

Abstract
Interior topological features, such as pockets and channels, have evolved in proteins to regulate biological functions by facilitating the diffusion of biomolecules. Decades of research using the globins as model heme proteins have clearly highlighted the importance of gas pockets around the heme in controlling the capture and release of O2. However, much less is known about how ligand migration contributes to the diverse functions of other heme protein scaffolds. Heme nitric oxide/oxygen binding (H-NOX) domains are a conserved family of gas-sensing heme proteins with a divergent fold that are critical to numerous signaling pathways. Utilizing X-ray crystallography with xenon, a tunnel network has been shown to serve as a molecular pathway for ligand diffusion. Structure-guided mutagenesis results show that the tunnels have unexpected effects on gas-sensing properties in H-NOX domains. The findings provide insights on how the flux of biomolecules through protein scaffolds modulates protein chemistry.
Keywords: diatomic gas signaling, hemoprotein, gas channels
Protein scaffolds are exquisitely tuned to carry out essential processes in biology. The structures of enzyme active sites, for example, have evolved to both catalyze and lend specificity to numerous chemical transformations. In addition to structural features that directly participate in active site chemistry, proteins often have interior cavities that provide spatial and temporal regulation. These structural features guide small molecule entry and exit as well as link intermediates between reactive centers. For example, channels for gas transport have been identified in enzymes utilizing O2 (e.g., oxidases and oxygenases) (1, 2), N2 (nitrogenases) (3), and H2 (hydrogenases) (4) for critical biological redox reactions. In enzymes with multiple active sites, tunnel networks shunt oftentimes volatile or reactive intermediates between reactive centers for processes such as bacterial carbon fixation (5) and the biosynthesis of essential cofactors (6) and amino acids (7, 8).
Interior structural features have evolved in heme proteins to facilitate diverse types of chemistry with an identical heme cofactor. Historically, globins have served as a model system for understanding how ligand flux through heme protein scaffolds controls gas-binding properties. Over the past 50 y, studies on myoglobin have utilized X-ray crystallography (9), time-resolved absorption techniques (10), CO photolysis of protein crystals (11, 12), and computational methods (13, 14) to provide a detailed molecular picture of ligand migration through the globin fold. Experimental approaches have implicated a histidine residue, the “His(E7) gate” (15), as the primary route for ligand entry and exit, in contrast to computational studies that have identified multiple ligand migration pathways (14). Internal cavities around the porphyrin in globins transiently hold O2 as it migrates to and from the heme iron. These structural features work in concert with residues directly involved in ligand coordination to efficiently capture and release O2 for gas transport, storage, and delivery in desired physiological contexts (16, 17).
Recent structural studies conducted on noncanonical globins have greatly expanded our knowledge of how globins function (18–23). Alternate topological features such as interior gas tunnels in truncated hemoglobins, for example, have been shown to be mechanistically important for modulating ligand migration (18, 19). However, further studies are needed to fully understand how these related protein frameworks within the globin family contribute to variations in ligand-binding properties. In addition, little is known about how ligand diffusion affects reversible gas binding within a broader spectrum of heme proteins.
Heme nitric oxide/oxygen binding (H-NOX) domains are a recently discovered family of gas-sensing heme proteins with a unique fold found in organisms from bacteria to humans (24). The best characterized H-NOX–containing protein is soluble guanylate cyclase (sGC), which is the principle mammalian receptor for NO and a central regulator of NO/cGMP-dependant signaling pathways (24). Members of the H-NOX family exhibit divergent ligand-binding properties within the identical protein fold. Some H-NOX domains bind NO, O2, and CO like the globins, whereas others (including sGC) discriminate against O2 binding, allowing them to serve as specific NO sensors even under aerobic conditions (24). The presence of a distal pocket tyrosine residue has been shown to be important for stabilizing O2 binding in the H-NOX family (25). However, it is apparent that additional structural and electronic features contribute to ligand-binding properties (25–30).
Surface representations of available H-NOX crystal structures (31, 32) indicate that, unlike globins, the heme in H-NOX domains is completely buried within the protein matrix (Fig. 1A). Close inspection of a non-O2-binding H-NOX domain, however, reveals a putative tunnel network (Fig. 1B) that extends between the solvent and interior heme site (33). This tunnel network is comprised of shorter (approximately 15 Å) and longer (approximately 22 Å) branches that are lined by well-conserved, hydrophobic residues and appear to be void of ordered solvent molecules (32). This indicates that the apparent tunnels may serve as potential conduits for gas diffusion. The tunnels are not evident in O2-binding H-NOX domains, suggesting that differential ligand flux through H-NOX scaffolds could be important for divergent functions (33).
Fig. 1.

Surface representations of WT Ns H-NOX depicting (A) the protein exterior and (B) a cross-sectional view where putative channels between the solvent and heme pocket are evident.
In the present study, direct experimental evidence is provided for the existence of a tunnel network in H-NOX proteins. High-resolution crystal structures were obtained of the non-O2-binding H-NOX domain from Nostoc sp. (Ns H-NOX) (32) pressurized with xenon, establishing a molecular route for ligand diffusion between the solvent and heme site. Structure-guided mutagenesis in combination with time-resolved absorption techniques were employed to probe the functional role of the tunnel network in modulating ligand-binding kinetics at the heme. Together, the data has led to an unexpected mechanism for how ligand diffusion regulates ligand-binding properties in H-NOX proteins. This work provides fundamental insights into the functional role of protein scaffolds in controlling the flux of biological molecules.
Results
Probing Gas Migration in the Predicted Tunnels with Xenon.
X-ray diffraction utilizing xenon is a powerful structural tool to observe gas diffusion through proteins (34). Xenon has a similar polarity and van der Waals radius to diatomic gases (2.16 Å versus 1.50–1.55 Å for O2, CO, and NO) (35), is freely diffusible in protein crystals, and weakly interacts with protein molecules (34). Importantly, xenon also possesses an appreciable anomalous signal, allowing for unambiguous localization of xenon atoms in protein crystal structures (34). Although xenon has high polarizability, which can bias interactions toward regions in proteins with more polarizable side chains (36), numerous studies have confirmed that the locations of xenon binding sites correlate well with areas that are favorable for diatomic gas migration (12, 13).
Crystals of ferrous-unligated Ns H-NOX were obtained for xenon pressurization via sitting drop vapor diffusion. A 2.13-Å structure of the native protein was solved by molecular replacement using the previously published structure of ferrous-unligated Ns H-NOX [Protein Data Bank (PDB) ID code 2O09] (32), which was obtained under different crystallization conditions, as the search model (Table 1). The Ns H-NOX crystal structure solved here was at a similar resolution (2.13 Å vs. 2.10 Å) in the identical space group (P213) and had comparable unit cell dimensions (a = b = c = 123.7 Å vs. 123.4 Å) as the previous structure (32). The two structures were found to superimpose well with an rmsd of 0.197 Å (Cα–Cα alignment).
Table 1.
