

Abstract
The combination of Ni(0) and an N-heterocyclic carbene act as a precatalyst for the cycloisomerization of enynes to afford 1,3-dienes. During the course of the reaction, a nickel hydride is formed from oxidative addition of the ortho C-H on the carbene ligand. Deuteriumn labeling studies are presented.
1. Introduction
During the development of Ni/NHC (NHC = N-heterocyclic carbene) catalyst systems for various cycloaddition reactions,1 we found that the Ni(COD)2/NHC system was capable of catalyzing the cycloisomerization of enyne 1 to diene 2 (eq 1). While this reaction is typically performed with “noble” metals (Pd, Ru, Rh, Ir),2 we were intrigued with the possibility of accessing the highly versatile 1,3-diene with a more economical Ni catalyst.3 As a key component of the Diels-Alder reaction, 1,3-dienes of varying complexity are important compounds.4 We now report the combination of Ni/NHC catalyzes the cycloisomerization of enynes. We also present evidence that suggests that catalyst generation occurs through C-H activation of the bound NHC ligand.
2. Results and discussion
Initially, we discovered the formation of moderate amounts of 2 as a side product in Ni/IPr catalyzed cycloadditions of 1 and ketones.1e Furthermore, reactions run with 1 alone led to relatively clean conversion to 2. In an effort to further enhance yields, a series of NHC ligands in combination with Ni(COD)2 were evaluated for catalytic activity in the cycloisomerization of 1 (Table 1).5 Interestingly, 1,3-diene (2) formation was only observed in reactions involving N-aryl-imidazol-2-ylidene ligands (entries 1–3). IDTB gave the best yield-to-conversion ratio (entry 1). Reactions run with the saturated NHC analogs failed to produce any of the desired product (entries 4–6). Although reactions with N-alkyl ligands displayed high enyne conversions, only substrate oligomerization occurred6 and no 1,3-diene was obtained. Ultimately, further optimization with IDTB led to general reaction conditions that employed 5 mol% Ni(COD)2, 10 mol% IDTB, and enyne concentrations of 0.1M in toluene at 60 °C (Table 2).
Table 1.
Ligand Screen for Enyne Cycloisomerizationa
| entry | NHC | conversion (%)b of 1 | yield (%)b of 2 |
|---|---|---|---|
| 1 | IDTB | 26 | 30 |
| 2 | IPr | 38 | 20 |
| 3 | IMes | 52 | 20 |
| 4 | SIDTB | 13 | 0 |
| 5 | SIPr | 40 | 0 |
| 6 | SIMes | 52 | 0 |
| 7 | IAd | 100 | 0 |
| 8 | IiBu | 100 | 0 |
Reaction Conditions: 5 mol% Ni(COD)2, 10 mol% NHC, 0.1M 1, rt.
Uncorrected GC yield based on napthalene internal standard.
Table 2.
Scope of Enyne Cycloisomerizationa
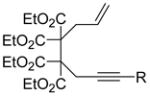
| entry | enyne | product | yield (%)b,c |
|---|---|---|---|
| 1 |
|
||
| 1 R = Me | 2 | 90 | |
| 3 R = Et | 4 | 92 | |
| 5 R = nPr | 6 | 97 | |
| 7 R = iPr | 8 | 99 | |
| 2 |
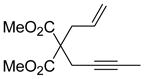 9 |
10 | 82 |
Reaction Conditions: 5 mol% Ni(COD)2, 10 mol% IDTB, 0.1M 1, 60 °C, 1h.
Isolated yield, average of two runs.
>95:5 E:Z based on 1H NMR analysis of crude reaction mixture.
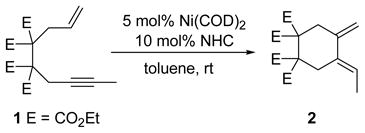 |
(1) |
A cursory examination of the substrate scope under optimized conditions showed that a variety of enynes participated in the reaction. Substituent groups at the terminal position of the alkyne of varying lengths were tolerated well as Me, Et and nPr substituted enynes (Table 2, entry 1) produced the corresponding dienes in excellent yields. The reaction of a more sterically demanding substrate such as the iPr substituted enyne (7) also gave excellent diene formation. The synthesis of a five-member ring was also successful as demonstrated by the formation of 9 in good yield (entry 2).
Based on the previously reported Pd catalyzed process, two general mechanisms for cycloisomerization are depicted in Scheme 1.2c In mechanism A, the enyne undergoes oxidative coupling with the Ni0 catalyst to generate a metallacyclopentene. This metallacyclopentene undergoes β-hydride elimination to generate a vinyl nickel hydride. The vinyl nickel hydride then reductively eliminates to yield the observed 1,3-diene. Alternatively, in mechanism B, the alkyne component of the enyne undergoes hydro-metallation with a Ni-H complex to generate a vinyl nickel species. The pendant olefin then inserts into the vinyl nickel bond thereby forming an alkyl nickel species. β-Hydride elimination and reductive elimination affords the observed 1,3-diene.
Scheme 1.

Proposed Mechanisms for Cycloisomerization.
