessed SIRT2 expression in the mouse cortex in late embryonic and early postnatal development and compared it with the pattern observed in adult animals (Fig. 2C). Our results confirm that SIRT2 is expressed at very low levels in the developing CNS, as has been reported previously (16), and that accumulation of SIRT2.2 in the cortex is a relatively late postnatal event. Further, these studies show that acetylated α-tubulin levels increase dramatically in the early postnatal cortex (Fig. 2C) and then exhibit a modest reduction in samples from late postnatal and adult animals, when SIRT2 is abundant. In contrast, levels of the HDAC6 MT deacetylase remained largely similar throughout the developmental stages examined. These results suggest that high-level SIRT2 expression and preferential accumulation of SIRT2.2 are markers of the mature CNS.
In previous reports, SIRT2 has been alternately described as both an oligodendroglial (16,33) and a neuronal (12,30) protein; therefore, we next wished to assess the distribution of SIRT2 immunoreactivity in specific CNS cell types. In late-stage primary cultures of embryonic cortical neurons, SIRT2-specific antibodies label both neuronal perikarya and processes (Fig. 2D), and SIRT2 staining in neuronal processes is generally associated with MTs. In accordance with previous reports (15,16), we detect robust SIRT2 labeling of CNPase-positive oligodendrocytes in primary mixed cortical cultures (Supplementary Material, Fig. S4A), whereas GFAP-stained astrocytes in these cultures were at best weakly positive for SIRT2 (Supplementary Material, Fig. S4B).
Embryonic cortical cultures cannot accurately represent mature CNS cells; we therefore extended our in vitro findings to confirm neuronal expression of SIRT2 in adult mouse brain sections (Fig. 3). Our results show that SIRT2 antibodies brightly label both fiber tracts and neuronal (i.e. NeuN-positive) cell bodies throughout the cortex (Fig. 3A) and in the cerebellum stain Purkinje cells (Fig. 3B), molecular layer neurons and fiber tracts (Fig. 3B–D). As expected, SIRT2 labeling overlapped with that of the oligodendroglial marker CNPase in fiber tracts (shown in the granule cell layer of the cerebellum, Fig. 3C); however, we did not detect robust SIRT2 labeling of oligodendroglial cell bodies (Fig. 3C, merged inset and Supplementary Material, Fig. S5). Further, in accordance with previous reports (15,16,33), in our studies SIRT2 staining did not coincide with that of the astrocyte marker GFAP (Fig. 3D and Supplementary Material, Fig. S5). Overall, these results suggest that the SIRT2 species that is most abundant in CNS tissues is predominantly a neuronal protein.
Figure 3.
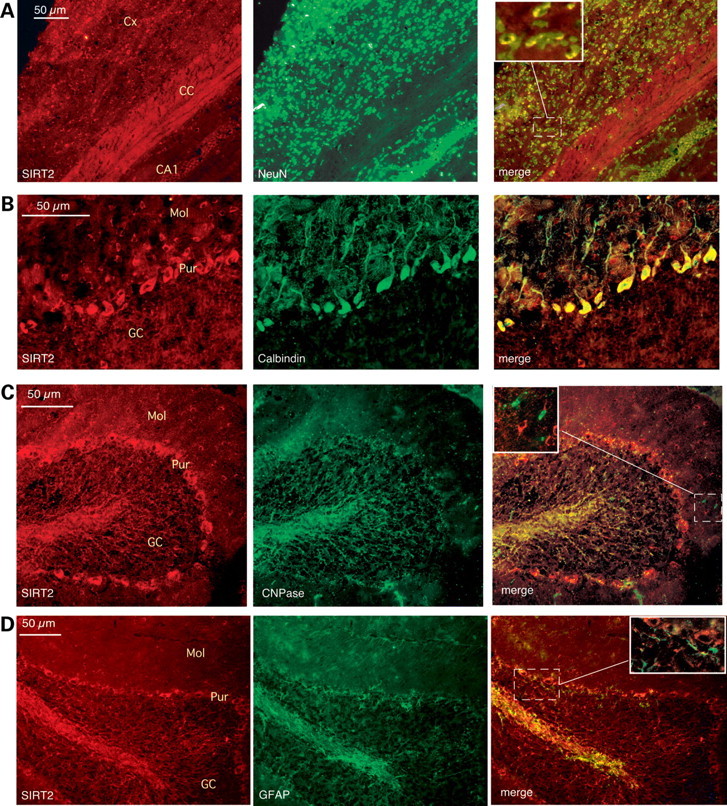
SIRT2 in the adult brain is predominantly a neuronal protein. (A–D). Immunofluorescence microscopy of midsagittal sections from the adult (5-month-old) mouse brain, showing SIRT2 immunoreactivity (red in all panels) in perikarya of NeuN-positive cells in the mouse cortex (A) and in Purkinje cells, molecular layer neurons and fiber tracts in the cerebellum (B and C). (A) Double-immunolabeling of SIRT2 (red) and the neuronal marker NeuN (green) in the cortex. Cx, cortex; CC, corpus callosum; CA1, hippocampal CA1 region. (B) Double-immunolabeling of SIRT2 (red) and the Purkinje cell marker calbindin (green) in the cerebellum. Mol, molecular layer; Pur, Purkinje neurons; GC, granule cell layer. (C) SIRT2 staining of fiber tracts, but not cell bodies, in the cerebellum overlaps with the oligodendroglial marker CNPase (green). (D) SIRT2 immunoreactivity is not detected in astrocytes labeled with GFAP (green). Individual color images for the insets in (C) and (D) are shown in Supplementary Material, Figure S5.