Ns H-NOX crystallography data collection and refinement statistics
| WT Ns H-NOX |
L66W |
L67W |
L66W/L67W |
||||
| Native | 1 atm Xe | 6 atm Xe | Native | 6 atm Xe | Native | Native | |
| Wavelength, Å | 0.97 | 1.40 | 1.40 | 0.97 | 1.40 | 0.97 | 0.97 |
| Space group | P213 | P213 | P213 | P213 | P213 | P213 | P213 |
| Cell dimensions | |||||||
| a = b = c, Å | 123.7 | 124.4 | 123.9 | 123.2 | 123.3 | 123.2 | 123.1 |
| α = β = γ, ° | 90 | 90 | 90 | 90 | 90 | 90 | 90 |
| Resolution, Å | 50.00–2.13 (2.17–2.13) | 50.00–2.59(2.63–2.59) | 50.00–2.27(2.31–2.27) | 50.00–1.96(1.99–1.96) | 50.00–1.99(2.02–1.99) | 50.00–1.94(1.97–1.94) | 50.00–1.90(1.93–1.90) |
| Redundancy | 5.6 (5.6) | 6.3 (6.1) | 6.3 (5.9) | 5.5 (5.6) | 6.8 (6.5) | 6.8 (6.8) | 6.8 (6.8) |
| Completeness*, % | 99.9 (100.0) | 99.5(100.0) | 99.5(97.9) | 100.0(100.0) | 100.0(100.0) | 100.0(100.0) | 100.0(100.0) |
| Rsym, % | 6.5 (63.4) | 7.3 (61.6) | 5.4 (28.1) | 4.9 (64.6) | 5.6 (68.3) | 5.5 (61.5) | 6.2 (62.5) |
| I/σ | 23.6 (3.2) | 20.4 (2.78) | 30.2 (5.76) | 29.9(2.28) | 28.7(2.91) | 29.8(3.70) | 28.2(3.32) |
| No. of reflections | 38,528 | 40,549 | 55,256 | 43,497 | 79,661 | 48,228 | 51,187 |
| Rwork/Rfree, % | 16.4/20.0 | 17.0/20.5 | 16.2/19.0 | 16.3/18.7 | 15.6/17.3 | 16.5/18.8 | 16.0/18.3 |
| Molecules in ASU† | 2 | 2 | 2 | 2 | 2 | 2 | 2 |
| No. atoms | |||||||
| Protein‡ | 2,886 | 2,819 | 2,779 | 2,890 | 2,875 | 3,038 | 3,022 |
| Heme | 96 | 96 | 96 | 96 | 96 | 96 | 96 |
| Xenon | 0 | 2 | 6 | 0 | 4 | 0 | 0 |
| Solvent | 224 | 20 | 90 | 205 | 191 | 247 | 262 |
| Rms deviation | |||||||
| Bond lengths, Å | 0.010 | 0.021 | 0.010 | 0.009 | 0.009 | 0.009 | 0.018 |
| Bond angles, ° | 1.233 | 1.881 | 1.267 | 1.290 | 1.227 | 1.251 | 1.661 |
| Xenon occupancy | |||||||
| Molecule A | 0.3§ | 0.6§, 0.3¶, 0.4|| | 0.3§, 0.1¶ | ||||
| Molecule B | 0.3§ | 0.7§, 0.4,¶, 0.5|| | 0.4§, 0.1¶ | ||||
*Overall completeness.
†ASU, asymmetric unit.
‡Non-hydrogen atoms.
§Xe1.
¶Xe2.
||Xe3.
To map the putative tunnels in Ns H-NOX, native Ns H-NOX crystals were subsequently exposed to 1–6 atm of xenon for various times using a xenon pressurization chamber (Hampton Research). A crystal structure of Ns H-NOX pressurized with 1 atm of xenon is reported to 2.59-Å resolution (Table 1). As shown in Fig. 2A, one xenon atom (blue mesh) was observed per protein molecule at this pressure. The xenon atom is located at the intersection of the apparent tunnels near the distal face of the heme where diatomic ligands bind. The presence of xenon is indicated by spherical electron density in the 2mFo–DFc map generated for Ns H-NOX (Fig. 2A) and unambiguously confirmed by the presence of xenon electron density in the anomalous difference map at the same position (Fig. S1). The xenon-binding cavity is formed by several hydrophobic contacts. These include the faces of the porphyrin macrocycle and W74 indole ring, the V5 and L148 side chains, the M1 and M144 sulfur atoms, as well as the F70 phenyl ring edge (Fig. S2).
Fig. 2.

Crystal structures of WT Ns H-NOX following xenon pressurization. Structures are reported for crystals pressurized with (A) 1 atm of xenon for 1 min and (B) 6 atm of xenon for 1 min. 2mFo–DFc maps (mesh surface) for xenon atoms are contoured at 1σ (see Fig. S1 for anomalous xenon density). (C) A detailed view of the 6-atm crystal structure showing numerous residues that line the tunnel network. Key residues that were selected for structure-based rational mutagenesis are indicated. See Fig. S2 for views of the individual xenon-binding sites (Xe1, Xe2, and Xe3). The tunnel network was modeled using CAVER with the heme iron as the point of origin and a probe radius of 1.5 Å.
To test if Ns H-NOX has other accessible xenon-binding sites, a crystal structure of Ns H-NOX under 6 atm of xenon was obtained (Table 1). The structure was solved to 2.27-Å resolution and found to align well with the native structure (0.127-Å Cα rmsd). At this higher pressure, three xenon atoms were observed that extend between the heme and the end of tunnel 1 near the solvent interface. The presence of xenon at these locations is evident from electron density in the 2mFo–DFc map (Fig. 2 B and C) and the superposition of xenon electron density in the anomalous difference map (Fig. S1). Xenon binding in the site identified at lower pressure (herein referred to as “Xe1”) is still observed and occurs with higher occupancy in the 6-atm structure (occupancy of 0.6 vs. 0.3 in molecule A). The newly identified sites (referred to as “Xe2” and “Xe3”) are also predominately composed of hydrophobic residues (Fig. 2C). For the Xe2 site, the side chains of V5, I9, L67, and L141 as well as the T48 methyl group and face of the F70 aromatic ring contribute to form the xenon-binding pocket. For the Xe3 site at the end of tunnel 1, the binding pocket opens into solvent and is formed at its base by the side chains of I9, M12, I13, V52, and L66, as well as the face of the F70 aromatic ring (Fig. S2). Notably, no xenon occupancy was observed in tunnel 2. This smaller channel is predicted to be accessible to diatomic gases in the crystal structure based on molecular modeling (37). However, it may be too narrow to effectively accommodate xenon and/or may lack sufficient contacts to stabilize xenon binding.
The crystallographic data suggest a dynamic picture for xenon movement in the tunnels. Xenon flux is supported by anisotropic electron density at the Xe2 and Xe3 sites (Fig. 2C) and residual density that bridges these sites in the 2mFo–DFc map for the structure at lower contour levels. Xenon flux in the structure is correlated to xenon occupancy, which decreases from 0.6 to 0.3 to 0.4 for Xe1, Xe2, and Xe3, respectively (using molecule A) (Table 1). Together, the data indicate that xenon moves most dynamically in the tunnel near the protein exterior and more stably interacts with the protein distal to the heme at the Xe1 site. Based on these observations, the tunnels appear to funnel gases between the solvent and protein interior where they are preferentially trapped in the heme distal pocket.