In an effort to differentiate between the mechanisms proposed in Scheme 1, the reaction of enyne 1-d1 (95% deuterium incorporation) was evaluated. If diene formation occurred through mechanism A, no deuterium washing would be observed since the deuterium would be shifted from the olefin to the alkyne of the same molecule. Conversely, if diene product was formed through mechanism B, any protium in the product would come from a source other than the substrate. The reaction of enyne 1-d1 produced diene 2-d1 in 65% isolated yield (eq 2).7 However, only 80% deuterium was incorporated in the product which immediately suggested that mechanism A was not the operative pathway.8
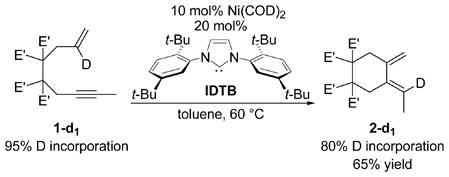 |
(2) |
It seemed likely that protium incorporation in 2-d1 arose from the formation of a reactive Ni-H species (Mechanism B). It was possible that this species could originate from solvent activation. However, reactions run with enyne 1 in deuterated toluene showed no deuterium incorporation. Furthermore, the cycloisomerizations are unaffected by protic additives, unlike many notable Pd catalyzed enyne rearrangement reactions.3 For example, the addition of water (either H2O or D2O) had no effect on protium incorporation.
An alternative route to a Ni-H species is the orthometallation of the pendant NHC ligand (Scheme 2). Yet unlike the orthometallation of Pd complexes to form Pd-H species,9,10 C-H oxidative addition by Ni complexes is rare.11,12 Nevertheless, when super stoichiometric amounts of Ni/IDTB were employed (200 mol% Ni(COD)2 and 400 mol% IDTB), a protium incorporation of 50% was observed in the 1,3-diene product 2-d1.13 Thus, an increase in ligand (and in catalyst) did indeed facilitate an increase in hydrogen incorporation.
Scheme 2.

C-H Activation of Ni/Carbene Complex.
 |
(3) |
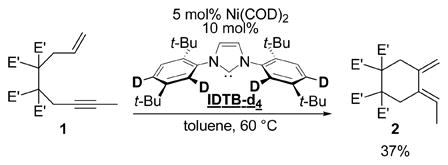
Additionally, we reasoned that if orthometalation of the NHC ligand was operative,14 then replacing the IDTB ligand with its deuterium labeled analog IDTB-d4 would have a profound affect on the cycloisomerization reaction.15 The IDTB-d4 ligand was prepared (>95% D incorporation) and evaluated as a ligand in Ni/NHC catalyzed reactions. Importantly, no difference in yields was observed between IDTB and IDTB-d4 when either of these ligands were employed in our other [2+2+2] cycloaddition reactions (98% vs 96%, respectively),1 suggesting that deuteration of the IDTB has a negligible effect on the steric and electronic properties of the NHC ligand.16 In contrast, severely depressed yields were obtained in the cycloisomerization of 2 run under otherwise identical conditions (90% yield with IDTB vs. 37% with IDTB-d4, eq 3).17 In addition, subjecting 1-d1 to the Ni(0)/IDTB-d4 catalyst afforded 2-d1 in only 21% yield.
IDTB-d4 possesses a stronger Caryl-D bond relative to the Caryl-H bond in IDTB.18 As such, generation of Ni-D is more difficult with IDTB-d4 than generation of Ni-H from IDTB. Yet, in reactions employing IDTB-d4, Ni-H can be formed, though in significantly lower concentrations than in IDTB reactions (Scheme 3). Thus, in a competition between diene formation, which is Ni-H catalyzed, and oligomerization, which is Ni(0) catalyzed, lower Ni-H concentrations would lead to a diminished yield of 1a as observed in equation 3. A similar yet less pronounced phenomenon is observed in the Ni/IDTB catalyzed cycloisomerization of 1-d1. Moreover, the Ni/IDTB-d4 catalyzed reaction of 1-d1 is even more difficult than the parent cycloisomerization (i.e., eq 1) and only a fraction of the product yield is observed.
Scheme 3.

Regeneration of a Ni-H from Ni(IDTB-d4)
3. Conclusion
The combination of Ni(COD)2 and IDTB catalyzes the cycloisomerization of enynes to synthetically valuable cyclic 1,3-dienes. Deuterium labeling studies suggest that the active catalyst species is a Ni-H species which is generated via a rare Ni(0) C-H activation. More extensive mechanistic studies are currently underway.
4. Experimental
General information
All reactions were conducted under an atmosphere of N2 using standard Schlenk techniques or in a N2 filled glove-box unless otherwise noted. Toluene was dried over neutral alumina under N2 using a Grubbs type solvent purification system. THF was freshly distilled from Na/benzophenone. Ni(COD)2 was purchased from Strem and used without further purification. The IPr, SIPr, IMes, SIMes, IAd, and ItBu ligands were prepared as previously reported.19 Sodium hydride was thoroughly washed with pentane and dried in vacuo prior to use. The compounds 1-bromo-4-methylpentyne and 1-bromo-2-hexyne were prepared from the corresponding alcohols by the method described by Brandsma.20 Enynes 1, 3, 5, 7, and 91e,21 as well as vinyl bromide 1422 and alkyne 171e were prepared by known literature procedures. Glyoxal was purchased from Aldrich Chemical Company as a 40% wt. solution in water. All other reagents were purchased and used without further purification unless otherwise noted.
1H and 13C Nuclear Magnetic Resonance spectra of pure compounds were acquired at 500 and 125 MHz, respectively unless otherwise noted. All spectra are referenced to a singlet at 7.27 ppm for 1H and to the center line of a triplet at 77.23 ppm for 13C. The abbreviations s, d, dd, dt, dq, t, td, tq, q, qt, quint, sept, septd, septt, m, brm, brd, brt, and brs stand for singlet, doublet, doublet of doublets, doublet of triplets, doublet of quartets, triplet, triplet of doublets, triplet of quartets, quartet, quartet of triplets, quintet, septet, septet of doublets, septet of triplets, multiplet, broad multiplet, broad doublet, broad triplet, and broad singlet, in that order. All 13C NMR spectra were proton decoupled. IR spectra were recorded on a Bruker Tensor 27 FT-IR spectrometer. HRMS were performed at the mass spectrometry facility at The University of Utah.