Our previous studies demonstrated that pharmacologic inhibition of SIRT2 exerts neuroprotective effects in invertebrate and cell-based models of PD and HD (11,12). Given that advent and progression of symptoms in these neurodegenerative diseases are age-dependent and that the sirtuin family of deacetylases is implicated in the regulation of longevity, we wished to determine whether SIRT2 expression levels vary with age. We therefore analyzed spinal cord extracts from young adult (4–5 month, n = 9) and aged (19–22 month, n = 11) C57BL6 mice, and these studies yielded two unexpected findings (Fig. 4A). First, although we had not previously detected appreciable amounts of the SIRT2.3 isoform in extracts from cultured cells or in tissues from young mice, a third protein species that likely represents SIRT2.3 is clearly present in the CNS of aged animals. Further, we detect significant accumulation of this faster migrating SIRT2 species in both the spinal cords (Fig. 4A and C) and cortices (not shown) of 19-month-old mice (Fig. 4C, P= 0.001 for spinal cord SIRT2.3). We did not detect statistically significant changes in the levels of either SIRT2.1 or SIRT2.2 in these studies; however, age-dependent accumulation of the SIRT2.3 isoform results in a modest but significant increase in overall SIRT2 levels in the CNS of aged mice (Fig. 4C, P= 0.014 for total spinal cord SIRT2). Because SIRT2 has been shown to function as an MT deacetylase, we assessed overall levels of acetylated α-tubulin in a subset of these animals (Fig. 4B). We detected a trend suggesting decreased α-tubulin acetylation in aged animals (from 0.94 ± 0.2 to 0.84 ± 0.06, Fig. 4C, right); however, this modest decrease was not statistically significant.
Figure 4.

SIRT2 accumulates in the CNS of aged mice. (A–C). SIRT2 expression (A and C) and acetylated α-tubulin levels (B and C) in spinal cords from 4-month and 19-month C57BL6/J mice. (D and E) SIRT2 and HDAC6 expression in cortices from 3-month and 18-month B6CBA mice. Lower panel in each shows GAPDH. (A and D). Representative immunoblots showing accumulation of the faster migrating SIRT2 species in aged mice. Prolonged exposures of the SIRT2 blots are shown to illustrate differences in the less-abundant SIRT2.3 isoform. (C and E) Quantification of SIRT2.3 (left, expressed as a fraction of total SIRT2) and total SIRT2 protein (right), from lighter exposures of blots as in (A) and (D), shows a statistically significant increase in SIRT2 levels in young (gray bars) versus aged (black bars) animals. Aged animals also exhibit a modest decrease in acetylated α-tubulin (C, right) that is not statistically significant (P = 0.19). Protein levels for each sample were normalized to GAPDH and results are shown as relative percentages. Total SIRT2 in spinal cords increased from 0.91 ± 0.03 (4–5 months, n = 9) to 1.15 ± 0.12 (19–22 months, n = 11); P = 0.014. Total SIRT2 in cortices increased from 0.90 ± 0.04 (3 months, n = 10) to 1.12 ± 0.09 (18 months, n = 10); P = 0.003. Error bars indicate standard deviation; two-tailed P-values were calculated using Student's t-test with α = 0.05.
To ensure that the observed age-dependent differences in SIRT2 levels were not limited to a particular group of animals, we next analyzed cortices from young (3 month, n = 10) and aged (18 month, n = 10) animals of a hybrid strain background (B6CBA) from an unrelated mouse colony. We again observed significant accumulation of SIRT2.3 in cortices from aged animals (Fig. 4D and E, P= 0.001 for cortex SIRT2.3; P= 0.003 for total cortex SIRT2). In contrast, we did not detect any significant age-dependent differences in levels of the MT deacetylase HDAC6 (Fig. 4D). Analysis of overall levels of acetylated α-tubulin in cortical samples from these animals again showed a modest decrease in aged animals that was not statistically significant (Supplementary Material, Fig. S6); nonetheless, this small decrease was consistent with that observed in spinal cord samples. Finally, retrospective analysis (not shown) of immunoblot data for a different group of mice [generated in a separate study (34)] revealed that this third SIRT2 isoform was readily detectable in all cortical samples from older animals (15–24 months, n = 20) but not in those from young animals (3–5 months, n = 8). Taken together, our results strongly suggest that accumulation of SIRT2.3 is an age-dependent marker in the CNS.
Although HDAC6 is generally considered to be the principal tubulin deacetylase in most cells, as noted above HDAC6 levels are low in the CNS compared with SIRT2 (30) and are higher in some peripheral tissues [Fig. 2A and (32)]. Further, HDAC6 knockout mice show increased α-tubulin acetylation in all tissues examined except the brain (32). Intriguingly, although SIRT2 is expressed abundantly and preferentially in the CNS, it is not known whether endogenous SIRT2 modulates MT acetylation patterns in neurons. To investigate this possibility, we assessed SIRT2 and total and acetylated α-tubulin staining patterns in late-stage cultured primary neurons (Fig. 5A–C). Although SIRT2 generally co-localizes with α-tubulin staining in neurons (Fig. 5A), our results show non-uniform distribution of endogenous SIRT2 in neuronal perikarya and processes (Fig. 5A–C). Further, focal accumulation of this protein is associated with areas of reduced α-tubulin acetylation in both cell bodies (Fig. 5B) and neurites (Fig. 5C).