To compare to O2-binding H-NOX proteins, which do not appear to display a tunnel network (Fig. S3) (33), crystals of the H-NOX protein from Thermoanaerobacter tengcongensis (Tt H-NOX) were pressurized with 6 atm of xenon. The resulting crystal structure of Tt H-NOX was solved to 2.03-Å resolution (Table S1) and found to overlay well with the native structure (0.129-Å Cα rmsd). Under these pressurization conditions, two xenon atoms were bound to each protein molecule (Fig. S3). However, unlike Ns H-NOX, a continuous pathway could not be traced in Tt H-NOX, either between the xenon atoms or to the heme pocket. This finding supports the prediction that gas diffusion in O2-binding H-NOX domains may not occur through discrete, preformed tunnels.
Structure-Guided Design of Mutations to Block the Tunnel Network.
To examine the role of the Ns H-NOX tunnels in modulating ligand migration, structure-guided mutants were designed in an attempt to block each branch of the tunnel network. Although numerous positions are potentially available for mutagenesis, two candidates (L66W to block tunnel 1 and L67W to block tunnel 2) were selected following modeling of allowed side-chain rotamers (38) (Fig. 2C). Leucine-to-tryptophan mutations were chosen principally to provide sufficient steric bulk to interact tightly with surrounding tunnel residues. Additionally, these mutations are predicted to maintain the hydrophobicity of the site (leucine and tryptophan partition coefficients differ by only 0.07) (39). The L66W, L67W, and L66W/L67W variants were generated using standard protocols and purified in a manner analogous to WT Ns H-NOX. Ferrous complexes (FeII–unligated, FeII–CO, and FeII–NO) of the isolated proteins were characterized with steady-state UV-visible spectroscopy. UV-visible spectra of the mutants were very similar to those of WT Ns H-NOX (Table S2), indicating that incorporation of tryptophan residues in the selected positions did not significantly alter the electronic properties of the heme.
Crystal Structures of Predicted Tunnel-Blocking Tryptophan Mutants.
High-resolution crystal structures of the Ns H-NOX variants were pursued to establish the effectiveness of the tryptophan residues in blocking the tunnels. Crystals of L66W, L67W, and the L66W/L67W double mutant were obtained in similar conditions to the WT protein (SI Materials and Methods). The structures were solved to 1.96-, 1.94-, and 1.90-Å resolution, respectively (Fig. 3). Notably, the structures were obtained in the same space group and found to have nearly identical unit cell dimensions as WT Ns H-NOX (Table 1), providing initial evidence that introduction of the tryptophan residues did not significantly alter the native fold. The final crystal structures were found to superimpose well with the WT protein (Fig. S4). Cα rmsd values of 0.273, 0.286, and 0.158 Å were obtained for L66W, L67W, and L66W/L67W, respectively (using molecule A of each structure).
Fig. 3.

Crystal structures of tunnel-blocking variants in Ns H-NOX. Structures of L66W (pressurized with 6 atm of xenon for 1 min), L67W, and L66W/L67W are shown. L66W blocks tunnel 1 and L67W blocks tunnel 2. Zoom-in views looking from the solvent to heme pocket depict van der Waals contacts with surrounding tunnel residues: I9, M12, I13, V52, A55, L59, L67, and F70 (for L66W); and T48, V52, F70, L141, M144, and Y49 (for L67W). Simulated-annealing composite omit maps (mesh surface) are shown for the tryptophan side chains. 2mFo–DFc maps (mesh surface) for xenon atoms are contoured at 1σ (see Fig. S1 for anomalous xenon density). The tunnel network was modeled with CAVER using the WT Ns H-NOX structure.
Closer inspection of the tryptophans in each crystal structure shows that the side chains were incorporated within the tunnel network as expected. Simulated-annealing composite omit maps for the L66W and L67W side chains show unambiguous electron density from the tryptophan indole rings (Fig. 3). These side chains make tight van der Waals contact with the surrounding, predominately hydrophobic residues in the tunnels. The tryptophan indole ring in the L66W single mutant is packed tightly by I9, M12, I13, V52, A55, L59, L67, and F70 at the top of tunnel 1. The tryptophan indole ring in the L67W single mutant is tightly contacted by T48, V52, F70, and L141 as well as the sulfur atom of M144 and the face of the Y49 phenol ring. Notably, the large tryptophan side chains appear to fit into the open space in the tunnel network and sterically seal tunnel 1 and tunnel 2. Decreased B-factors in the crystal structures at these sites support tight van der Waals contacts between the tryptophan side chains and tunnel residues and suggest that introduction of the tryptophan residues locally enhances conformational rigidity.
The tryptophan side chains in molecule A of the double mutant (L66W/L67W) are in the same orientation as the single mutants, and contacts with surrounding residues in the tunnels are preserved (Fig. 3). Additionally, the edge of the L66W residue is in van der Waals contact with the face of the L67W indole ring. In molecule B of the double mutant, the L66W side chain is in at least one different rotameric position than in molecule A but still functions to partially seal the tunnel (Fig. S5). The different observable rotamers of L66W suggest that this side chain has enhanced flexibility in the double mutant structure.
To examine the effect of blocking the tunnels on gas accessibility in Ns H-NOX, crystals of L66W were subsequently pressurized with xenon. L66W was selected as a representative mutant because it is predicted to block the main pathway (tunnel 1) for xenon diffusion between the solvent and protein interior. The structure of L66W under 6 atm of xenon was obtained for direct comparison to WT Ns H-NOX (Table 1). As seen in the 1.99-Å resolution structure (0.071-Å Cα rmsd vs. native L66W), introduction of the tryptophan residue in tunnel 1 sterically blocks xenon binding at the former Xe3 position but preserves the Xe1 and Xe2 sites (Fig. 3 and Fig. S1). Retention of xenon binding in the single mutant reflects the ability of xenon to access the base of the channel through other pathway(s). This suggests that tunnel 2 could serve as a possible conduit for gases even though xenon occupancy remains to be observed in that tunnel directly.
Together, the structural data indicate that the Ns H-NOX tunnel network was blocked by introducing tryptophan residues and that these residues sterically control gas migration in the protein interior. The ability to modify the protein without significant structural changes suggests that this approach represents a unique opportunity to systematically dissect the contribution of the H-NOX tunnels to ligand-binding kinetics at the heme.
Blocking the Tunnels Affects the CO kon.
To examine how blocking the tunnel network affects ligand association rates, time-resolved absorption spectroscopies using CO gas were employed with the tryptophan variants. CO has several important advantages over NO as a spectroscopic tool to probe ligand migration in heme proteins: (i) CO reacts more slowly (approximately 100- to 1,000-fold) with ferrous heme (40), (ii) CO has been previously shown to both bind to and dissociate from the Ns H-NOX heme in a simple one-step process (40), and (iii) CO does not participate in redox chemistry with proteins (41). Two likely outcomes were predicted for blocking the tunnels with respect to ligand binding. Blocking the tunnels could decrease CO association rate constants (kon values or “on-rates”) if diffusion into the heme pocket were dramatically slowed. Alternatively, on-rates could increase if ligand trapping in the heme pocket were enhanced (following ligand diffusion through the protein matrix) due to the blocking of route(s) for ligand escape.