Gas Chromatographies were performed on an Agilent 6890 gas chomatograph with a 30 meter HP-5 column using the following conditions: initial oven temperature: 100 °C; temperature ramp rate 50 °C/min.; final temperature: 300 °C held for 7 minutes; detector temperature: 250 °C.
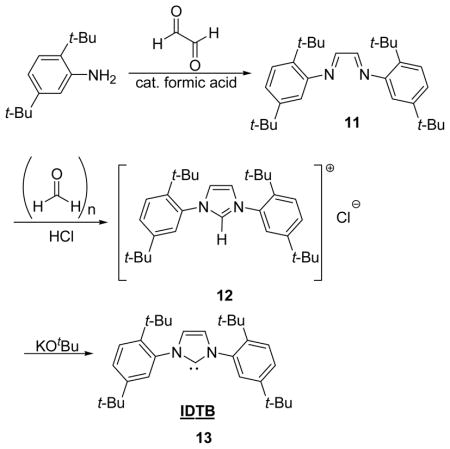
Preparation of (N, N′E, N, N′E)-N, N′-(ethane-1,2-diylidene)bis(2,5-di-tert-butylaniline) (11)
To a stirring solution of 2,5-di-tert-butyl aniline (13.95 g, 68 mmol) in 130 mL EtOH was added glyoxal (3.9 mL 34 mmol) and 4 drops of formic acid. The resulting yellow solution was stirred at room temperature for 2 h at which time a yellow precipitate formed. The mixture was stirred for an additional 12 h and then cooled to −78 °C. The cold mixture was then quickly filtered and the collected yellow solids were washed with cold MeOH (−78 °C) until the filtrate ran clear. The yellow solid was dried in vacuo to yield 11 (12.21 g, 83%). mp: 186 °C. 1H NMR (500 MHz, CDCl3): δ (ppm) 8.28 (s, 2H), 7.36 (d, J = 8.3 Hz, 2H), 7.26 (dd, J1 = 2.0, J2 = 8.3 Hz), 6.87 (d, J = 2.0 Hz), 1.44 (s, 18H), 1.36 (s, 18H). 13C {1H} NMR (125 MHz, CDCl3): δ (ppm) 158.8, 150.3, 150.2, 140.7, 126.2, 124.0, 116.2, 35.5, 34.7, 31.6, 30.7. IR (neat): 2959, 2872, 1608, 1554, 1262, 926, 880, 827 cm−1. HRMS(FAB): calcd for C30H44N2 (M+) 432.3504, obsd 432.3468.
Preparation of 1,3-bis(2,5-di-tert-butylphenyl)-1H-imidazol-3-ium chloride (12)
A stirring suspension of bisimine 11 (6.43 g, 14.88 mmol) and paraformaldehyde (447.0 mg, 14.88 mmol) in toluene (35 mL) was heated at 80 °C for 1 h. The solution was then cooled to room temperature and HCl (5.6 mL, 22.3 mmol, 4M in dioxane) was added dropwise at a rate such that the previous drop had fully dispersed before addition of the next drop. The resulting dark red/brown solution was stirred at room temperature for 8 h and then filtered. The collected solids were washed with cold THF (−78 °C) until the filtrate ran clear. The tan solid was further purified by dissolving the solid in hot acetone and re-precipitating it by the addition of hexanes. The beige solids were collected by vacuum filtration, washed with Et2O (30 mL), and dried in vacuo to yield crude imidazolium salt 12 (2.51 g, 35%). HRMS(FAB): calcd for C31H45N2 (M+) 445.3583, obsd 445.3587.
Preparation of IDTB (13)
In a glove box, imidazolium salt 7 (534.1 mg, 1.11mmol) and KOtBu (186.9 mg, 1.67 mmol) were weighed into a vial and suspended in toluene (4 mL). The resulting suspension was stirred at room temperature for 4 h and the solvent was removed in vacuo. The solids were then suspended in 2 mL toluene and the excess salts were precipitated by the addition of 7 mL pentane. The mixture was then vacuum filtered and the collected solids were subjected to the suspension, precipitation, filtration sequence two more times. The collected filtrate was concentrated in vacuo to yield IDTB as a beige solid (412.4 mg 84%). Analytically pure sample could be obtained by cooling a saturated solution of IDTB in Et2O to −40 °C. mp: Decomp. 206 °C. 1H NMR (500 MHz, C6D6): δ 7.47 (d, J = 8.3 Hz, 2H), 7.38 (d, J = 2.0 Hz), 7.27 (dd, J1 = 2.0, J2 = 8.3 Hz, 2H), 6.77 (s, 2H), 1.47 (s, 18H), 1.16 (s, 18H). 13C {1H} NMR (125 MHz, C6D6): δ (ppm) 221.4, 149.9, 144.0, 142.5, 129.0, 125.8, 123.0, 36.3, 34.5, 32.6, 31.5. HRMS(EI): calcd for C31H44N2 (MH+) 445.3583, obsd 445.3580.
General Procedure for the Cycloisomerization of Enynes
To a stirring solution of enyne in toluene (~0.4 M) at 60 °C was added the catalyst solution (Ni(COD)2 and IDTB which was previously equilibrated for at least 8 h at room temperature at a concentration of ~0.04 M). The resulting solution was stirred for 1 h at 60 °C, cooled to room temperature, and quenched with the addition of MeOH (0.5 mL). The crude mixture was then concentrated in vacuo and the residue was purified by flash column chromatography on SiO2 to yield the 1,3-diene.
