

Abstract
Periods of oceanic anoxia have had a major influence on the evolutionary history of Earth and are often contemporaneous with mass extinction events. Changes in global (as opposed to local) redox conditions can be potentially evaluated using U system proxies. The intensity and timing of oceanic redox changes associated with the end-Permian extinction horizon (EH) were assessed from variations in 238U/235U (δ238U) and Th/U ratios in a carbonate section at Dawen in southern China. The EH is characterized by shifts toward lower δ238U values (from -0.37‰ to -0.65‰), indicative of an expansion of oceanic anoxia, and higher Th/U ratios (from 0.06 to 0.42), indicative of drawdown of U concentrations in seawater. Using a mass balance model, we estimate that this isotopic shift represents a sixfold increase in the flux of U to anoxic facies, implying a corresponding increase in the extent of oceanic anoxia. The intensification of oceanic anoxia coincided with, or slightly preceded, the EH and persisted for an interval of at least 40,000 to 50,000 y following the EH. These findings challenge previous hypotheses of an extended period of whole-ocean anoxia prior to the end-Permian extinction.
Keywords: carbonates, uranium isotopes, paleoredox
The end-Permian extinction represents the largest mass extinction in Earth history, with the demise of an estimated 90% of all marine species (1). While it has been extensively studied, the exact nature and cause of the end-Permian extinction remains the subject of intense scientific debate. Proposed kill mechanisms have included a nearby supernova, bolide impacts, periods of extreme volcanism (e.g., Siberian Traps), extensive glaciation, and widespread oceanic anoxia (2). Evidence for shallow-ocean anoxia in conjunction with the end-Permian mass extinction is widespread (3–6), but the intensity and timing of oceanic redox changes remain uncertain (7–10). Recent hypotheses have invoked the release of hydrogen sulfide gas (H2S) from seawater as a kill mechanism (11–13). Such models call upon strong expansion of oceanic anoxia below the oxygenated surface layer to allow buildup of H2S, followed by an upward excursion of the chemocline that releases the poisonous gas into the atmosphere (13). In this study, we examine the 238U/235U and Th/U (thorium/uranium) ratios in a carbonate section spanning the end-Permian extinction horizon (EH) to evaluate the timing and scale of these possibilities. Samples for this study were collected from the Dawen section of the Yangtze Block in southern China (Fig. 1), which has been correlated with the global stratotype section and point (GSSP) of the Permian-Triassic boundary at Meishan (14).
Fig. 1.

Location of South China at ∼252 Ma, the time of the end-Permian extinction [Fig. 1A, modified base map from R. Blakey (http://jan.ucc.nau.edu/~rcb7/260moll.jpg)] and present-day location of the Dawen section (Fig. 1B, modified from ref. 14). The location of the Meishan GSSP is shown for reference. Lithostratigraphy of the Dawen section is shown in Fig. 1C; see ref. 14 for its correlation to the Meishan GSSP.
Due to the geochemical properties of U, the ratio of 238U/235U can be used as a tool to investigate the history of ocean oxygenation at a global scale, as opposed to the local redox information provided by most commonly used proxies. The long residence time (∼500 ky) of U in the oceans leads to a homogeneous U concentration in seawater (15, 16), as well as to a homogenous U isotopic composition (17–19). The low-temperature redox transition of U (from U6+ to U4+) is the primary cause of 238U/235U fractionation on Earth, with the reduced species preferentially enriched in 238U (18–22). During times of oceanic anoxia, the flux of reduced U to anoxic facies (such as black shales) increases, preferentially removing 238U from seawater. The loss of isotopically heavy U drives seawater to lighter isotopic compositions (23). Changes in the U isotope ratios of organic-rich sediments have been used to study oceanic redox conditions during the Cretaceous (23). Here, we apply this isotope system to Late Permian and Early Triassic carbonate rocks. Existing evidence indicates that carbonates record the 238U/235U ratio of the seawater in which they were deposited (18, 19), suggesting that ancient carbonates that retain a primary signal of U isotopes can be used to estimate relative changes in ocean oxygenation.