CO on-rates for the tryptophan variants were measured with stopped-flow spectroscopy in a manner similar to that described previously (40). Ferrous protein was rapidly mixed with increasing CO concentrations, and CO binding was followed using a diode array detector on a stopped-flow instrument (Fig. 4A). Plots of observed rate (kobs) versus CO concentration (Fig. S6) were used to calculate kon values from the slope of the linear fits. WT Ns H-NOX had a CO kon of 1.52 × 106 M-1 s-1, which is comparable to the previously reported value (40). To our initial surprise, all the tunnel-blocking variants exhibited faster CO kon values compared to the WT protein. The on-rate for L67W (blocking tunnel 2) increased by 50%, while the on-rates for L66W (blocking tunnel 1) and L66W/L67W increased by approximately 100% relative to the WT protein (Fig. 4B and Table 2). These data indicate that even with blocking the tunnels, CO is still able to rapidly diffuse through the protein matrix, which likely occurs within the dead time of the stopped-flow measurement (< 1.5 ms). Therefore, CO on-rates determined with stopped-flow spectroscopy appear to reflect CO binding following diffusion from both the solvent and protein scaffold. In agreement with the xenon pressurization data, the most significant changes in rate are observed upon blocking tunnel 1, which suggests that it is a more favorable ligand migration pathway.
Fig. 4.

Determination of CO kon values (× 106 M-1 s-1) for the Ns H-NOX variants. (A) Time-resolved UV-visible spectra of CO binding to WT ferrous Ns H-NOX in a stopped-flow spectrometer (11.9 μM CO data is shown). (A, Inset) Single wavelength stopped-flow traces of the absorbance at 424 nm versus time for WT Ns H-NOX in the presence of varying CO concentrations. (B) Summary of CO kon values for the Ns H-NOX variants determined with stopped-flow spectroscopy (see Fig. S6 for a plot of kobs versus CO concentration). N = 3–5. Error represents the standard deviation. (C) Transient absorption traces of CO binding to WT ferrous Ns H-NOX under 1 atm of CO following laser photolysis. The trace at 440 nm represents the loss of the FeII-unligated species, and the trace at 415 nm represents the formation of the FeII–CO species. (D) Summary of CO kon values for all the Ns H-NOX variants determined with laser photolysis. N = 2. Error represents the standard deviation.
Table 2.
CO binding constants for Ns H-NOX and selected H-NOX proteins
| Protein | CO kon, × 106 M-1 s-1 |
CO koff, s-1 | CO KD*, μM | Ref.† | |
| Laser photolysis | Stopped flow | ||||
| Ns H-NOX | |||||
| Wild-type | 3.7 ± 0.1‡ | 1.52 ± 0.01 | 6.73 ± 0.42 | 4.42 ± 0.28 | |
| L67W | 4.4 ± 0.2 | 2.42 ± 0.04 | 6.71 ± 0.15 | 2.78 ± 0.08 | |
| L66W | 5.1 ± 0.3 | 2.84 ± 0.12 | 7.36 ± 0.62 | 2.59 ± 0.24 | |
| L66W/L67W | N/D§ | 2.74 ± 0.13 | 5.36 ± 0.20 | 1.96 ± 0.12 | |
| T48W | 5.0 ± 0.2 | 2.49 ± 0.07 | 4.13 ± 0.54 | 1.66 ± 0.22 | |
| I9W | 4.3 ± 0.3 | 2.12 ± 0.07 | 7.00 ± 0.18 | 3.30 ± 0.14 | |
| Tt H-NOX | 4.1 ± 0.2 | 2.18 ± 0.09 | 3.56 ± 0.42 | 1.63 ± 0.20 | |
| sGC | N/D | 0.0358 ± 0.0015 | 3.5 ± 0.5 | 98 ± 15 | 54 |
*Calculated from koff/kon using stopped-flow data.
†This work unless otherwise noted.
‡Error (±) represents the standard deviation.
§N/D, not determined.
To confirm the increases observed in CO on-rates when the tunnels are blocked, tryptophan residues were also engineered at the I9 and T48 positions in the tunnel network. I9 and T48 are located on different α-helices than L66W and L67W (Fig. 2). Based on modeling of allowed tryptophan rotamers (38), the T48W side chain is predicted to be incorporated near the branch point between tunnel 1 and tunnel 2, and the I9W side chain is predicted to be incorporated further away from the heme in tunnel 1 in-between the locations of T48W and L66W (Fig. S7). The I9W and T48W single mutants were generated as described above. As with the other variants, UV-visible spectra obtained for T48W and I9W heme–ligand complexes were very similar to the WT protein (Table S2), again suggesting that the tryptophan residues did not greatly alter the electronic properties of the heme. Characterization of CO on-rates with stopped-flow spectroscopy illustrated that the CO kon values for I9W and T48W are faster than WT. The T48W variant in particular exhibited a greater increase in rate that is similar to L66W and the L66W/L67W double mutant (Fig. 4B and Table 2). The increases in ligand on-rates across a broad panel of tunnel-blocking mutants suggest that the kinetic trend is a general phenomenon.
To eliminate the requirement for CO diffusion into the heme pocket during data acquisition, association kinetics were also evaluated in the tryptophan variants under a CO-equilibrated atmosphere using laser photolysis (42). Ferrous Ns H-NOX was incubated anaerobically overnight under 1 atm of CO to ensure full sample equilibration. Photolysis of the FeII–CO bond was carried out using 560-nm laser irradiation to transiently form the FeII–unligated heme. CO binding was subsequently followed over a 10-ms timescale (Fig. 4C) (26, 42). A CO kon value of 3.7 × 106 M-1 s-1 was obtained for WT Ns H-NOX. This rate is somewhat faster than the CO on-rate (1.52 × 106 M-1 s-1) observed in the stopped-flow experiment, which could reflect different protein conformational changes formed upon photodissociation of the CO ligand. Importantly, the relative trend in CO on-rates for the tunnel-blocking variants obtained with laser photolysis parallels what was observed in the stopped-flow experiment. WT Ns H-NOX exhibited the slowest on-rate, and the tunnel-blocking mutants exhibited faster rates with L66W showing the greatest increase (Fig. 4D and Table 2). The lack of significant structural changes in the tryptophan variants suggests that the faster ligand on-rates are due to more efficient ligand trapping in the protein interior. Blocking the tunnels both individually and together appears to eliminate routes of ligand diffusion from the protein to the solvent, increasing the effective CO concentration near the heme and the subsequent rate of CO binding to the iron.
Blocking the Tunnels Decreases the CO koff and Enhances CO Binding Affinity.
To examine if increased ligand trapping in the heme pocket also affects CO dissociation kinetics, dissociation rate constants (koff values or “off-rates”) were measured for the Ns H-NOX variants using stopped-flow spectroscopy in the presence of an NO trap (40, 42). An NO trap was utilized in order to limit CO rebinding following CO release from the heme. NO reacts quickly (approximately 108 M-1 s-1) with ferrous heme and binds with high affinity (KD ∼ pM) such that CO is not able to displace NO once it is coordinated to the iron (40, 42). WT Ns H-NOX was found to have a CO koff value of 6.73 s-1 (Fig. 5A), which is comparable to that reported previously (40). Several of the single tryptophan tunnel-blocking mutants (I9W, L66W, and L67W) had no significant change in CO off-rate. However, the L66W/L67W double mutant and the T48W mutant exhibited slower rates than WT (Fig. 5B and Table 2). It appears that blocking both tunnels is necessary to elicit an effect on CO koff values. These tryptophan residues likely decrease the CO off-rate by increasing rapid CO recombination to the heme following transient dissociation.
