

Background
The mammalian target of rapamycin (mTOR) protein complex belongs to the phosphatidylinositol 3-kinase (PI3K)-related family of kinases. Activation occurs through a complex signaling network in which activation of a transmembrane receptor leads to activation of PI3K, subsequently stimulating Akt phosphorylation and signaling through mTOR to initiate protein synthesis (Figure 1). Signaling through mTOR can have multiple functions, in that mTOR complex 1 (mTORC1) is responsive to rapalogues, whereas mTOR complex 2 (mTORC2) is relatively insensitive to rapalogues, and both are involved in negative feedback loops which modulate signaling. The mTOR pathway is constitutively activated in NSCLC as evidenced by phosphorylation of mTOR (69%), p70 s6K (81%), and p4EBP-1 (79%) in tumor tissue. In addition, activation of Akt occurs frequently in NSCLC, and has been associated with tobacco carcinogen-induced cellular transformation, promotion of tumor invasion, angiogenesis and resistance to therapy (1, 2). More than 70% of non-small cell lung cancer (NSCLC) tumors demonstrate activation of Akt at both the ser473 and thr308 phosphorylation sites, which is associated with a shorter survival (3). Furthermore, phosphorylation of Akt can be inhibited by the phosphatase and tensin homologue gene (PTEN), and loss of PTEN is also associated with poor prognosis in NSCLC (4). Therapy with rapalogues as single agents results in limited tumor responses in lung cancer, and prolonged treatment induces resistance, which appears to be mediated by Akt signaling (5). Blocking PI3K may decrease the upregulation of Akt signaling induced by mTOR inhibition. Thus, combined blockade of PI3K/Akt and mTOR may result in enhanced antitumor activity.
Figure 1. PI3K/Akt/mTOR signaling cascade.
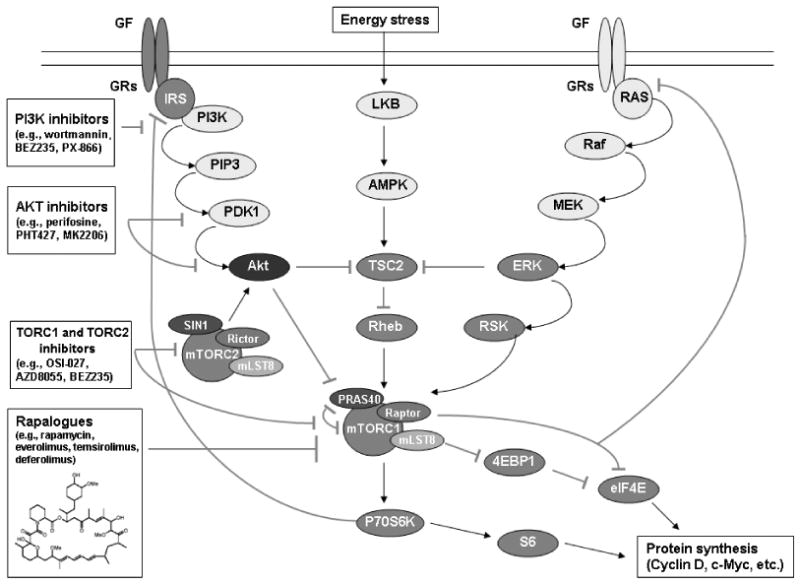
Signaling through a transmembrane receptor activates the PI3K signaling network to phosphorylate Akt and promote cell proliferation and invasion through mTOR. Multiple feedback loops exist within this signaling cascade, and a number of inhibitors are in development to target this pathway in cancer.
mTOR inhibition
Sirolimus (rapamycin) is an oral rapalogue which has demonstrated synergism in combination with pemetrexed in vitro and in vivo in NSCLC models. Pemetrexed is an antifolate drug that blocks multiple pathways in folate metabolism. Recently, a downstream target has been described, aminoimidazolecarboxamide ribonucleotide formyltransferase (AICART), which results in inhibition of mTOR through increased cellular ZMP (6). Accumulation of ZMP activates AMP-activated protein kinase, which in turn, blocks mTOR and subsequent protein synthesis and cell growth. Therefore, the combination of pemetrexed and mTOR inhibition may further decrease signaling through the mTOR pathway in NSCLC. A phase I/II trial evaluating pemetrexed and sirolimus in advanced NSCLC patients with tumors that demonstrate activation of mTOR is ongoing. A phase I dose escalation will be followed by a phase II portion which requires a biopsy sample to establish mTOR activation prior to drug administration and following cycle 2 of therapy. The endpoints include determination of dose limiting toxicities and maximum tolerated dose in the phase I portion; and response rate, progression free survival and modulation of mTOR activity in the phase II portion. Twelve patients are evaluable to date, with 3 partial responses.