Results and Discussion
In the Dawen section, the average U isotopic composition of samples deposited prior to the EH (δ238U = -0.37‰) is very close to that of modern seawater [δ238U = -0.41 ± 0.03‰ (19)]. This observation suggests that the fraction of U removed to reducing sinks during the late Permian was similar to that of the modern ocean. The Dawen section exhibits an abrupt and significant change in δ238U at the EH (Fig. 2) to values averaging -0.65‰. The δ238U ratios of pre- and post-EH samples are significantly different (p < 0.0001; two-tailed student’s t-test with a significance level of α = 0.01). A few isotopically light samples are present below the EH (-118 and -97 cm), which may provide evidence of brief episodes of transient intensification of oceanic anoxia preceding the end-Permian mass extinction. This inference is supported by evidence from additional geochemical proxies in other studies (7) (see SI Text for further discussion).
Fig. 2.

Geochemical profiles for the Dawen section. The vertical dashed lines represent average values of δ238U and Th/U for pre-EH and post-EH samples. The 238U/235U ratios are reported using standard δ-notation, where δ238U = [Ratiomeas/Ratiostd(SRM950a) - 1] × 1,000. Average 2 × standard deviation (2SD) uncertainty of δ238U values is shown on the top data point only for clarity. δ13C and stratigraphic data from ref. 14.
The shift toward lighter U isotopic compositions after the extinction event is consistent with an increase in the deposition of isotopically heavy U in anoxic facies. The isotopic composition of U in seawater is ultimately controlled by the relative sizes and isotopic signatures of the major sources and sinks of U to the ocean. A simple box model of the oceanic U budget for the modern and end-Permian oceans is shown in Fig. 3. Invoking mass balance, we calculated the approximate increase in anoxic sedimentation in the end-Permian ocean as:
| [1] |
Here, fanoxic represents the fraction of U deposited in anoxic facies and δ238U represents the δ238U values of the anoxic and “other” (i.e., nonanoxic) sinks. Following Montoya-Pino, et al. (23), we assume: (1) isotopically constant U input from rivers [the largest source of U to the ocean (24)] over geologic time with a value of -0.3‰; (2) a constant isotope fractionation between seawater and anoxic/euxinic environments of +0.5‰; and (3) a constant (+0.1‰) isotope fractionation between seawater and the sum of other sinks, including ferromanganese oxide, hydrothermal, and suboxic sediments. Suboxic sediments, which are defined by their low oxygen concentrations in the bottom water [e.g., 0.2–2 mL O2 per l H2O (25)], are likely to represent the largest nonanoxic sink (24) and are known to accumulate U with a small fractionation (+0.1‰) relative to seawater (19). Based on the assumptions above, 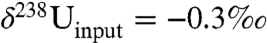 ,
, 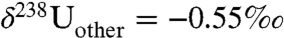 (i.e., -0.65 + 0.1‰), and
(i.e., -0.65 + 0.1‰), and 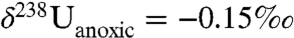 (i.e., -0.65 + 0.5‰). These values yield an estimated sixfold increase in the flux of U to anoxic facies in conjunction with the EH.
(i.e., -0.65 + 0.5‰). These values yield an estimated sixfold increase in the flux of U to anoxic facies in conjunction with the EH.
Fig. 3.

Box models representing the modern ocean (left) and the hypothesized end-Permian anoxic ocean on the right. Estimates of the percentage of U flux for each sink are shown above the arrows. All data are displayed using standard δ-notation, as defined above. Modern δ238U values are taken from Weyer, et al. (19). The values from the PTB ocean (right) are based on calculations using the average δ238U of carbonates after the EH in our sample set (see Eq. 1) and assuming a constant isotope fractionation between seawater and anoxic sediments.
Th/U ratios serve as an additional and independent line of evidence for oceanic redox changes in conjunction with the end-Permian extinction. Previous workers have used Th/U ratios in reduced sediments as a proxy for ocean redox chemistry (5). As Th has only one redox state (Th4+), its concentration in sediments is unaffected by redox conditions. On the other hand, U is a redox-sensitive metal and is readily removed from seawater as insoluble U4+ under reducing conditions (26–28), thus concentrating U relative to Th in anoxic facies. An increase in anoxic sedimentation reduces the concentration of U in seawater as more U is sequestered in organic-rich sediments. Because the U concentration of carbonates is related to the U concentration of the seawater in which they are deposited (29, 30), an increase in anoxic sedimentation results in an increase in the Th/U ratio of carbonate sediments. At Dawen, average Th/U ratios increase from 0.06 below the EH to 0.42 above the EH (Fig. 2). This increase reflects a decrease in the U content of seawater, possibly by a factor of ∼7× if Th concentrations remained constant. A change in seawater U concentrations of this magnitude is consistent with the sixfold expansion of oceanic anoxic inferred from our δ238U data, and is also consistent with the sharp decrease in U concentrations across the Permian-Triassic boundary (PTB) previously reported from a carbonate section in Oman (10). Although a lower U concentration would imply a shorter ocean residence time, even if reduced tenfold the U residence time would have been 50–100 times longer than the time scale for ocean mixing. Therefore, it is reasonable to assume that δ238U and Th/U provide information at global scale.