Fig. 5.

Determination of CO koff and KD values for the Ns H-NOX variants. (A) Time-resolved UV-visible spectra of CO dissociation from FeII–CO WT Ns H-NOX in the presence of a NO trap in a stopped-flow spectrometer. (A, Inset) Absorbance at 424 nm vs. time for CO dissociation from WT Ns H-NOX. (B) Summary of CO koff values. N≥3. Error represents the standard deviation. (C) Summary of CO KD values for all the Ns H-NOX variants. KD values were obtained from koff/kon.
Calculation of CO KD values (koff/kon) shows that blocking the tunnels increases CO binding affinity up to approximately 2.5-fold or approximately 2.2-fold in the T48W mutant based on stopped-flow and photolysis data, respectively (Fig. 5C and Table 2). The enhancement in ligand affinity is due to an increase in CO on-rate and decrease in CO off-rate, which appears to be associated with better ligand trapping in the heme pocket (Fig. 6). Additionally, the enhanced ligand affinity in the smaller cavity is likely thermodynamically favorable because of a decreased entropic penalty associated with ligand binding (16). Together, these data attest to the ability of H-NOX protein scaffolds to modulate ligand flux through the protein interior and implicate a role for protein tunnels in tuning ligand-binding properties in the H-NOX family.
Fig. 6.

Tunnel-blocking mutants modulate heme ligand-binding properties in H-NOX proteins by altering ligand migration. (A) A native H-NOX protein and (B) an H-NOX protein with tunnel 1 blocked are shown. A tryptophan residue is shown in blue, and diatomic ligands are shown in red. Blocking the tunnels individually (L66W and L67W mutants) and together (L66W/L67W mutant) in Ns H-NOX increases ligand trapping in the protein interior. This functions to both increase the CO kon following CO diffusion through the protein scaffold and decrease the CO koff. These kinetic trends appear to be due to the elimination of route(s) for ligand escape.
Discussion
Extensive studies on the globins have provided a model for how heme protein scaffolds control ligand binding (16, 17). Myoglobin, for example, has a heme group surrounded by deep gas pockets that work to efficiently capture and release O2 (16, 17). H-NOX protein scaffolds possess a unique fold and, unlike the globins, bind a heme cofactor that is completely buried within the protein interior. This framework appears to funnel gases toward the heme pocket and tune gas-sensing properties for involvement in diverse signaling pathways.
Much work has elucidated important determinants of ligand binding in the H-NOX family (25–30). However, the direct role of the protein scaffold in modulating ligand binding from sites beyond the heme pocket has not been previously addressed. Utilizing X-ray crystallography with xenon and kinetic measurements, a tunnel network has been mapped in a non-O2-binding H-NOX protein (Ns H-NOX) that appears to be conserved in H-NOX proteins with similar ligand-binding properties. The tunnel network consists of a Y-shaped, hydrophobic channel that serves as a pathway for gas diffusion to and from the heme (Figs. 1 and 2). Blocking the tunnel network influences heme ligand-binding kinetics—increasing ligand on-rates and decreasing ligand off-rates. These changes appear to be due to better ligand trapping near the heme, which results from removing pathways for ligand escape to the solvent (Fig. 6).
Although globins and H-NOX domains have unique interior routes for ligand diffusion, comparison between these protein families shows that divergent topological features may tune reversible ligand binding in parallel ways. Similar to Ns H-NOX, mutagenesis studies in myoglobin have found that mutations that vary the size and character of cavities affect affinity by modifying the occupation and migration of ligands (16, 17). This is reflected in O2 photolysis experiments on short time scales (nanosecond to microsecond) where blocking the gas pocket distal to the heme increases the fraction and rate of geminate ligand rebinding (16, 17). However, unlike Ns H-NOX, decreasing the interior cavity volume in myoglobin with structure-guided mutagenesis is associated with slowed ligand on-rates (16, 17). This is likely due to less efficient ligand capture in myoglobin’s more open heme site where decreasing the interior cavity volume appears to preferentially favor ligand escape (16, 17).
To provide initial insight into ligand migration in H-NOX proteins, a molecular dynamics (MD) study was conducted (33). The results of the MD simulations are in agreement with the ligand diffusion pathways observed here. Additionally, the calculations show that the presence of preformed pathways in Ns H-NOX increases the rate of CO escape to solvent relative to Tt H-NOX, which does not have an open tunnel network (Fig. S3) (33). This observation is consistent with the experimentally determined CO dissociation rates for both proteins reported in the present study (Table 2) and supports the hypothesis that blocking protein tunnels increases ligand capture.
Protein dynamics in H-NOX domains could work in concert with tunnels to control ligand diffusion to and from the heme pocket. An MD simulation carried out on Tt H-NOX, for example, led to the conclusion that the protein fluctuates near the heme edge to permit ligand flux (33). The potential role of transiently formed pathways in facilitating ligand access is particularly evident in Ns H-NOX when both tunnels are blocked. It is possible that the introduction of bulky residues could alter protein dynamics in a manner not evident in the structural data to form additional routes for ligand diffusion. Potential changes in dynamics in the mutants could also dampen our observed kinetic effects. Importantly, these transient pathways appear less important in the native Ns H-NOX scaffold where both computational and experimental data suggest that the open tunnel network represents the most favorable route for gas diffusion.
Differences in ligand flux between divergent classes of H-NOX proteins may influence ligand specificity. In H-NOX proteins that bind O2, the absence of preformed tunnels could work in concert with H-bonding residues to trap O2 at the heme iron and tighten O2 affinity through enhancing ligand rebinding (33). Most bacterial O2-binding H-NOX domains identified to date are in obligate anaerobes where a high O2 affinity would be important to sense and respond to low amounts of environmental O2 (24). In non-O2-binding H-NOX proteins, the tunnel network appears to permit more ligand flux through the heme pocket. This could favor NO binding under aerobic conditions because NO binds to ferrous heme with higher intrinsic affinity than O2 (42). The potential ability to tune ligand-binding properties with distinct interior architectures may reflect the coevolution of ligand diffusion pathways and diverse H-NOX protein functions.