Everolimus has been studied extensively in NSCLC as monotherapy and in combination with chemotherapy and epidermal growth factor receptor (EGFR) tyrosine kinase inhibition (TKI). A phase I study assessed the combination of gefitinib and everolimus in former smokers, which resulted in 2 partial responses in eight evaluable patients (7). This led to a phase II trial that enrolled patients who were current or former smokers into 2 cohorts, untreated versus prior chemotherapy, and the primary endpoint was objective response rate. 62 patients were enrolled, and 8 (13%) patients had partial or complete response, 5 untreated and 3 previously treated. Two responders in the untreated cohort harbored KRAS mutations (both G12F), 2 carried EGFR mutations and 1 had neither. In the previously treated cohort, one patient harbored an EGFR mutation and 2 were wild type for both EGFR and KRAS. The most common drug-related toxicities included rash, diarrhea, oral ulcerations and fatigue. Two patients were removed from study for pulmonary toxicity. The role of mTOR inhibition in G12F KRAS mutated NSCLC is under investigation. Additional studies of everolimus have attempted to define molecular endpoints through pre-operative evaluation in NSCLC tumors. A study evaluating everolimus given for 3 weeks pre-operatively has enrolled 12 patients to date, and has found a reduction in pS6 with upregulation of pAkt following therapy.
Temsirolimus is an ester of sirolimus, and has shown minimal activity as monotherapy in lung cancer. Combination therapy with EGFR TKI, chemotherapy, vascular endothelial growth factor (VEGF) inhibitors and VEGF receptor (VEGFR) inhibitors have demonstrated the potential for augmented tumor responses in a variety of tumor types, although combination trials in NSCLC remain in early phases.
TORC1 and TORC2 inhibition
OSI-027 attenuates Akt activation through inhibition of both mTORC1 and mTORC2. The compound has been shown to induce apoptosis in multiple solid tumor and hematologic malignancy models, including those resistant to rapamycin. It has been shown to potentiate chemotherapy-induced apoptosis and to decrease VEGF production and blood vessel formation. A phase I trial is ongoing evaluating weekly, intermittent and continuous dosing of OSI-027, and the recommended phase 2 dose has been determined for all cohorts. Pharmacokinetics indicate a dose-response for increasing concentrations, and pharmacodynamic data evaluating p4EBP-1 levels in peripheral blood mononuclear cells (PBMCs) demonstrate inhibition of mTOR signaling.
AZD8055 inhibits both mTORC1 and mTORC2, resulting in increased tumor apoptosis and decreased cell proliferation. It has been shown to induce dose-dependent anti-tumor activity and to modulate pS6 and pAkt. A phase I trial is ongoing and has enrolled 38 patients, showing a dose-dependent pharmacodynamic modulation of pAkt and p4EBP-1 in PBMCs. The most common toxicities include rash, neutropenia, mucositis, hyperinsulinemia and mild hyperglycemia.
PI3K inhibition
PI3K pathway inhibition may inhibit tumor growth and proliferation and sensitize cancer cells to programmed cell death. BEZ235 is an imidazo[4,5-c]quinoline derivative, and acts as a dual PI3K and mTORC1 and mTORC2 inhibitor. The compound binds the ATP-binding cleft of the p110α subunit of PI3K and mTORC1 and mTORC2. It induces G1 cell cycle arrest and apoptosis and inhibits downstream effector activation in multiple malignancies. It has demonstrated anti-tumor activity in xenograft models harboring PI3K alterations. It has been shown to inhibit VEGF-induced angiogenesis and microvessel permeability with blockade of endothelial nitric oxide synthase. A phase I trial has been completed with 59 subjects, and 2 partial responses. The drug demonstrated dose-dependent PI3K inhibition as measured by elevation of plasma C-peptide levels.
PX-866 is a PI3K inhibitor which induces anti-tumor effects through the inhibition pAkt. Evaluation of markers to determine response demonstrates resistance in tumors with mutant KRAS, while loss of PTEN and alterations in PI3K predict for sensitivity.
Akt inhibition
Combinations of Akt and EGFR inhibition may prove beneficial in EGFR TKI resistant NSCLC. PHT427 is an Akt inhibitor which binds to the phosphorylation domains of Akt and phosphoinositol-dependent kinase 1 (PDK1), inhibits phosphorylation at ser473 and decreases tumor growth. Its effects have been most pronounced in KRAS mutant NSCLC when used in combination with erlotinib.
Hepatocyte growth factor (HGF) can induce EGFR TKI resistance through c-met, and co-expression of c-met and EGFR can stimulate synergistic tumor cell growth. Erlotinib and MK2206 have been shown to augment anti-tumor effects in both KRAS/EGFR wild type and EGFR mutant NSCLC models. Furthermore, MK2206 can reverse HGF-induced resistance to erlotinib. A phase II trial is planned to evaluate patients with advanced NSCLC who have progressed on erlotinib after prior response to EGFR TKI therapy. Patients will be stratified by EGFR mutation status, and will receive erlotinib daily and MK2206 on an every other day schedule. Tumor tissue will be evaluated for Akt pathway inhibition.