Preparation of (E)-tetraethyl 4-ethylidene-5-methylenecyclohexane-1,1,2,2-tetracarboxylate (2)
The general procedure was used with enyne 1 (200.0 mg, 0.4873 mmol), Ni(COD)2 (6.7 mg, 0.0244 mmol), IDTB (21.7 mg, 0.0588 mmol) and 4.87 mL toluene. The reaction mixture was purified by flash column chromatography on SiO2 eluting with 10% EtOAc/hexanes to yield diene 2 (191.2 mg, 96%) as a sticky, pale yellow oil. 1H NMR (500 MHz, CDCl3): δ (ppm) 5.75 (q, J = 6.83 Hz, 1H), 5.00 (brs, 1H), 4.66 (brs, 1H), 4.27-4.16 (m, 8H), 3.07 (s, 2H), 2.94 (s, 2H), 1.68 (d, J = 6.83, 3H), 1.27 (app. td, 12H). 13C {1H} NMR (125 MHz, CDCl3): δ (ppm) 169.9, 169.7, 144.7, 134.7, 121.4, 110.3, 61.9, 61.7, 59.6, 59.2, 38.1, 31.6, 14.0, 13.4. IR (neat): 2983, 2938, 2907, 1738, 1300, 1266, 1202, 1042, 865 cm−1. HRMS(CI): calcd for C21H31O8 (MH+) 411.2019, obsd 411.2023. COSY summary, the following pertinent cross-peaks were observed: H(4) with H(1) and H(3), H(5) with H(6) and H(2), H(6) with H(5) and H(2). NOE summary; the following pertinent enhancements were observed: irradiation of H(4) showed enhancement of H(3) and H(5); irradiation of H(5) showed enhancement of H(4); irradiation of H(1) showed enhancement of H(3); irradiation of H(3) showed enhancement of H(1).
Preparation of (E)-tetraethyl 4-methylene-5-propylidenecyclohexane-1,1,2,2-tetracarboxylate (4)
The general procedure was used with enyne 3 (200.0 mg, 0.471 mmol), Ni(COD)2 (6.5 mg, 0.0236 mmol), IDTB (20.9 mg, 0.0471 mmol) and 4.71 mL toluene. The reaction mixture was purified by flash column chromatography on SiO2 eluting with 10% EtOAc/hexanes to yield diene 4 (189.5 mg, 95%) as a sticky, pale yellow oil. 1H NMR (500 MHz, CDCl3): δ (ppm) 5.66 (t, J = 7.32 Hz, 1H), 5.02 (brs, 1H), 4.67 (brs, 1H), 4.27-4.16 (m, 8H), 3.05 (s, 2H), 2.97 (s, 2H), 2.10 (quint, J = 7.32 Hz, 2H), 1.27 (app. td, 12H), 0.99 (t, J = 7.32 Hz). 13C {1H} NMR (125 MHz, CDCl3): δ (ppm) 169.9, 169.7, 144.7, 133.2, 129.1, 110.5, 61.9, 61.8, 59.5, 59.4, 38.2, 31.9, 21.1, 14.2, 14.07, 14.05. IR (neat): 2983, 2938, 2907, 1738, 1300, 1266, 1202, 1042, 865 cm−1. HRMS(CI): calcd for C22H33O8 (MH+) 425.2175, obsd 425.2164. COSY summary, the following pertinent cross-peaks were observed: H(4) with H(1) and H(3), H(5) with H(6) and H(2), H(6) with H(5) and H(2). NOE summary; the following pertinent enhancements were observed: irradiation of H(4) showed enhancement of H(3) and H(5); irradiation of H(5) showed enhancement of H(4); irradiation of H(1) showed enhancement of H(3); irradiation of H(3) showed enhancement of H(1).
Preparation of (E)-tetraethyl 4-butylidene-5-methylenecyclohexane-1,1,2,2-tetracarboxylate (6)
The general procedure was used with enyne 5 (150.0 mg, 0.342 mmol), Ni(COD)2 (4.7 mg, 0.017 mmol), IDTB (15.2 mg, 0.0342 mmol) and 3.42 mL toluene. The reaction mixture was purified by flash column chromatography on SiO2 eluting with 10% EtOAc/hexanes to yield diene 4a (147.6 mg, 98%) as a sticky, pale yellow oil. 1H NMR (500 MHz, CDCl3): δ (ppm) 5.68 (t, J = 7.32 Hz, 1H), 5.02 (brs, 1H), 4.66 (brs, 1H), 4.27-4.16 (m, 8H), 3.05 (s, 2H), 2.97 (s, 2H), 2.06 (q, J = 7.32, 2H), 1.40 (sext, J = 7.32, 2H) 1.31-1.23 (m, 12H), 0.92 (t, J = 7.32 Hz, 3H). 13C {1H} NMR (125 MHz, CDCl3): δ (ppm) 169.8, 169.7, 144.7, 133.8, 127.3, 110.4, 61.9, 61.7, 59.4, 59.3, 38.1, 32.0, 29.8, 22.9, 14.1, 14.008, 13.997. IR (neat): 2982, 2934, 2873, 1738, 1367, 1266, 1159, 1040, 865 cm−1. HRMS(CI): HRMS(CI): calcd for C23H35O8 (MH+) 439.2332, obsd 439.2350. COSY summary, the following pertinent cross-peaks were observed: H(4) with H(1) and H(3), H(5) with H(6) and H(2), H(6) with H(5) and H(2). NOE summary; the following pertinent enhancements were observed: irradiation of H(4) showed enhancement of H(3) and H(5); irradiation of H(5) showed enhancement of H(4); irradiation of H(1) showed enhancement of H(3); irradiation of H(3) showed enhancement of H(1).