Internal topological features in numerous proteins are essential for controlling the capture and release of biomolecules. For example, in NiFe hydrogenase, tunnels have been shown to promote H2 back-exchange with solvent but also facilitate the escape of inhibitory amounts of CO (4); in superoxide dismutase, a positively charged cleft steers 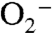 to the active site, enabling nearly diffusion-limited turnover rates (43); and in soybean lipoxygenase, the route of O2 migration into the active site influences product regiochemistry (2). Results reported here show that a tunnel network in H-NOX heme sensor domains guides gases to the heme iron and also tunes reversible gas binding by establishing routes for ligand diffusion. Together, our findings reflect the tightly coordinated relationship between protein frameworks and protein function. Structural features are employed in proteins not only to directly participate in active site chemistry but also control the flux of biomolecules. A deeper understanding of this complex relationship is essential to unraveling protein mechanisms and predicting biochemical properties across diverse protein families.
to the active site, enabling nearly diffusion-limited turnover rates (43); and in soybean lipoxygenase, the route of O2 migration into the active site influences product regiochemistry (2). Results reported here show that a tunnel network in H-NOX heme sensor domains guides gases to the heme iron and also tunes reversible gas binding by establishing routes for ligand diffusion. Together, our findings reflect the tightly coordinated relationship between protein frameworks and protein function. Structural features are employed in proteins not only to directly participate in active site chemistry but also control the flux of biomolecules. A deeper understanding of this complex relationship is essential to unraveling protein mechanisms and predicting biochemical properties across diverse protein families.
Materials and Methods
Plasmids for Protein Expression.
The gene for Ns H-NOX (residues 1–189) from Nostoc sp PCC 7120 (32) was purchased codon-optimized in the pUC57 vector from GenScript. The gene was PCR amplified and cloned into the pCW plasmid. DNA primers for PCR amplification and site-directed mutagenesis were synthesized by Integrated DNA Technologies. Sequencing of all Ns H-NOX variants was carried out by Elim Biopharmaceuticals, Inc. The gene for untagged Tt H-NOX (residues 1–188 of TtTar4H from Thermoanaerobacter tengcongensis) (44) and the construct with a C-terminal His6 tag (26) were used as described previously.
Protein Expression.
Ns H-NOX constructs were transformed into the RP523 strain of Escherichia coli (45). Expression cultures were grown at 37 °C in Terrific Broth in the presence of 30 μg/mL hemin (approximately 200× stock in DMSO) and 75–100 μg/mL ampicillin. Expression was induced with 1 mM IPTG, and induction was allowed to occur for 18–22 h at room temperature. Tt H-NOX constructs were expressed as described previously (26).
Protein Purification.
All Ns H-NOX variants were purified using anion exchange chromatography followed by size-exclusion chromatography (SI Materials and Methods). Proteins were isolated in the FeII-unligated state (Soret maxima of 429–430 nm). Tt H-NOX constructs were purified as described previously (26, 45) and isolated in the FeII–O2 ligation state (Soret maximum of approximately 416 nm).
UV-Visible Spectral Characterization.
Heme–ligand complexes were generated as described previously using established methods (SI Materials and Methods) (44).
Protein Crystallization.
Crystallization conditions for the Ns H-NOX variants and WT Tt H-NOX are reported in the SI Materials and Methods. All crystals appeared within 12 h. Cryoprotection was achieved by transferring the crystals stepwise into mother liquor solutions containing 5%, 10%, and 15% glycerol for Ns H-NOX variants and 5%, 10%, 15%, and 20% for Tt H-NOX. The crystals for native datasets were flash frozen in liquid N2 for storage without further manipulation.
Xenon Derivatization.
Crystals were derivatized with xenon (research grade; Praxair, Inc.) using a Xenon Chamber (Hampton Research) apparatus following standard protocols (46). Briefly, the wick in the provided Mini-Vial/Wick system was saturated with cryoprotectant, and an additional approximately 75 μL of cryoprotectant was added to the bottom of the vial to prevent crystal dehydration. The cryoprotectant in the Mini-Vial/Wick system was preequilibrated with xenon by pressurizing the chamber three times at each xenon pressure to be subsequently used for crystal derivatization. Crystals underwent cryoprotection as described above and were immediately pressurized in the chamber with 1 to 8 atm xenon for 0.5, 1, 5, 10, 15, or 30 min. Following derivatization, the chamber was rapidly depressurized (< 10 s), and the crystals were immediately frozen (approximately 5 s) in liquid N2 for storage.
X-Ray Data Collection and Structure Refinement.
For native crystals, X-ray data were collected using synchrotron radiation at beamlines 5.0.2 or 8.3.1 at the Advanced Light Source, Lawrence Berkeley National Laboratory (Berkeley, CA). Diffraction images were collected at 100 K with exposure times of 0.1 to 1 s and 0.35° to 1° oscillations per frame at a wavelength of λ = 0.97 Å. Data integration and scaling were performed using the HKL2000 (47) suite, and structure solution was performed using Phaser (48) with Ns H-NOX (PDB ID code 2O09) or Tt H-NOX (PDB ID code 1U55) as the search model. Rebuilding of the model was performed using ARP/wARP (49) following molecular replacement, and manual model rebuilding was performed using Coot (38). Iterative model refinement was performed using PHENIX (50) with TLS refinement parameters incorporated. Refinement statistics are listed in Table 1 and Table S1. Stereochemical properties were assessed by MOLPROBITY (51) and PROCHECK (52). Data collection and structure refinement for xenon-pressurized crystals was carried out as described in SI Materials and Methods.
Stopped-Flow Spectroscopy.
CO association (kon) and dissociation (koff) rate constants were determined at 20 °C using a HiTech KinetAsyst stopped-flow instrument equipped with a diode array detector (SI Materials and Methods). Briefly, for CO association rate constants, ferrous protein (1 μM) was mixed with varying amounts of CO (24, 48, 95, and 143 μM) that were generated from a saturated CO solution (99.99%; Praxair, Inc.) in the presence of approximately 10 mM dithionite. UV-visible spectra were recorded for 450 ms (300 scans, 1.5-ms integration time) from 400–700 nm using the Kinetic Studio program (TgK Scientific). The spectral transition was fit to a two-state model using the SPECFIT Global Analysis System (version 3.0.14). Replicate CO titrations (N = 3–5) were carried out on different days utilizing protein from different preparations whenever possible. Average observed rates (kobs) for each CO concentration are reported for > 30 sample shots on the stopped flow. For CO dissociation rate constants, ferrous-CO protein (2 μM) was mixed with an NO (99.5%; Praxair, Inc.) trap as described previously (40, 42). Rates were found to be independent of trap concentration. UV-visible spectra were recorded for 4.5 s from 350–700 nm using the Kinetic Studio program as above. The difference in absorbance (424 and 416 nm) versus time was fit to a single exponential equation using Igor Pro (version 5.01). Measurements were recorded from ≥3 samples on different days and represent ≥18 sample shots on the stopped flow.
Flash Photolysis.
CO association rate constants were also determined using transient absorption spectroscopy at the Beckman Institute Laser Resource Center (California Institute of Technology, Pasadena, CA) with an experimental setup that has been described previously (53). Proteins were reduced with dithionite and desalted into spectral buffer (50 mM HEPES, pH 7.4, 50 mM NaCl) as described in the stopped-flow experiment (SI Materials and Methods). The proteins were subsequently sparged with 1 atm CO in a sealed 1-cm path-length quartz cuvette and incubated under a CO atmosphere overnight at 4 °C with gentle rocking to ensure full sample equilibration prior to data acquisition. The protein FeII–CO bond was photolyzed using 560 nm irradiation from a Nd:YAG laser (8-ns pulse). Data were collected at 20 °C over 10 ms at 415 and 440 nm from two samples on different days. Traces were fit to a single exponential equation using Igor Pro.
Supplementary Material
Acknowledgments.