Future Directions
Selection of patients who are responsive to the mTOR/PI3K/Akt pathway is essential as these agents are developed for NSCLC therapy. Studies are underway to identify key molecular and genetic changes which may be important in regulating this pathway. Two tumor suppressor genes have been linked to promoting NSCLC, and mTOR signaling. LKB1 has serine threonine kinase activity and controls cell differentiation, cell polarity and energy control (8), and alterations occur in approximately 39% of NSCLC. BRG1 is important in ATP-dependent chromatin remodeling, cell cycle arrest, DNA repair, cell differentiation and regulation of apoptosis (9), and 33% of NSCLC harbor mutations. LKB1 and BRG1 inactivating mutations correlate with a unique gene expression profile in NSCLC that affects many crucial signaling pathways. Importantly, BRG1 plays an essential role in peripheral airway development while LKB1 inhibits proliferation and invasion through the mTOR pathway. Further delineation of these genes and others involved in signaling through mTOR will enhance our understanding of lung cancer pathogenesis. Opportunities for targeting this axis requires an improved understanding of the biology and signaling within the individual tumors, and combination treatments with rapalogues and PI3K inhibitors or novel agents may provide new approaches.
References
- 1.Balsara BR, Pei J, Mitsuuchi Y, Page R, Klein-Szanto A, Wang H, Unger M, Testa JR. Frequent activation of AKT in non-small cell lung carcinomas and preneoplastic bronchial lesions. Carcinogenesis. 2004 November 1;25(11):2053–9. doi: 10.1093/carcin/bgh226. [DOI] [PubMed] [Google Scholar]
- 2.West KA, Linnoila IR, Belinsky SA, Harris CC, Dennis PA. Tobacco carcinogen-induced cellular transformation increases activation of the phosphatidylinositol 3′-kinase/Akt pathway in vitro and in vivo. Cancer Res. 2004 Jan 15;64(2):446–51. doi: 10.1158/0008-5472.can-03-3241. [DOI] [PubMed] [Google Scholar]
- 3.Tsurutani J, Fukuoka J, Tsurutani H, Shih JH, Hewitt SM, Travis WD, Jen J, Dennis PA. Evaluation of two phosphorylation sites improves the prognostic significance of Akt activation in non-small-cell lung cancer tumors. J Clin Oncol. 2006 Jan 10;24(2):306–14. doi: 10.1200/JCO.2005.02.4133. [DOI] [PubMed] [Google Scholar]
- 4.Tang JM, He QY, Guo RX, Chang XJ. Phosphorylated Akt overexpression and loss of PTEN expression in non-small cell lung cancer confers poor prognosis. Lung Cancer. 2006;51(2):181–91. doi: 10.1016/j.lungcan.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 5.Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, Khuri FR. Activation of Akt and eIF4E Survival Pathways by Rapamycin-Mediated Mammalian Target of Rapamycin Inhibition. Cancer Res. 2005 August 15;65(16):7052–8. doi: 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- 6.Racanelli AC, Rothbart SB, Heyer CL, Moran RG. Therapeutics by cytotoxic metabolite accumulation: pemetrexed causes ZMP accumulation, AMPK activation, and mammalian target of rapamycin inhibition. Cancer Res. 2009 Jul 1;69(13):5467–74. doi: 10.1158/0008-5472.CAN-08-4979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Milton DT, Riely GJ, Azzoli CG, Gomez JE, Heelan RT, Kris MG, Krug LM, Pao W, Pizzo B, Rizvi NA, Miller VA. Phase 1 trial of everolimus and gefitinib in patients with advanced nonsmall-cell lung cancer. Cancer. 2007;110(3):599–605. doi: 10.1002/cncr.22816. [DOI] [PubMed] [Google Scholar]
- 8.Sanchez-Cespedes M, Parrella P, Esteller M, Nomoto S, Trink B, Engles JM, Westra WH, Herman JG, Sidransky D. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002 Jul 1;62(13):3659–62. [PubMed] [Google Scholar]
- 9.Wong AK, Shanahan F, Chen Y, Lian L, Ha P, Hendricks K, Ghaffari S, Iliev D, Penn B, Woodland AM, Smith R, Salada G, Carillo A, Laity K, Gupte J, Swedlund B, Tavtigian SV, Teng DH, Lees E. BRG1, a component of the SWI-SNF complex, is mutated in multiple human tumor cell lines. Cancer Res. 2000 Nov 1;60(21):6171–7. [PubMed] [Google Scholar]