Preparation of (E)-tetraethyl 4-methylene-5-(2-methylpropylidene)cyclohexane-1,1,2,2-tetracarboxylate (8)
The general procedure was used with enyne 7 (150.0 mg, 0.342 mmol), Ni(COD)2 (4.7 mg, 0.0171 mmol), IDTB (15.2 mg, 0.0342 mmol) and 3.42 mL toluene. The reaction mixture was purified by flash column chromatography on SiO2 eluting with 10% EtOAc/hexanes to yield diene 8 (149.0 mg, 99%) as a sticky, pale yellow oil. (1H NMR (500 MHz, CDCl3): δ (ppm) 5.50 (d, J = 9.27 Hz, 1H), 5.02 (brs, 1H), 4.66 (brs, 1H), 4.27-4.16 (m, 8H), 3.05 (s, 2H), 2.99 (s, 2H), 2.59 (m, 1H), 1.31-1.23 (m, 12H), 0.97 (d, J = 6.83 Hz, 6H). 13C {1H} NMR (125 MHz, CDCl3): δ (ppm) 169.9, 169.7, 144.7, 134.7, 131.6, 110.6, 61.9, 61.8, 59.45, 59.36, 38.1, 32.1, 26.8, 23.1, 14.04, 14.02. IR (neat): 2982, 2961, 2907, 2869, 1738, 1267, 1234, 1202, 1040, 865 cm−1. HRMS(CI): HRMS(CI): calcd for C23H35O8 (MH+) 439.2332, obsd 439.2335. COSY summary, the following pertinent cross-peaks were observed: H(4) with H(1) and H(3), H(5) with H(6) and H(2), H(6) with H(5) and H(2). NOE summary; the following pertinent enhancements were observed: irradiation of H(4) showed enhancement of H(3) and H(5); irradiation of H(5) showed enhancement of H(4); irradiation of H(1) showed enhancement of H(3); irradiation of H(3) showed enhancement of H(1).
Preparation of (E)-dimethyl 3-ethylidene-4-methylenecyclopentane-1,1-dicarboxylate (10)
The general procedure was used with enyne 9 (150.0 mg, 0.669 mmol), Ni(COD)2 (9.2 mg, 0.0334 mmol), IDTB (29.7 mg, 0.0668 mmol) and 6.69 mL toluene. The reaction mixture was purified by flash column chromatography on SiO2 eluting with 6% EtOAc/hexanes to yield diene 5a (125.2 mg, 83%) as a pale yellow oil. (1H NMR (500 MHz, (ppm) 5.94 (qt, J1 = 2.4, J2 = 7.3 Hz, 1H), 5.23 CDCl3): δ (s, 1H), 4.81 (s, 1H), 3.73 (s, 6H), 3.01 (t, J = 2.0 Hz, 2H), 2.98 (s, 2H), 1.71 (d, J = 7.3 Hz, 3H). 13C {1H} NMR (125 MHz, CDCl3): δ (ppm) 172.1, 145.3, 137.0, 117.0, 102.8, 57.7, 53.0, 41.7, 37.7, 15.0. IR (neat): 2956, 2917, 2855, 1737, 1436, 1248, 1202, 1073, 880 cm−1. HRMS(CI): calcd for C12H17O4 (MH+) 225.1127, obsd 225.1098.
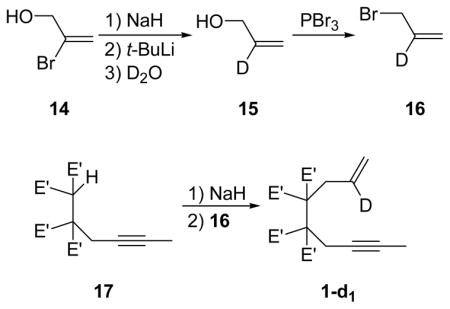
Preparation of deuterated enyne 1-d1
To a stirring suspension of NaH (2.412 g, 100.5 mmol) in Et2O (250 mL) was added vinyl bromide 14 dropwise (10.59 g, 77.27 mmol). The resulting brown solution was stirred at room temperature for 1 h. The solution was then cooled to −78 °C and t-BuLi was added dropwise. The resulting pale yellow solution was warmed to −10 °C for 2 h and then cooled to −78 °C at which time D2O (20.00, 1000 mmol) was added. The reaction mixture was warmed to room temperature and stirred for an additional 2 h. The reaction mixture was then quenched by the addition of saturated NH4Cl solution (50 mL) and the two phases were separated. The aqueous phase was extracted with Et2O (3 × 10 mL) and the collected organics were washed with brine (15 mL) and dried with MgSO4. The mixture was concentrated via careful distillation through a Vigreux column. The concentrate was further purified via fractional distillation through a Vigreux column. The fraction boiling from 46 °C to 94 °C was collected to yield the crude alcohol 15 as a colorless oil (2.2147 g, 49%). To a stirring solution of PBr3 (1.35 mL, 14.2 mmol) in Et2O (15 mL) was added crude alcohol 15 (2.10 g, 35.5 mmol) dropwise at 0 °C. The resulting solution was stirred at 0 °C for 1 h then carefully quenched by the addition brine (7 mL). The layers were separated and the organics were washed with a saturated solution of NaHCO3 (3 × 5 mL), brine (5 mL) and dried over MgSO4. The volatiles were carefully removed via distillation through a Vigreux column. Crude allyl bromide 16 was obtained as yellow oil (1.9712 g, 46%). To