We thank beamline scientists at Lawrence Berkeley National Laboratory (in particular Dr. Corie Y. Ralston and Dr. James M. Holton) for use of equipment and technical assistance. We are exceptionally grateful to Dr. Charlotte A. Whited and Prof. Harry B. Gray at the Beckman Institute Laser Resource Center (California Institute of Technology) for acquisition of laser photolysis data. We also thank Dr. Eric S. Underbakke for generating a graphical illustration of our model and Dr. Emily E. Weinert for providing Tt H-NOX with a His6 tag for kinetic measurements. We thank current and past members of the Marletta laboratory (in particular Dr. Emily E. Weinert, Dr. Eric S. Underbakke, Dr. Christine M. Phillips-Piro, and Dr. Steven Y. Reece) for frequent helpful discussions and their invaluable insight. We thank the National Institutes of Health Grant GM070671 (M.A.M), the Aldo DeBenedictis Fund (M.B.W.), and a National Institutes of Health Molecular Biophysics Training grant (M.A.H.) for financial support.
Footnotes
Conflict of interest statement: M.A.M. is a cofounder of Omniox, Inc. This company is developing H-NOX domains for oxygen therapy.
See Author Summary on page 17577.
Data deposition: The atomic coordinates have been deposited in the Research Collaboratory for Structural Bioinformatics Protein Data Bank, http://www.rcsb.org/pdb/home/home.do [PDB ID codes 3TF8 (WT Ns H-NOX), 3TF9 (WT Ns H-NOX under 1 atm Xe), 3TFA (WT Ns H-NOX under 6 atm Xe), 3TFD (L66W), 3TFE (L66W under 6 atm Xe), 3TFF (L67W), 3TFG (L66W/L67W), 3TF0 (WT Tt H-NOX), and 3TF1 (WT Tt H-NOX under 6 atm Xe)].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1114038108/-/DCSupplemental.
References
- 1.Baron R, et al. Multiple pathways guide oxygen diffusion into flavoenzyme active sites. Proc Natl Acad Sci USA. 2009;106:10603–10608. doi: 10.1073/pnas.0903809106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Knapp MJ, Seebeck FP, Klinman JP. Steric control of oxygenation regiochemistry in soybean lipoxygenase-1. J Am Chem Soc. 2001;123:2931–2932. doi: 10.1021/ja003855k. [DOI] [PubMed] [Google Scholar]
- 3.Igarashi RY, Seefeldt LC. Nitrogen fixation: The mechanism of the Mo-dependent nitrogenase. Crit Rev Biochem Mol Biol. 2003;38:351–384. doi: 10.1080/10409230391036766. [DOI] [PubMed] [Google Scholar]
- 4.Leroux F, et al. Experimental approaches to kinetics of gas diffusion in hydrogenase. Proc Natl Acad Sci USA. 2008;105:11188–11193. doi: 10.1073/pnas.0803689105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doukov TI, Iverson TM, Seravalli J, Ragsdale SW, Drennan CL. A Ni-Fe-Cu center in a bifunctional carbon monoxide dehydrogenase/acetyl-CoA synthase. Science. 2002;298:567–572. doi: 10.1126/science.1075843. [DOI] [PubMed] [Google Scholar]
- 6.Manjasetty BA, Powlowski J, Vrielink A. Crystal structure of a bifunctional aldolase-dehydrogenase: Sequestering a reactive and volatile intermediate. Proc Natl Acad Sci USA. 2003;100:6992–6997. doi: 10.1073/pnas.1236794100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lund L, Fan Y, Shao Q, Gao YQ, Raushel FM. Carbamate transport in carbamoyl phosphate synthetase: A theoretical and experimental investigation. J Am Chem Soc. 2010;132:3870–3878. doi: 10.1021/ja910441v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hyde CC, Ahmed SA, Padlan EA, Miles EW, Davies DR. Three-dimensional structure of the tryptophan synthase alpha 2 beta 2 multienzyme complex from Salmonella typhimurium. J Biol Chem. 1988;263:17857–17871. [PubMed] [Google Scholar]
- 9.Tilton RF, Jr, Kuntz ID, Jr, Petsko GA. Cavities in proteins: Structure of a metmyoglobin-xenon complex solved to 1.9 A. Biochemistry. 1984;23:2849–2857. doi: 10.1021/bi00308a002. [DOI] [PubMed] [Google Scholar]
- 10.Austin RH, Beeson KW, Eisenstein L, Frauenfelder H, Gunsalus IC. Dynamics of ligand binding to myoglobin. Biochemistry. 1975;14:5355–5373. doi: 10.1021/bi00695a021. [DOI] [PubMed] [Google Scholar]
- 11.Schlichting I, Berendzen J, Phillips GN, Jr, Sweet RM. Crystal structure of photolysed carbonmonoxy-myoglobin. Nature. 1994;371:808–812. doi: 10.1038/371808a0. [DOI] [PubMed] [Google Scholar]
- 12.Schotte F, et al. Watching a protein as it functions with 150-ps time-resolved X-ray crystallography. Science. 2003;300:1944–1947. doi: 10.1126/science.1078797. [DOI] [PubMed] [Google Scholar]
- 13.Cohen J, Arkhipov A, Braun R, Schulten K. Imaging the migration pathways for O2, CO, NO, and Xe inside myoglobin. Biophys J. 2006;91:1844–1857. doi: 10.1529/biophysj.106.085746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Elber R. Ligand diffusion in globins: Simulations versus experiment. Curr Opin Struct Biol. 2010;20:162–167. doi: 10.1016/j.sbi.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ringe D, Petsko GA, Kerr DE, Ortiz de Montellano PR. Reaction of myoglobin with phenylhydrazine: A molecular doorstop. Biochemistry. 1984;23:2–4. doi: 10.1021/bi00296a001. [DOI] [PubMed] [Google Scholar]
- 16.Olson JS, Soman J, Phillips GN., Jr Ligand pathways in myoglobin: A review of Trp cavity mutations. IUBMB Life. 2007;59:552–562. doi: 10.1080/15216540701230495. [DOI] [PubMed] [Google Scholar]
- 17.Scott EE, Gibson QH, Olson JS. Mapping the pathways for O2 entry into and exit from myoglobin. J Biol Chem. 2001;276:5177–5188. doi: 10.1074/jbc.M008282200. [DOI] [PubMed] [Google Scholar]
- 18.Salter MD, et al. The apolar channel in Cerebratulus lacteus hemoglobin is the route for O2 entry and exit. J Biol Chem. 2008;283:35689–35702. doi: 10.1074/jbc.M805727200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Milani M, et al. Heme-ligand tunneling in group I truncated hemoglobins. J Biol Chem. 2004;279:21520–21525. doi: 10.1074/jbc.M401320200. [DOI] [PubMed] [Google Scholar]
- 20.de Sanctis D, et al. Crystal structure of cytoglobin: The fourth globin type discovered in man displays heme hexa-coordination. J Mol Biol. 2004;336:917–927. doi: 10.1016/j.jmb.2003.12.063. [DOI] [PubMed] [Google Scholar]
- 21.Pesce A, et al. Human brain neuroglobin structure reveals a distinct mode of controlling oxygen affinity. Structure. 2003;11:1087–1095. doi: 10.1016/s0969-2126(03)00166-7. [DOI] [PubMed] [Google Scholar]