Figure 5.

Focal accumulation of SIRT2 coincides with reduced MT acetylation in neurons. (A). SIRT2 antibodies label MTs in processes of cultured primary cortical neurons (DIV 10). Panels show SIRT2 (left), total α-tubulin (middle) and merged image (right) indicating overlapping staining of SIRT2 and α-tubulin. (B and C). Areas of high-level endogenous SIRT2 expression (arrows) overlap with reduced α-tubulin acetylation in perikarya (B) and processes (C) of cultured primary neurons (DIV 11). Panels show SIRT2 (left), acetylated α-tubulin (middle) and merged images. (D). Fractionation of proteins from cultured primary neurons shows that endogenous SIRT2.2 is found primarily in the insoluble pellet (P) fraction; in contrast, SIRT2.1 is found largely in the soluble (S) fraction. Samples from five independent cultures are shown. (E). Brain-enriched SIRT2 isoforms are associated with the insoluble fraction in transfected cells. Like SIRT2.2, a significant amount of SIRT2.3 is found in the NP40-insoluble (P) fraction of transiently transfected N2a cells. Endogenous SIRT2.1 in these cells is found almost exclusively in the soluble (S) fraction.
To extend these observations, we investigated whether the brain-enriched isoforms of SIRT2 are stably associated with neuronal MTs. Crude biochemical fractionation of endogenous SIRT2 proteins from cultured primary cortical neurons reveals that the SIRT2.2 isoform is preferentially associated with the NP40-insoluble (i.e. MT-containing) fraction, whereas SIRT2.1 is found almost exclusively in the soluble fraction (Fig. 5D). Moreover, on blots such as that shown in Figure 5D, we noted the presence of a faint third band migrating similarly to SIRT2.3 in the pellet fractions only. Because SIRT2.3 levels are low-to-undetectable in primary neurons, however, we were unable to confirm the fractionation profile of endogenous SIRT2.3 in these cells. As an alternative approach, we compared fractionation profiles of SIRT2.2 and SIRT2.3 in transfected N2a cells and found significant association of both isoforms with the insoluble fraction (Fig. 5E); again, endogenous SIRT2.1 in these samples was detected only in the soluble fraction, as has been previously reported (10). Taken together, these results suggest that the brain-enriched SIRT2 isoforms—SIRT2.2 and SIRT2.3—are normally associated with MTs in neurons.
DISCUSSION
The studies presented here were designed to investigate the distribution and function of the SIRT2 MT deacetylase in the mammalian CNS. Our results describe the isolation and initial characterization of three distinct SIRT2 isoforms and demonstrate strikingly abundant and preferential expression of the SIRT2.2 species in the brain and spinal cord. Moreover, these studies indicate that high-level SIRT2 expression is a marker of the adult CNS and show that SIRT2 is predominantly a neuronal protein in the adult brain. Finally, our results imply that the brain-enriched isoforms of SIRT2 are MT-associated proteins in mature neurons.
The different cell- and tissue-type expression patterns of SIRT2 isoforms, together with their biochemical fractionation properties, strongly suggest that these proteins may exert distinct biological functions in specific cell types. Several substrates other than α-tubulin have been suggested for SIRT2 in non-neuronal cells, including histone H4 (35) and the forkhead transcription factors FoxO1 (36,37) and FoxO3a (38). In addition, our recent studies suggested a link between SIRT2 activity and regulation of neuronal cholesterol levels (12). The specific biological activities mediated by SIRT2 in a given cell likely depend both on the SIRT2 isoform and substrate combinations expressed in that cell type and on the proximity of the SIRT2 deacetylase(s) to those substrates. The larger SIRT2.1 isoform appears to predominate in non-neural tissues and in immortalized cells in culture and is easily extracted by using non-ionic detergents. In contrast, our results suggest that the brain-enriched isoforms of SIRT2 are normally associated with neuronal MTs. In addition, our own unpublished studies (not shown) and previous reports (31) indicate that the two smaller SIRT2 isoforms cannot be maintained by dividing cells in culture and, taken together with the results described here, suggest that these proteins are normally active in mature, fully differentiated neurons. In future experiments, it will be of great interest to investigate whether the three SIRT2 isoforms described here exhibit distinct subcellular localizations and/or substrate preferences, and whether any observed differences are specific to neurons versus non-neuronal cells.
Surprisingly, our results showed significant age-dependent accumulation of a novel SIRT2 species in the mouse brain and spinal cord; to our knowledge, these findings represent the first documented age-dependent change in a mammalian sirtuin. We note that, although the experiments presented here do not permit unequivocal identification of the observed age-associated SIRT2 species as SIRT2.3, this supposition is supported both by the demonstrated specificity for SIRT2 of the antibody used in these studies and by the fact that the novel species migrates on SDS–PAGE at the expected size for SIRT2.3. However, we acknowledge that this protein could represent another as-yet-uncharacterized variant of SIRT2 or a post-translational modification of SIRT2.1 or (more likely) SIRT2.2. The precise molecular nature of the age-associated SIRT2 protein described here will be elucidated in follow-up studies using mass spectrometry; nevertheless, our results strongly suggest that increased overall SIRT2 levels are associated with aging in the CNS.