a stirring suspension of NaH (194.4 mg, 8.100 mmol) in 18 mL THF was added tetra-ester 17 (1.000 g, 2.702 mmol) in 2 mL THF. The resulting solution was stirred at room temperature for 1 h at which time crude allyl bromide 16 (1.000 g, 8.197 mmol) was added in a single portion via syringe. The flask was the equipped with a reflux condenser and the mixture was stirred at reflux until no starting material was observed by GC analysis (~36 h). The mixture was then cooled to room temperature and quenched with 20 mL of a saturated NH4Cl solution. The layers were separated and the aqueous layer was extracted with Et2O (3 × 10 mL). The combined organics were washed with brine (10 mL), dried over Na2SO4, and concentrated in vacuo to yield a yellow oil. The crude oil was purified by flash column chromatography eluting with 15% EtOAc/hexanes producing a yellow oil which was then triturated with cold hexanes to yield enyne 1-d1 (892.3 mg, 80%) as a white solid. The degree of deuteration was ~95% as determined by 1H analysis. mp: 67 °C. 1H NMR (500 MHz, CDCl3): δ (ppm) 5.10 (brs, 1H), 5.05 (brs, 1H), 4.31-4.12 (m, 8H), 3.07 (brs, 2H), 2.85 (brs, 2H), 1.75 (t, J = 2.4 Hz, 3H), 1.31-1.25 (m, 12H). 13C {1H} NMR (125 MHz, CDCl3): δ (ppm) 169.2, 161.1, 133.7 (t, J = 24.0 Hz), 119.0, 78.1, 74.8, 62.5, 62.3, 61.9, 61.7, 36.1, 22.8, 14.01, 13.99, 3.8. IR (neat): 3458, 2984, 1739, 1446, 1214, 1042, 923 cm−1. HRMS(CI): calcd for C21H30DO8 (MH+) 412.2082, obsd 412.2078.
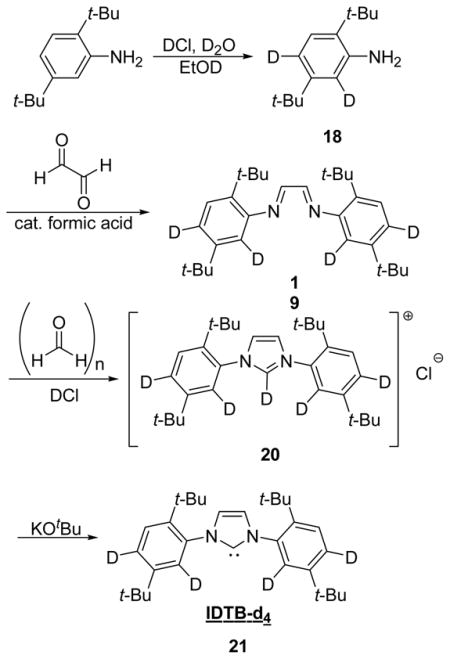
Preparation of deuterated aniline (18)
The following is modification of a published procedure.23 To a N2 flushed thick walled bomb with a Teflon screw top was added 2,5-di-tert-butylaniline (10.0 g, 48.7 mmol), DCl (10 mL, 20 mmol, 2N in D2O), D2O (20 mL) and EtOD (20 mL). The flask was sealed and heated at 105 °C for 3 days at which time the flask was cooled to room temperature and the solvents were removed in vacuo. The flask was flushed with N2 and DCl (10 mL, 20 mmol), D2O (20 mL), and EtOD (20 mL) were added. The flask was resealed and heated to 105 °C for 5 days. The flask was cooled to room temperature and the solvents were removed in vacuo. The flask was flushed with N2 and DCl (10 mL, 20mmol), D2O (20 mL), and EtOD (40 mL) were added. The flask was resealed and heated to 105 °C for another 5 days. The flask was cooled to room temperature and the reaction mixture was quenched by the addition of 2 N NH4OH to pH 10. The mixture was then extracted with Et2O (3 × 75 mL) and the collected organics were washed with brine (20 mL) then dried of MgSO4. The volatiles were removed in vacuo to yield 18 as an off-white solid (8.93 g, 89%). The degree of deuteration was >95% at the ortho and para positions as determined by 1H analysis. mp: 104 °C 1H NMR (500 MHz, CDCl3): δ (ppm) 7.18 (s, 1H), 3.98 (brs, 2H), 1.42 (s, 9H), 1.29 (s, 9H). 13C {1H} NMR (125 MHz, CDCl3): δ (ppm) 149.9, 144.2, 131.1, 126.4, 115.7 (t, J = 23.8 Hz), 115.0 (t, J = 22.9 Hz), 34.2, 34.1, 31.5, 29.9. IR (neat): 3470, 2957, 2872, 1620, 1548, 1470, 792 cm−1. HRMS(CI): calcd for C14H21D2N (M+) 207.1956, obsd 207.1966.
Preparation of deuterated bisimine (19)
The procedure was identical to the procedure used in the synthesis of bisimine 11 using deuterated aniline 18 (7.038 g, 33.94 mmol), glyoxal (1.95 mL, 17.0 mmol), formic acid (4 drops) in 65 mL EtOH. The bisimine 19 was isolated as a yellow solid in 93% yield. mp: 182 °C. 1H NMR (500 MHz, CDCl3): δ (ppm) 8.28 (s, 2H), 1.44 (s, 18H), 1.35 (s, 18H). 13C {1H} NMR (125 MHz, CDCl3): δ (ppm) 158.8, 150.2, 150.1, 140.7, 126.1, 123.7 (t, J = 22.4 Hz), 115.9 (t, J = 20.6 Hz), 35.5, 34.6, 31.5, 30.7. IR (neat): 2959, 2872, 1612, 1539, 1362, 1265, 909, 873 cm−1. HRMS(CI): calcd for C30H41D4N2 (MH+) 437.3834, obsd 437.3845.