Although the study section has undergone burial diagenesis, the U and carbonate C isotopic profiles are unlikely to have been altered greatly. For example, neither δ238U nor Th/U shows a correlation with degree of dolomitization or with terrigenous input; see SI Text for discussion.
The timing for the onset of widespread oceanic anoxia implied by our results from Dawen is difficult to reconcile with previous hypotheses of persistent anoxia for hundreds of thousands or millions of years prior to the end-Permian extinction event (3, 5, 6, 8). The abrupt increase in Th/U ratios and decrease in δ238U that begin at or just below the EH indicate that sustained expanded anoxia did not exist in the Late Permian ocean until immediately prior to the extinction event. This global redox signal is consistent with proxies recording local redox conditions within the Panthalassic Ocean (31, 32). In contrast, the steady and prolonged decline of carbonate δ13C seen in many stratigraphic sections prior to the EH has been used to argue for an extended period of ocean stagnation and whole-ocean anoxia (2). If so, δ238U and Th/U in carbonates should track δ13C. This behavior is not observed (Fig. 2).
The existence of an unconformable surface in the Dawen section at the level of the EH (Fig. 2) makes exact assessment of the timing of global redox changes unattainable. If the missing section is equivalent to beds 25 and 26 at Meishan, it would represent a hiatus of about 50,000–75,000 y (14). However, we note that the δ13C curve for Dawen (see Fig. 5 in ref. 14) shows an almost unbroken shift toward more negative values across the contact, suggesting that the hiatus was of limited duration. These data indicate that the abruptness of the shifts in δ238U and Th/U ratios at the EH (Fig. 2) was the result not of a missing section but, rather, of a rapid and sustained change in oceanographic conditions. The persistence of low δ238U and high Th/U ratios through the 10 m of section above the EH that were analyzed in this study (Fig. 2) indicates that intensified anoxia persisted a minimum of ∼40,000–50,000 y following the end-Permian extinction (14, 33, 34).*
While our data do not support the extended period of whole-ocean anoxia prior to the EH inferred from δ13C records, they do not invalidate the idea that the end-Permian mass extinction was caused by oceanic oxygen depletion and a subsequent buildup and release of H2S from the oceans, as inferred on the basis of geochemical, isotopic, and biomarker studies (3–6, 8). Commonly, models of this process invoke an extended period of sluggish ocean circulation, producing deep-ocean anoxia and accumulation of H2S. This interpretation was previously challenged by numerical models of the ocean-climate system suggesting that the deep ocean was most likely well ventilated throughout the Late Permian-Early Triassic interval (35). We propose that the geochemical data and numerical models can be reconciled by hypothesizing expanded and more intense oxygen-minimum zones at middepths in the late-Permian ocean (7, 9, 32). Suboxic deep-ocean conditions during the Late Permian prior to the EH (9, 32) would have decreased the U concentration of the ocean, lowering the residence time of U in seawater and setting the stage for the rapid shift in Th/U at the EH observed at Dawen (Fig. 2). Th/U ratios of pre-EH carbonates at Dawen are significantly higher than Th/U ratios in modern marine carbonates, supporting the hypothesis that U was substantially drawn down in latest Permian seawater relative to modern seawater. Suboxic deep-ocean conditions would not have markedly altered the U isotope budget of the global ocean, as suboxic sedimentation does not fractionate U isotopes with the same magnitude as anoxic sedimentation (19, 23). Uranium isotopes would have shifted measurably only with an increase in anoxic sedimentation. However, transient disturbances to Late Permian oceans [e.g., warming or an increase in continentally derived nutrients (9)] may have resulted in brief episodes of expansion of oxygen-minimum zones before the end-Permian extinction, as reflected in light δ238U ratios below the EH (-118 and -97 cm). Expansion of oxygen-minimum zones could have introduced H2S into the photic zone (9) if the upper boundary of the former shallowed sufficiently (to 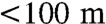 water depth). This could then result in release of toxic gasses into shallow-marine environments and the atmosphere (12), similar to the degassing of H2S in modern oxygen-minimum zones in Namibia (36).
water depth). This could then result in release of toxic gasses into shallow-marine environments and the atmosphere (12), similar to the degassing of H2S in modern oxygen-minimum zones in Namibia (36).