Although often described as a marker of stable MTs, acetylation of α-tubulin is a non-uniform posttranslational modification that occurs at discrete regions along cellular MTs, and appears to be a consequence rather than a cause of MT stability (19,20). Our finding that strong SIRT2 immunoreactivity is associated with focal areas of decreased acetylated α-tubulin staining in neurons implies a key role for the brain-enriched isoforms of SIRT2 in modulating neuronal MT acetylation patterns. Moreover, the extremely high levels of SIRT2 protein in CNS tissues, in contrast to the low levels observed in non-neural tissues and in cultured cells, suggest that SIRT2 may function as the predominant MT deacetylase in mature neurons. The striking abundance of the SIRT2 deacetylase in the CNS might also account for the observation that tubulin is not hyperacetylated in the brains of HDAC6-deficient mice (32), since SIRT2 activity could potentially compensate for loss of HDAC6 in these tissues.
The precise functions of tubulin acetylation in neurons are not entirely clear, but are beginning to be elucidated from new studies identifying the molecular players that regulate this modification. Previous key reports have documented a role for acetylated MTs in directing trafficking of motor proteins (24,39,40) and in establishing and maintaining synaptic targets (17), processes that exhibit deficits in both normal aging and in neurodegenerative disorders (22,41). For example, post-mortem cortical tissue from HD patients displays reduced levels of acetylated α-tubulin (24,42), and impairment of intracellular trafficking in HD model neurons is attenuated by pharmacologic inhibition of HDAC6 (24). Perhaps most significantly, recent works have identified the elongation protein 3 (Elp3) subunit of the Elongator complex as an α-tubulin acetyltransferase and shown that activity of this complex is required for neuronal development and maturation in both mice (17) and worms (18). Moreover, defects in elongation proteins are associated with neurodegenerative disease: mutations affecting expression of the catalytic subunit, Elp3, show genetic association with sporadic amyotrophic lateral sclerosis (43), and mutations in the structural subunit cause familial dysautonomia (44,45).
These recent findings imply that establishment and maintenance of appropriate MT acetylation patterns are critical to normal neuronal function. Thus, the age-dependent accumulation of the SIRT2 MT deacetylase in the mouse brain and spinal cord, which we have detected in all samples examined to date, could potentially have significant functional consequences for older animals. Although the observed increase in SIRT2 is associated with only a modest reduction in overall levels of acetylated α-tubulin, it is conceivable that moderate or focal decreases in MT acetylation could exert profound effects on localized acetylation-dependent cellular pathways. Viewed in light of the exciting new discoveries documenting the importance of MT acetylation for neuronal function, our finding that MT-associated species of SIRT2 accumulate in the CNS of aged mice suggests a tantalizing link between MT acetylation and the aging brain. In addition, our results implicate a specific molecular mechanism that could account, at least in part, for the previously reported beneficial effects of SIRT2 inhibition in diverse models of neurodegenerative disease (11,12). Taken together, the studies presented here highlight a previously unexplored cellular pathway that might underlie age- or disease-dependent impairment of neuronal function, and further underscore the potential of SIRT2 as a viable molecular target for neuroprotective therapy.
MATERIALS AND METHODS
SIRT2 cDNA cloning
Full-length SIRT2 cDNA clones were obtained by 5′ RNA ligase-mediated RACE using the GeneRacer™ Kit (Invitrogen), which permits selective amplification of full-length, 5′-capped mRNAs. One microgram of total RNA isolated from mouse cortex was used as the starting material for the RACE procedure. RACE PCR reactions were performed using the GeneRacer 5′ oligos provided and SIRT2-specific 3′ primers (first-round primer 5′-CTGCATGTTAAGAGGGCCCAGTGC-3′; nested primer 5′-CTCGAGCTGTCCTGCGGGAGGTCATGGTTA-3′). PCR products were directionally cloned into the mammalian expression vector pcDNA3.1 (Invitrogen). Because we observed that addition of an epitope-tag can alter biochemical characteristics of SIRT2, all expression constructs used in these studies contained untagged SIRT2 cDNAs. Constructs were sequence-verified prior to use.
Cell culture and transfections
N2a murine neuroblastoma cells were obtained from the ATCC (CCL-131) and cultured according to standard procedures. N2a cells were maintained in Eagle's minimal essential medium (EMEM) supplemented with 10% fetal bovine serum, 2 mm l-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. For differentiation experiments, cells were plated at low density and 24 h later were switched to EMEM containing 1% donor horse serum, 2 mm L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, and supplemented with 10 µm all-trans retinoic acid (Sigma). Plasmid DNA and DNA-based shRNA transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's recommendations and published procedures (46). Transfection efficiency was estimated using a pcDNA-EGFP expression construct. These parameters routinely yield transfection efficiencies of >90% in N2a cells.