Preparation of deuterated imidazolium salt (20)
The procedure was identical to the procedure used in the synthesis of imidazolium salt 12 using bisimine 19 (6.500 g, 14.88 mmol), paraformaldehyde (447.0 mg, 14.88 mmol), and DCl (1M in Et2O, 22.3 mL, 22.3 mmol) in 35 mL toluene. The crude imidazolium salt 20 was isolated as a light tan solid in 42% yield. HRMS(FAB): calcd for C31H40D5N2 (M+) 450.3897, obsd 450.3923.
Preparation of IDTB-d4 (21)
The procedure used was identical to the procedure used in the synthesis of the IDTB carbene (13) using deuterated imidazolium salt 20 (508 mg, 1.04 mmol) and KOtBu (176 mg, 1.57 mmol) in 4 mL toluene. The title compound was isolated as a beige solid in 24% yield. Analytically pure sample could be obtained by cooling a saturated solution of the carbene in Et2O to −40 °C. mp: Decomp. 206 °C. 1H NMR (500 MHz, C6D6): δ 7.47 (s, 2H), 6.77 (s, 2H), 1.48 (s, 18H), 1.16 (s, 18H). 13C {1H} NMR (125 MHz, C6D6): δ (ppm) 221.4, 149.2, 143.5, 141.9, 125.0 (t, J = 23.3 Hz), 122.5, 35.8, 33.9, 32.1, 31.0. HRMS(EI): calcd for C31H40D4N2 (MH+) 449.3834, obsd 449.3837.
Supplementary Material
Acknowledgments
We gratefully acknowledge Merck, NSF (Career Award), the NIH-NIGMS, and the Alfred P. Sloan Foundation for supporting this research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.(a) Louie J, Gibby JE, Farnworth MV, Tekavec TN. J Am Chem Soc. 2002;124:15188. doi: 10.1021/ja027438e. [DOI] [PubMed] [Google Scholar]; (b) Tekavec TN, Arif AM, Louie J. Tetrahedron. 2004;60:7431. [Google Scholar]; (c) Duong HA, Cross MJ, Louie J. J Am Chem. 2004;126:11438. doi: 10.1021/ja046477i. [DOI] [PubMed] [Google Scholar]; (d) McCormick MM, Duong HA, Zuo G, Louie J. J Am Chem Soc. 2005;127:5030. doi: 10.1021/ja0508931. [DOI] [PubMed] [Google Scholar]; (e) Tekavec TN, Louie J. J Org Chem. ASAP; 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reviews: Trost BM, Toste FD, Pinkerton AB. Chem Rev. 2001;101:2067. doi: 10.1021/cr000666b.Aubert C, Buisine O, Malacria M. Chem Rev. 2002;102:813. doi: 10.1021/cr980054f.Trost BM, Krische MJ. Synlett. 1998:1.Lloyd-Jones GC. Org Biomol Chem. 2003;1:215. doi: 10.1039/b209175p.Fairlamb IJS. Angew Chem, Int Ed. 2004;43:1048. doi: 10.1002/anie.200301699.Select Reports: [Ir]: Kezuka S, Okado T, Niou E, Takeuchi R. Org Lett. 2005;7:1711. doi: 10.1021/ol050085e.Chatani N, Inoue H, Morimoto T, Muto T, Murai S. J Org Chem. 2001;66:4433. doi: 10.1021/jo010091y.[Pt]: Trost BM, Chang VK. Synthesis. 1993:824.[Ru]: Chatani N, Morimoto T, Muto T, Murai S. J Am Chem Soc. 1994;116:6049.Mori M, Kozawa Y, Nishida M, Kanamaru M, Onozuka K, Takimoto M. Org Lett. 2000;2:3245. doi: 10.1021/ol006510f.[Pd]: Trost BM, Lee DC, Rise F. Tetrahedron Lett. 1989;30:651.
- 3.(a) Trost BM, Tour J. J Am Chem Soc. 1987;109:5268. [Google Scholar]; (b) Ikeda SI, Daimon N, Sanuki R, Odashima K. Chem Eur J. 2006;12:1797. doi: 10.1002/chem.200500894. [DOI] [PubMed] [Google Scholar]
- 4.Reviews: Takao K, Munakata R, Tadano K. Chem Rev. 2005;105:4779. doi: 10.1021/cr040632u.Winkler JD. Chem Rev. 1996;96:167. doi: 10.1021/cr950029z.Recent reports: Nicolaou KC, Harrison ST. Angew Chem Int Ed. 2006;45:3256. doi: 10.1002/anie.200601116.Leduc AB, Kerr MA. Eur J Org Chem. 2007:237.Pandey SK, Orellana A, Greene AE, Poisson JF. Org Lett. 2006;8:5665. doi: 10.1021/ol062419l.Crimmins MT, Brown BH, Plake HR. J Am Chem Soc. 2006;128:1371. doi: 10.1021/ja056334b.Caussanel F, Wang K, Ramachandran SA, Deslongchamps P. J Org Chem. 2006;71:7370. doi: 10.1021/jo061230k.