A model of ocean chemistry with widespread regions of relatively warm and poorly oxygenated deep water and localized intermittent sulfide maxima at midwater depths (i.e., within the oxygen-minimum zone) satisfies not only δ13C evidence previously used to argue for sustained oceanic anoxia prior to the EH, but also explains the observed geochemical and biogeochemical signatures associated with anoxia/euxinia at the close of the Permian. Development of middepth sulfide maxima poised on the edge of expansion into the surface water layer could account for the presence of biomarkers indicative of photic-zone euxinia in shallow-marine sections prior to the EH (4, 6) without requiring anoxia of the deep ocean, which would alter the U isotope budget of seawater. Evidence from the U system indicates widespread oceanic anoxia only became pronounced and persistent at, or just preceding the EH. Thus, this study supports the possibility of H2S as a killing mechanism, but calls for buildup of H2S in the oxygen-minimum zone rather than prolonged accumulation in the deep ocean. This interpretation should be tested by investigating U proxies across other Permian-Triassic sections and by examining the fidelity with which carbonate sediments record and preserve primary U proxy signatures.
Methods
Study samples were powdered and dissolved using dilute (∼1 M) hydrochloric acid, leaving any noncarbonate species present (e.g., organics, pyrite, siliciclastics, etc…) intact and nonparticipatory in subsequent procedures. The dissolved material was dried and reconstituted in 3 M HNO3. Approximately 10% of the material was used for trace element analyses, with data obtained using a Thermo X-series quadrupole ICP-MS at the W. M. Keck Laboratory for Environmental Biogeochemistry at Arizona State University (ASU). The remaining 90% of the dissolved carbonate material was passed through a column containing Eichrom® UTEVA resin, following the procedure outlined in Weyer, et al. (19) to separate uranium from the matrix. Uranium isotope measurements were performed on a ThermoFinnigan Neptune MC-ICP-MS instrument at Arizona State University (ASU, W.M. Keck Laboratory for Environmental Biogeochemistry), utilizing a 236U∶233U double-spike MC-ICP-MS procedure described in Weyer, et al. (19). The isotopic composition of the double spike used is 236U/233U = 1.00494, 238U/233U = 0.000958, 235U/233U = 0.000108. Samples were spiked to achieve 236U and 233U signals of ∼2.5 times the voltage on the least abundant measured isotope, 235U. This spiking technique maximizes the counting statistics on the spike masses, while minimizing the tailing contributions at mass 235. All measured isotopes of U were collected by a Faraday cup collector array, utilizing 1011 ohm resistors for all masses. Samples dissolved in 2% HNO3 were introduced with an Apex-Q sample introduction system. Optimum precision was obtained running samples at ∼100 ppb U. The U isotope standards SRM950a and CRM129a were measured bracketing samples as checks for run reproducibility and consistency. External reproducibility based on multiple runs of the SRM950a and CRM129a standards over the course of this study is shown in SI Text. The U isotopic compositions of the samples are reported as relative to the U isotope standard SRM950a.
Supplementary Material
Acknowledgments.
We thank the W.M. Keck Laboratory for Environmental Biogeochemistry and G. Gordon for technical assistance with U, Th, and δ238U analyses. We are grateful to L. Borg and R. Williams for assistance with the U double spike and to S. Romaniello for many helpful discussions. We also thank two anonymous reviewers for helpful comments that greatly improved the manuscript. This work was supported by the NASA Astrobiology Institute, the NASA Exobiology Program, and by National Science Foundation (NSF) grant OCE-0952394.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1106039108/-/DCSupplemental.