Protein extraction from cultured cells
Dishes were rinsed with PBS and a suitable volume of 1× lysis buffer was added directly to the culture dish. For total cell extracts (TCEs), 1× TCE buffer (50 mm Tris–Cl at pH 7.0, 2% SDS, 10% glycerol and 1 mm DTT) was preheated to 65°C before use. Lysates were collected by scraping, transferred to microtubes and subjected to two rounds of boiling at 100°C for 5 min followed by sonication for 1 min. For crude fractionation experiments, we used NP40 lysis buffer containing 50 mm Tris–Cl, pH 7.5, 150 mm NaCl and 1% NP-40, supplemented with 1 mm DTT, 1 mm PMSF and Complete™ Protease Inhibitor Cocktail (Roche). NP-40 lysates were collected from culture dishes by scraping and transferred to microtubes. Soluble and insoluble protein fractions were obtained by rotating samples at 4°C for 15 min, followed by centrifugation at 4°C for 10 min at 8000g. Supernatants, containing NP40-soluble proteins, were transferred to fresh tubes, and pellet fractions were solubilized by addition of 1/10-volume TCE buffer, followed by two rounds of boiling and sonication as described above. Proteins were quantified using the BCA Protein Assay Kit (Pierce).
qRT-PCR
SIRT2 RNA levels were measured by quantitative RT-PCR using TaqMan primers and probes (Applied Biosystems). Total cellular RNA was isolated using the RNeasy Kit (Qiagen), and qPCR was performed using the Superscript III Platinum Two-Step qRT-PCR Kit (Invitrogen). For relative quantification, reactions were multiplexed with either mouse β-actin or GAPDH, as indicated in the text, as an internal control. Assays were run on a 7500 Fast Real-Time PCR System (Applied Biosystems).
Mouse and human tissue samples
Mice used in this study were wild-type C57BL6/J inbred or B6CBA hybrid animals and were maintained in a specific pathogen-free facility at the Massachusetts General Hospital. Tissues were collected from groups of animals at the ages indicated in the text; both male and female animals were used and no differences between the sexes were observed. All animal experiments were carried out in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals and were approved by the MGH Subcommittee on Research Animal Care.
Human cortical tissues were obtained from the Tissue Resource Center of the Alzheimer Disease Research Center (ADRC) at the Massachusetts General Hospital and used according to institutional regulations.
Primary cultures
Primary mouse neuronal cultures were prepared from E15.5–16.5 mouse embryos via published procedures (47), which yield a population comprised mostly of NeuN-positive neurons and containing a few residual glial cells. For microscopy experiments, neurons were plated at 8 × 104 cells/cm2 onto glass cover slips, which had previously been coated overnight with poly-L-lysine solution followed by 1h with ECL Cell Attachment Matrix (Millipore). Cultures were maintained in neurobasal medium (Invitrogen) supplemented with B-27, N2, 2 mm L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin and 6 mm glucose. On DIV3, cultures were treated with 10 µm AraC for 24 h to inhibit glial cell proliferation. For the remainder of the culture period, one-half volume of medium was replaced every third day. Neurons were fixed in 4% paraformaldehyde (PFA) for 15 min and permeabilized using 0.1% Triton-X100 in PBS. Immunostaining and fluorescence microscopy were performed as described for tissue sections.
Protein extraction from tissues
For preparation of mouse protein samples, tissues were rapidly dissected and flash-frozen in liquid nitrogen prior to extraction. Human cortical extracts were prepared from frozen post-mortem specimens. Frozen tissue samples were homogenized in 5–10 volumes of either PBS containing 1 mm PMSF and Complete™ Protease Inhibitor Cocktail (Roche) or modified RIPA buffer (50 mm Tris, pH 8, 150 mm NaCl, 1% NP40, 0.5% NaDeoxycholate, 0.1% SDS, supplemented with 5 mm EDTA, 1 mm PMSF and Complete™ Protease Inhibitor). PBS homogenates were used to prepare TCEs as follows: aliquots of total tissue homogenate were removed and proteins extracted by addition of an equal volume of 2× TCE buffer (1× buffer contains 50 mm Tris–Cl at pH 7.0, 2% SDS, 10% glycerol and 1 mm DTT). TCEs were subjected to two rounds of boiling at 100°C for 5 min followed by sonication for 1 min. RIPA extracts were incubated at 4°C for 30 min and clarified by centrifugation at 4°C for 15 min at 20 000g. Proteins were quantified using the BCA Protein Assay Kit (Pierce).
Immunoblotting
For immunoblotting, 3–15 µg of total cell or tissue extract was fractionated by SDS–PAGE and transferred to PVDF membrane (Immobilon-P, Millipore). Maximum resolution of SIRT2 isoforms was achieved by electrophoresis of samples on 11.25% gels, which were then used for SIRT2 immunoblots throughout these studies. Primary antibodies used for immunoblotting were as follows: rabbit anti-SIRT2 (S8447, Sigma-Aldrich), mouse anti-GAPDH (clone 6C5, MAB374, Millipore), mouse anti-α-tubulin (T6074, Sigma-Aldrich), mouse anti-acetylated α-tubulin (clone 611B-1, T6793, Sigma-Aldrich), rabbit anti-actin (A2066, Sigma-Aldrich) and rabbit anti-HDAC6 (NB100-91805, Novus Biologicals). Secondary antibodies were horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG (Sigma-Aldrich). Bound antibodies were detected using the ECL Plus Kit (Amersham).