- 5.IDTB = 1,3-bis(2,5-di-tert-butylphenyl)-imidazol-2-ylidene; IPr = 1,3-bis(2,6-diisopropylphenyl)-imidazol -2-ylidene; IMes = 1,3-bis(2,4,6-trimethylphenyl)-imidazol -2-ylidene; SIDTB = 1,3-bis(2,5-di-tert-butylphenyl)-4,5-dihydroimidazolin-2-ylidene; SIPr = 1,3-bis(2,5-diisopropylphenyl)-4,5-dihydroimidazolin-2-ylidene; SIMes = 1,3-bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazolin-2-ylidene; IAd = 1,3-diadamantylimidazol-2-ylidene; ItBu = 1,3-di-tert-butylimidazol-2-ylidene.
- 6.The oligomerization of alkynes is a predominant side reaction that is catalyzed by Ni(0)/NHC complexes. See ref 1.
- 7.The depressed yield of 2 arises from an increased difficulty to undergo β-D elimination (the last step of either mechanism A or B).
- 8.All efforts to recovery the ITDB ligand after the cycloaddition led to intractable mixtures.
- 9.Reviews: Ritleng V, Sirlin C, Pfeffer M. Chem Rev. 2002;102:1731. doi: 10.1021/cr0104330.Naota T, Takaya H, Murahashi SI. Chem Rev. 1998;98:2599. doi: 10.1021/cr9403695.Dyker G. Angew Chem Int Ed. 1999;38:1699. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1698::AID-ANIE1698>3.0.CO;2-6.
- 10.Stylianides N, Danopoulos AA, Pugh D, Hancock F, Zanotti-Gerosa A. Organometallics. 2007;26:5627. [Google Scholar]
- 11.Theoretical calculations show that C-H activation is endothermic for Ni-complexes in comparison with the analogous Pt-complexes. Reinhold M, McGrady JE, Perutz RN. J Am Chem Soc. 2004;126:5268. doi: 10.1021/ja0396908.
- 12.C-H activation by Ni(0) species: Keen AL, Johnson SA. J Am Chem Soc. 2006;128:1806. doi: 10.1021/ja0572553.Brunkan NM, Brestensky DM, Jones WD. J Am Chem Soc. 2004;126:3627. doi: 10.1021/ja037002e.van der Boom ME, Liou SY, Shimon LJ, Ben-David Y, Milstein D. Inorg Chim Acta. 2004;357:4015.
- 13.Product yields were identical to reactions run with 1 and superstoichiometric amounts of Ni(COD)2 and IDTB.
- 14.It is interesting to note that N-alkyl NHC ligands do not facilitate the cycloisomerization. This may be due to a decreased propensity for orthometallation relative to NHC ligands that possess easily oxidizable ortho-aromatic C-H bonds (such as IDTB) or benzylic C-H bonds (such as IPr and IMes).
- 15.For a review of bond activation reactions with NHC’s, see: Crudden CM, Allen DP. Coord Chem Rev. 2004;248:2247.For recent examples of C-H activation: [Ir]: Hanasaka F, Tanabe Y, Fujita K, Yamaguchi R. Organometallics. 2006;25:826.Corberán R, Sanaú M, Peris E. Organometallics. 2006;25:4002.Corberán R, Sanaú M, Peris E. J Am Chem Soc. 2006;128:3974. doi: 10.1021/ja058253l.Scott NM, Pons V, Stevens ED, Heinekey DM, Nolan SP. Angew Chem Int Ed. 2005;44:2512. doi: 10.1002/anie.200463000.[Rh]: Huang J, Stevens ED, Nolan SP. Organometallics. 2000;19:1194.Scott NM, Dorta R, Stevens ED, Correa A, Cavallo L, Nolan SP. J Am Chem Soc. 2005;127:3516. doi: 10.1021/ja043249f.[Ru]: Cabeza JA, del Rio I, Miguel D, Sánchez-Vega MG. Chem Commun. 2005:3956. doi: 10.1039/b506287j.
- 16.In addition, the 1H NMR spectra of a Ni(COD)2/IDTB solution and a Ni(COD)2/IDTB-d4 solution were essentially identical.
- 17.Rate constants for reactions run with IDTB and IDTB-d4 are difficult to compare. Ni-H is formed in both reactions but in different and immeasurable amounts. Furthermore, Ni(0) catalyzed enyne oligomerization is a competitive side reaction in IDTB-d4 reactions.
- 18.For examples of kinetic isotope effect used to partition between two products in a synthesis see: Miyashita M, Sasaki M, Hattori I, Sakai M, Tanino K. Science. 2004;305:495. doi: 10.1126/science.1098851.Clive DLJ, Cantin M, Khodabocus A, Kong X, Tao Y. Tetrahedron. 1993;49:7917.
- 19.(a) Böhm VPW, Gstöttmayr CWK, Weskamp T, Herrmann WA. Angew Chem Int Ed. 2001;40:3387. doi: 10.1002/1521-3773(20010917)40:18<3387::aid-anie3387>3.3.co;2-y. [DOI] [PubMed] [Google Scholar]; (b) Jensen DR, Sigman MS. Org Lett. 2003;5:63. doi: 10.1021/ol027190y. [DOI] [PubMed] [Google Scholar]
- 20.Synthesis of Acetylenes, Allenes, and Cumulenes: Brandsma L, editor. Methods and Techniques. Elsevier; Oxford: 2004. p. 371.
- 21.Atkinson RS, Grimshire MJ. J Chem Soc Perkin Trans. 1986;1:1215. [Google Scholar]
- 22.Trost BM, Machacek MR, Tsui HC. J Am Chem Soc. 2005;127:7014. doi: 10.1021/ja050340q. [DOI] [PubMed] [Google Scholar]
- 23.Lersch M, Dalhus B, Bercaw JE, Labinger J, Tilset M. Organometallics. 2006;25:1055. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.