*The duration of the Griesbachian is constrained by refs. 33, 34. The ∼730-ky-long Griesbachian at Dawen is ∼160 m thick, therefore the first 10 m represents ∼40,000–50,000 y, assuming constant sedimentation rates. A similar result is achieved by using data from ref. 14, which estimates that the stratigraphic equivalent of Beds 25 through 28 at Meishan is the lower ∼30 m at Dawen. The duration of this interval was estimated at 150,000 y, therefore the lower 10 m at Dawen represent ∼50,000 y.
References
- 1.Stanley SM. An analysis of the history of marine animal diversity. Paleobiology. 2007;33:1–55. [Google Scholar]
- 2.Erwin DH, Bowring SA, Yugan J. In: Catastrophic Events and Mass Extinctions: Impacts and Beyond: Geological Society of America Special Paper. Koeberl C, MacLeod KG, editors. 2002. pp. 363–383. [Google Scholar]
- 3.Bond DPG, Wignall PB. Pyrite framboid study of marine Permian-Triassic boundary sections: a complex anoxic event and its relationship to contemporaneous mass extinction. Geol Soc Am Bull. 2010;122:1265–1279. [Google Scholar]
- 4.Grice K, et al. Photic zone euxinia during the Permian-Triassic superanoxic event. Science. 2005;307:706–709. doi: 10.1126/science.1104323. [DOI] [PubMed] [Google Scholar]
- 5.Wignall PB, Twitchett RJ. Ocean anoxia and the end-Permian mass extinction. Science. 1996;272:1155–1158. doi: 10.1126/science.272.5265.1155. [DOI] [PubMed] [Google Scholar]
- 6.Cao C, et al. Biogeochemical evidence for euxinic oceans and ecological disturbance presaging the end-Permian mass extinction event. Earth Planet SC Lett. 2009;281:188–201. [Google Scholar]
- 7.Algeo TJ, et al. Changes in productivity and redox conditions in the Panthalassic Ocean during the latest Permian. Geology. 2010;38:187–190. [Google Scholar]
- 8.Isozaki Y. Permian-Triassic boundary superanoxia and stratified superocean: records from lost deep sea. Science. 1997;276:235–238. doi: 10.1126/science.276.5310.235. [DOI] [PubMed] [Google Scholar]
- 9.Algeo TJ, Chen ZQ, Fraiser ML, Twitchett RJ. Terrestrial-marine teleconnections in the collapse and rebuilding of Early Triassic marine ecosystems. Palaeogeography Palaeoclimatology Palaeoecology. 2011;308:1–11. [Google Scholar]
- 10.Ehrenberg SN, Svånå TA, Swart PK. Uranium depletion across the Permian-Triassic boundary in Middle East carbonates: Signature of oceanic anoxia. AAPG Bull. 2008;92:691–707. [Google Scholar]
- 11.Kump LR, Pavlov A, Arthur MA. Massive release of hydrogen sulfide to the surface ocean and atmosphere during intervals of oceanic anoxia. Geology. 2005;33:397–400. [Google Scholar]
- 12.Meyer KM, Kump LR, Ridgwell A. The biogeochemical controls on photic-zone euxinia during the end-Permian mass extinction. Geology. 2008;36:747–750. [Google Scholar]
- 13.Riccardi A, Kump LR, Arthur MA, D'Hondt S. Carbon isotopic evidence for chemocline upward excursions during the end-Permian event. Palaeogeography, Palaeoclimatology, Palaeoecology. 2007;248:73–81. [Google Scholar]
- 14.Chen J, Beatty TW, Henderson CM, Rowe H. Conodont biostratigraphy across the Permian-Triassic boundary at the Dawen section, Great Bank of Guizhou, Guizhou Province, South China: implications for the Late Permian extinction and correlation with Meishan. J Asian Earth Sci. 2009;36:442–458. [Google Scholar]
- 15.Klinkhammer GP, Palmer MR. Uranium in the oceans: where it goes and why. Geochimica et Cosmochimica Acta. 1991;55:1799–1806. [Google Scholar]
- 16.Ku T.-L, Knauss K, Mathieu GG. Uranium in the open ocean: concentration and isotopic composition. Deep-sea res. 1977;24:1005–1017. [Google Scholar]