Quantification and statistical analysis of immunoblot data
Sample loading was estimated and data were normalized to control signal (GAPDH, β-actin and/or α-tubulin) obtained either from blots stripped and re-probed or from blots of identical samples run and probed in parallel. All samples were analyzed in at least three independent experiments. Band intensity on exposures showing linear signal range was quantified using ImageJ. Data were analyzed using Microsoft Excel; reported P-values are two-tailed and were obtained using Student's t-test with α = 0.05.
Immunohistochemistry of mouse brain sections
To prepare tissues for histology, mice were deeply anesthetized with isoflurane and transcardially perfused with 4% PFA in phosphate buffer. Whole brains were dissected, post-fixed for 24 h in 4% PFA and gradually equilibrated in 30% sucrose solution prior to being cut in half along the midline and mounted in OCT. Midsagittal sections were cut to 10 μm using a cryostat (Thermo-Shandon) and mounted onto glass slides. Sections were postfixed in 4% PFA for 10 min and nonspecific sites were blocked by incubation in PBS containing 10% normal goat serum and 0.3% Tween-20. Sections were incubated in primary antibody diluted in PBS + 0.1% Tween-20 for 3 h at room temperature or overnight at 4°C, washed in PBS and incubated for 1 h at room temperature in fluorochrome-conjugated anti-rabbit and/or anti-mouse secondary antibodies. ‘No primary’ controls were included in all immunofluorescence experiments. Slides were mounted using Vectashield HardSet™ Mounting Medium (Vector Laboratories). Primary antibodies used for immunostaining were as follows: rabbit anti-SIRT2 (Sigma-Aldrich), mouse anti-Neurofilament H (smi-32, Abcam); mouse anti-NeuN (clone A60, Millipore), mouse anti-CNPase (clone 11-5B, Millipore), mouse anti GFAP (clone GA5, Millipore), mouse anti-acetylated α-tubulin (clone 611B-1, Sigma-Aldrich) and rabbit anti-HDAC6 (Novus Biologicals). Secondary antibodies were AlexaFluor 488-conjugated goat anti-mouse IgG (Invitrogen) and either AlexaFluor 568-conjugated (Invitrogen) or cy3-conjugated (Jackson ImmunoResearch) goat anti-rabbit IgG. Immunolabeled sections were visualized and photographed using an Olympus BX70 fluorescent microscope and DP-70 digital camera. Images from individual color channels were acquired at maximum pixel depth; all subsequent manipulations, including cropping and generation of merged overlays, were performed on copies of original image files using Adobe Photoshop.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the MassGeneral Institute for Neurodegenerative Disease and by grants from the National Institutes of Health (R21-NS062189 to M.M.M.; P01-NS058793 to S.M.H.); the ALS Therapy Alliance (to M.M.M.); the R.J.G. Foundation (to A.G.K.); the Milton Carmen Foundation (to A.G.K.); the HDSA New England Center of Excellence (to S.M.H.); and the HDSA Coalition for the Cure (to S.M.H.).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Eric Wang and David Housman for mining of existing deep-sequencing data sets and for sharing the results of these analyses. We also thank Sarah Meade for technical assistance and are grateful to Antonio Valencia for advice regarding isolation and culture of primary mouse cortical neurons.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Blander G., Guarente L. The Sir2 family of protein deacetylases. Annu. Rev. Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- 2.Denu J.M. The Sir 2 family of protein deacetylases. Curr. Opin. Chem. Biol. 2005;9:431–440. doi: 10.1016/j.cbpa.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 3.Donmez G., Guarente L. Aging and disease: connections to sirtuins. Aging Cell. 2010;9:285–290. doi: 10.1111/j.1474-9726.2010.00548.x. [DOI] [PubMed] [Google Scholar]
- 4.Kaeberlein M., McVey M., Guarente L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999;13:2570–2580. doi: 10.1101/gad.13.19.2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rogina B., Helfand S.L. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc. Natl Acad. Sci. USA. 2004;101:15998–16003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Michan S., Sinclair D. Sirtuins in mammals: insights into their biological function. Biochem. J. 2007;404:1–13. doi: 10.1042/BJ20070140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor D.M., Maxwell M.M., Luthi-Carter R., Kazantsev A.G. Biological and potential therapeutic roles of sirtuin deacetylases. Cell. Mol. Life Sci. 2008;65:4000–4018. doi: 10.1007/s00018-008-8357-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harting K., Knoll B. SIRT2-mediated protein deacetylation: an emerging key regulator in brain physiology and pathology. Eur. J. Cell. Biol. 2010;89:262–269. doi: 10.1016/j.ejcb.2009.11.006. [DOI] [PubMed] [Google Scholar]
- 9.Pfister J.A., Ma C., Morrison B.E., D'Mello S.R. Opposing effects of sirtuins on neuronal survival: SIRT1-mediated neuroprotection is independent of its deacetylase activity. PLoS One. 2008;3:e4090. doi: 10.1371/journal.pone.0004090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suzuki K., Koike T. Mammalian Sir2-related protein (SIRT) 2-mediated modulation of resistance to axonal degeneration in slow Wallerian degeneration mice: a crucial role of tubulin deacetylation. Neuroscience. 2007;147:599–612. doi: 10.1016/j.neuroscience.2007.04.059. [DOI] [PubMed] [Google Scholar]