- 22.Hargrove MS, et al. Characterization of recombinant soybean leghemoglobin a and apolar distal histidine mutants. J Mol Biol. 1997;266:1032–1042. doi: 10.1006/jmbi.1996.0833. [DOI] [PubMed] [Google Scholar]
- 23.Vallone B, Nienhaus K, Matthes A, Brunori M, Nienhaus GU. The structure of carbonmonoxy neuroglobin reveals a heme-sliding mechanism for control of ligand affinity. Proc Natl Acad Sci USA. 2004;101:17351–17356. doi: 10.1073/pnas.0407633101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boon EM, Marletta MA. Ligand discrimination in soluble guanylate cyclase and the H-NOX family of heme sensor proteins. Curr Opin Chem Biol. 2005;9:441–446. doi: 10.1016/j.cbpa.2005.08.015. [DOI] [PubMed] [Google Scholar]
- 25.Boon EM, Huang SH, Marletta MA. A molecular basis for NO selectivity in soluble guanylate cyclase. Nat Chem Biol. 2005;1:53–59. doi: 10.1038/nchembio704. [DOI] [PubMed] [Google Scholar]
- 26.Weinert EE, Plate L, Whited CA, Olea C, Jr, Marletta MA. Determinants of ligand affinity and heme reactivity in H-NOX domains. Angew Chem Int Ed Engl. 2009;49:720–723. doi: 10.1002/anie.200904799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rothkegel C, et al. Identification of residues crucially involved in soluble guanylate cyclase activation. FEBS Lett. 2006;580:4205–4213. doi: 10.1016/j.febslet.2006.06.079. [DOI] [PubMed] [Google Scholar]
- 28.Martin E, Berka V, Bogatenkova E, Murad F, Tsai AL. Ligand selectivity of soluble guanylyl cyclase: Effect of the hydrogen-bonding tyrosine in the distal heme pocket on binding of oxygen, nitric oxide, and carbon monoxide. J Biol Chem. 2006;281:27836–27845. doi: 10.1074/jbc.M601078200. [DOI] [PubMed] [Google Scholar]
- 29.Derbyshire ER, Deng S, Marletta MA. Incorporation of tyrosine and glutamine residues into the soluble guanylate cyclase heme distal pocket alters NO and O2 binding. J Biol Chem. 2010;285:17471–17478. doi: 10.1074/jbc.M109.098269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olea C, Boon EM, Pellicena P, Kuriyan J, Marletta MA. Probing the function of heme distortion in the H-NOX family. ACS Chem Biol. 2008;3:703–710. doi: 10.1021/cb800185h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pellicena P, Karow DS, Boon EM, Marletta MA, Kuriyan J. Crystal structure of an oxygen-binding heme domain related to soluble guanylate cyclases. Proc Natl Acad Sci USA. 2004;101:12854–12859. doi: 10.1073/pnas.0405188101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma X, Sayed N, Beuve A, van den Akker F. NO and CO differentially activate soluble guanylyl cyclase via a heme pivot-bend mechanism. EMBO J. 2007;26:578–588. doi: 10.1038/sj.emboj.7601521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, Lu M, Cheng Y, Li Z. H-NOX domains display different tunnel systems for ligand migration. J Mol Graph Model. 2010;28:814–819. doi: 10.1016/j.jmgm.2010.02.007. [DOI] [PubMed] [Google Scholar]
- 34.Marassio G, et al. Pressure-response analysis of anesthetic gases xenon and nitrous oxide on urate oxidase: A crystallographic study. FASEB J. 2011;25:2266–2275. doi: 10.1096/fj.11-183046. [DOI] [PubMed] [Google Scholar]
- 35.Bondi A. van der Waals volumes and radii. J Phys Chem. 1964;68:441–451. [Google Scholar]
- 36.Quillin ML, Breyer WA, Griswold IJ, Matthews BW. Size versus polarizability in protein-ligand interactions: Binding of noble gases within engineered cavities in phage T4 lysozyme. J Mol Biol. 2000;302:955–977. doi: 10.1006/jmbi.2000.4063. [DOI] [PubMed] [Google Scholar]
- 37.Beneš P, et al. 2010. CAVER, Version 2.1.
- 38.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 39.Abraham DJ, Leo AJ. Extension of the fragment method to calculate amino acid zwitterion and side chain partition coefficients. Proteins. 1987;2:130–152. doi: 10.1002/prot.340020207. [DOI] [PubMed] [Google Scholar]
- 40.Tsai AL, et al. Is Nostoc H-NOX a NO sensor or redox switch? Biochemistry. 2010;49:6587–6599. doi: 10.1021/bi1002234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tannenbaum SR, White FM. Regulation and specificity of S-nitrosylation and denitrosylation. ACS Chem Biol. 2006;1:615–618. doi: 10.1021/cb600439h. [DOI] [PubMed] [Google Scholar]
- 42.Rohlfs RJ, et al. The effects of amino acid substitution at position E7 (residue 64) on the kinetics of ligand binding to sperm whale myoglobin. J Biol Chem. 1990;265:3168–3176. [PubMed] [Google Scholar]
- 43.Getzoff ED, et al. Faster superoxide dismutase mutants designed by enhancing electrostatic guidance. Nature. 1992;358:347–351. doi: 10.1038/358347a0. [DOI] [PubMed] [Google Scholar]
- 44.Karow DS, et al. Spectroscopic characterization of the soluble guanylate cyclase-like heme domains from Vibrio cholerae and Thermoanaerobacter tengcongensis. Biochemistry. 2004;43:10203–10211. doi: 10.1021/bi049374l. [DOI] [PubMed] [Google Scholar]
- 45.Winter MB, et al. Ru-porphyrin protein scaffolds for sensing O2. J Am Chem Soc. 2010;132:5582–5583. doi: 10.1021/ja101527r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Soltis SM, Stowell MHB, Wiener MC, Phillips GN, Rees DC. Successful flash-cooling of xenon-derivatized myoglobin crystals. J Appl Crystallogr. 1997;30:190–194. [Google Scholar]
- 47.Otwinowski A, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 48.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61:458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 49.Lamzin VS, Perrakis A, Wilson KS. In: International Tables for Crystallography. Rossmann MG, Arnold E, editors. Dordrecht, The Netherlands: Kluwer; 2001. pp. 720–722. [Google Scholar]
- 50.Adams PD, et al. PHENIX: Building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr. 2002;58:1948–1954. doi: 10.1107/s0907444902016657. [DOI] [PubMed] [Google Scholar]
- 51.Davis IW, et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 53.Dempsey JL, Winkler JR, Gray HB. Kinetics of electron transfer reactions of H2-evolving cobalt diglyoxime catalysts. J Am Chem Soc. 2010;132:1060–1065. doi: 10.1021/ja9080259. [DOI] [PubMed] [Google Scholar]
- 54.Stone JR, Sands RH, Dunham WR, Marletta MA. Spectral and ligand-binding properties of an unusual hemoprotein, the ferric form of soluble guanylate cyclase. Biochemistry. 1996;35:3258–3262. doi: 10.1021/bi952386+. [DOI] [PubMed] [Google Scholar]