- 11.Outeiro T.F., Kontopoulos E., Altmann S.M., Kufareva I., Strathearn K.E., Amore A.M., Volk C.B., Maxwell M.M., Rochet J.C., McLean P.J., et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson's disease. Science. 2007;317:516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- 12.Luthi-Carter R., Taylor D.M., Pallos J., Lambert E., Amore A., Parker A., Moffitt H., Smith D.L., Runne H., Gokce O., et al. SIRT2 inhibition achieves neuroprotection by decreasing sterol biosynthesis. Proc. Natl Acad. Sci. USA. 2010;107:7927–7932. doi: 10.1073/pnas.1002924107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.North B.J., Marshall B.L., Borra M.T., Denu J.M., Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell. 2003;11:437–444. doi: 10.1016/s1097-2765(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 14.Dryden S.C., Nahhas F.A., Nowak J.E., Goustin A.S., Tainsky M.A. Role for human SIRT2 NAD-dependent deacetylase activity in control of mitotic exit in the cell cycle. Mol. Cell. Biol. 2003;23:3173–3185. doi: 10.1128/MCB.23.9.3173-3185.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Werner H.B., Kuhlmann K., Shen S., Uecker M., Schardt A., Dimova K., Orfaniotou F., Dhaunchak A., Brinkmann B.G., Mobius W., et al. Proteolipid protein is required for transport of sirtuin 2 into CNS myelin. J. Neurosci. 2007;27:7717–7730. doi: 10.1523/JNEUROSCI.1254-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li W., Zhang B., Tang J., Cao Q., Wu Y., Wu C., Guo J., Ling E.A., Liang F. Sirtuin 2, a mammalian homolog of yeast silent information regulator-2 longevity regulator, is an oligodendroglial protein that decelerates cell differentiation through deacetylating alpha-tubulin. J. Neurosci. 2007;27:2606–2616. doi: 10.1523/JNEUROSCI.4181-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Creppe C., Malinouskaya L., Volvert M.L., Gillard M., Close P., Malaise O., Laguesse S., Cornez I., Rahmouni S., Ormenese S., et al. Elongator controls the migration and differentiation of cortical neurons through acetylation of alpha-tubulin. Cell. 2009;136:551–564. doi: 10.1016/j.cell.2008.11.043. [DOI] [PubMed] [Google Scholar]
- 18.Solinger J.A., Paolinelli R., Kloss H., Scorza F.B., Marchesi S., Sauder U., Mitsushima D., Capuani F., Sturzenbaum S.R., Cassata G. The Caenorhabditis elegans Elongator complex regulates neuronal alpha-tubulin acetylation. PLoS Genet. 2010;6:e1000820. doi: 10.1371/journal.pgen.1000820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Janke C., Kneussel M. Tubulin post-translational modifications: encoding functions on the neuronal microtubule cytoskeleton. Trends Neurosci. 2010;33:362–372. doi: 10.1016/j.tins.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 20.Perdiz D., Mackeh R., Pous C., Baillet A. The ins and outs of tubulin acetylation: more than just a post-translational modification? Cell Signal. 2011;23:763–771. doi: 10.1016/j.cellsig.2010.10.014. [DOI] [PubMed] [Google Scholar]
- 21.De Vos K.J., Grierson A.J., Ackerley S., Miller C.C. Role of axonal transport in neurodegenerative diseases. Annu. Rev. Neurosci. 2008;31:151–173. doi: 10.1146/annurev.neuro.31.061307.090711. [DOI] [PubMed] [Google Scholar]
- 22.Nguyen L., Humbert S., Saudou F., Chariot A. Elongator – an emerging role in neurological disorders. Trends Mol. Med. 2010;16:1–6. doi: 10.1016/j.molmed.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 23.Creppe C., Buschbeck M. Elongator: an ancestral complex driving transcription and migration through protein acetylation. J. Biomed. Biotechnol. 2011;2011:924898. doi: 10.1155/2011/924898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dompierre J.P., Godin J.D., Charrin B.C., Cordelieres F.P., King S.J., Humbert S., Saudou F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J. Neurosci. 2007;27:3571–3583. doi: 10.1523/JNEUROSCI.0037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tapia M., Wandosell F., Garrido J.J. Impaired function of HDAC6 slows down axonal growth and interferes with axon initial segment development. PLoS One. 2010;5:e12908. doi: 10.1371/journal.pone.0012908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen S., Owens G.C., Makarenkova H., Edelman D.B. HDAC6 regulates mitochondrial transport in hippocampal neurons. PLoS One. 2010;5:e10848. doi: 10.1371/journal.pone.0010848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hubbert C., Guardiola A., Shao R., Kawaguchi Y., Ito A., Nixon A., Yoshida M., Wang X.F., Yao T.P. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 28.Matsuyama A., Shimazu T., Sumida Y., Saito A., Yoshimatsu Y., Seigneurin-Berny D., Osada H., Komatsu Y., Nishino N., Khochbin S., et al. In vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J., 2002;21:6820–6831. doi: 10.1093/emboj/cdf682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Y., Li N., Caron C., Matthias G., Hess D., Khochbin S., Matthias P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003;22:1168–1179. doi: 10.1093/emboj/cdg115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pandithage R., Lilischkis R., Harting K., Wolf A., Jedamzik B., Luscher-Firzlaff J., Vervoorts J., Lasonder E., Kremmer E., Knoll B., et al. The regulation of SIRT2 function by cyclin-dependent kinases affects cell motility. J. Cell Biol. 2008;180:915–929. doi: 10.1083/jcb.200707126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nahhas F., Dryden S.C., Abrams J., Tainsky M.A. Mutations in SIRT2 deacetylase which regulate enzymatic activity but not its interaction with HDAC6 and tubulin. Mol. Cell. Biochem. 2007;303:221–230. doi: 10.1007/s11010-007-9478-6. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Y., Kwon S., Yamaguchi T., Cubizolles F., Rousseaux S., Kneissel M., Cao C., Li N., Cheng H.L., Chua K., et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol. Cell. Biol. 2008;28:1688–1701. doi: 10.1128/MCB.01154-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Southwood C.M., Peppi M., Dryden S., Tainsky M.A., Gow A. Microtubule deacetylases, SirT2 and HDAC6, in the nervous system. Neurochem. Res. 2007;32:187–195. doi: 10.1007/s11064-006-9127-6. [DOI] [PubMed] [Google Scholar]
- 34.Quinti L., Chopra V., Rotili D., Valente S., Amore A., Franci G., Meade S., Valenza M., Altucci L., Maxwell M.M., et al. Evaluation of histone deacetylases as drug targets in Huntington's disease models. Study of HDACs in brain tissues from R6/2 and CAG140 knock-in HD mouse models and human patients and in a neuronal HD cell model. PLoS Curr. 2010;2:RRN1172. doi: 10.1371/currents.RRN1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vaquero A., Scher M.B., Lee D.H., Sutton A., Cheng H.L., Alt F.W., Serrano L., Sternglanz R., Reinberg D. SirT2 is a histone deacetylase with preference for histone H4 Lys 16 during mitosis. Genes Dev. 2006;20:1256–1261. doi: 10.1101/gad.1412706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jing E., Gesta S., Kahn C.R. SIRT2 regulates adipocyte differentiation through FoxO1 acetylation/deacetylation. Cell Metab. 2007;6:105–114. doi: 10.1016/j.cmet.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang F., Tong Q. SIRT2 suppresses adipocyte differentiation by deacetylating FOXO1 and enhancing FOXO1's repressive interaction with PPARgamma. Mol. Biol. Cell. 2009;20:801–808. doi: 10.1091/mbc.E08-06-0647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang F., Nguyen M., Qin F.X., Tong Q. SIRT2 deacetylates FOXO3a in response to oxidative stress and caloric restriction. Aging Cell. 2007;6:505–514. doi: 10.1111/j.1474-9726.2007.00304.x. [DOI] [PubMed] [Google Scholar]
- 39.Reed N.A., Cai D., Blasius T.L., Jih G.T., Meyhofer E., Gaertig J., Verhey K.J. Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 2006;16:2166–2172. doi: 10.1016/j.cub.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 40.Hammond J.W., Huang C.F., Kaech S., Jacobson C., Banker G., Verhey K.J. Posttranslational modifications of tubulin and the polarized transport of kinesin-1 in neurons. Mol. Biol. Cell. 2010;21:572–583. doi: 10.1091/mbc.E09-01-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mattson M.P., Magnus T. Ageing and neuronal vulnerability. Nat. Rev. Neurosci. 2006;7:278–294. doi: 10.1038/nrn1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gauthier L.R., Charrin B.C., Borrell-Pages M., Dompierre J.P., Rangone H., Cordelieres F.P., De Mey J., MacDonald M.E., Lessmann V., Humbert S., et al. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- 43.Simpson C.L., Lemmens R., Miskiewicz K., Broom W.J., Hansen V.K., van Vught P.W., Landers J.E., Sapp P., Van Den Bosch L., Knight J., et al. Variants of the elongator protein 3 (ELP3) gene are associated with motor neuron degeneration. Hum. Mol. Genet. 2009;18:472–481. doi: 10.1093/hmg/ddn375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Slaugenhaupt S.A., Blumenfeld A., Gill S.P., Leyne M., Mull J., Cuajungco M.P., Liebert C.B., Chadwick B., Idelson M., Reznik L., et al. Tissue-specific expression of a splicing mutation in the IKBKAP gene causes familial dysautonomia. Am. J. Hum. Genet. 2001;68:598–605. doi: 10.1086/318810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gardiner J., Barton D., Marc J., Overall R. Potential role of tubulin acetylation and microtubule-based protein trafficking in familial dysautonomia. Traffic. 2007;8:1145–1149. doi: 10.1111/j.1600-0854.2007.00605.x. [DOI] [PubMed] [Google Scholar]
- 46.Maxwell M.M., Pasinelli P., Kazantsev A.G., Brown R.H., Jr. RNA interference-mediated silencing of mutant superoxide dismutase rescues cyclosporin A-induced death in cultured neuroblastoma cells. Proc. Natl Acad. Sci. USA. 2004;101:3178–3183. doi: 10.1073/pnas.0308726100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Valencia A., Reeves P.B., Sapp E., Li X., Alexander J., Kegel K.B., Chase K., Aronin N., DiFiglia M. Mutant huntingtin and glycogen synthase kinase 3-beta accumulate in neuronal lipid rafts of a presymptomatic knock-in mouse model of Huntington's disease. J. Neurosci. Res. 2010;88:179–190. doi: 10.1002/jnr.22184. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.