- 17.Delanghe D, Bard E, Hamelin B. New TIMS constraints on the uranium-238 and uranium-234 in seawaters from the main ocean basins and the Mediterranean Sea. Mar Chem. 2002;80:79–93. [Google Scholar]
- 18.Stirling CH, Anderson MB, Potter E.-K, Halliday A. Low-temperature isotopic fractionation of uranium. Earth Planet SC Lett. 2007;264:208–225. [Google Scholar]
- 19.Weyer S, et al. Natural fractionation of 238U/235U. Geochimica et Cosmochimica Acta. 2008;72:345–359. [Google Scholar]
- 20.Bopp CJ, Lundstrom CC, Johnson TM, Glessner JJG. Variations in 238U/235U in uranium ore deposits: isotopic signatures of the U reduction process? Geology. 2009;37:611–614. [Google Scholar]
- 21.Brennecka GA, Borg LE, Hutcheon ID, Sharp MA, Anbar AD. Natural variations in uranium isotope ratios of uranium ore concentrates: understanding the 238U/235U fractionation mechanism. Earth Planet SC Lett. 2010;291:228–233. [Google Scholar]
- 22.Bopp CJ, et al. Uranium 238U/235U isotope ratios as indicators of reduction: results from an in situ biostimulation experiment at Rifle, Colorado, USA. Environmental Science and Technology. 2010;44:5927–5933. doi: 10.1021/es100643v. [DOI] [PubMed] [Google Scholar]
- 23.Montoya-Pino C, et al. Global enhancement of ocean anoxia during Oceanic Anoxic Event 2: a quantitative approach using U isotopes. Geology. 2010;38:315–318. [Google Scholar]
- 24.Dunk RM, Mills RA, Jenkins WJ. A reevaluation of the oceanic uranium budget for the Holocene. Chem Geol. 2002;190:45–67. [Google Scholar]
- 25.Tyson RV, Pearson TH. Modern and ancient continental shelf anoxia: an overview. In: Tyson RV, Pearson TH, editors. Modern and Ancient Continental Shelf Anoxia. vol. 58. London: Geololgical Society Special Publications; 1991. pp. 1–26. [Google Scholar]
- 26.Anderson RF, Fleisher MQ, Lehuray AP. Concentration, oxidation-state, and particulate flux of uranium in the Black-Sea. Geochimica et Cosmochimica Acta. 1989;53:2215–2224. [Google Scholar]
- 27.McManus J, et al. Molybdenum and uranium geochemistry in continental margin sediments: paleoproxy potential. Geochimica et Cosmochimica Acta. 2006;70:4643–4662. [Google Scholar]
- 28.Morford JL, Emerson S. The geochemistry of redox sensitive trace metals in sediments. Geochimica et Cosmochimica Acta. 1999;63:1735–1750. [Google Scholar]
- 29.Gvirtzman G, Friedman GM, Miller DS. Control and distribution of uranium in coral reefs during diagenesis. J Sediment Res. 1973;43:985–997. [Google Scholar]
- 30.Shen GT, Dunbar RB. Environmental controls on uranium in reef corals. Geochimica et Cosmochimica Acta. 1995;59:2009–2024. [Google Scholar]
- 31.Wignall PB, et al. An 80 million year oceanic redox history from Permian to Jurassic pelagic sediments of the Mino-Tamba terrane, SW Japan, and the origin of four mass extinctions. Global and Planet Change. 2010;71:109–123. [Google Scholar]
- 32.Algeo TJ, et al. Spatial variation in sediment fluxes, redox conditions, and productivity in the Permian-Triassic Panthalassic Ocean. Palaeogeography, Palaeoclimatology, Palaeoecology. 2011;308:65–83. [Google Scholar]
- 33.Mundil R, Palfry J, Renne PR, Brack P. In: The Triassic Timescale Geological Society. Lucas SG, editor. London: Special Publications; 2010. pp. 41–60. [Google Scholar]
- 34.Guo G, Tong J, Zhang S, Zhang J, Bai L. Cyclostratigraphy of the Induan (Early Triassic) in West Pingdingshan Section, Chaohu, Anhui Province. Sci China Ser D. 2008;51:22–29. [Google Scholar]
- 35.Winguth AME, Maier-Reimer E. Causes of the marine productivity and oxygen changes associated with the Permian-Triassic boundary: a reevaluation with ocean general circulation models. Mar Geol. 2005;217:283–304. [Google Scholar]
- 36.Brüchert V, Currie B, Peard KR. Hydrogen sulphide and methane emissions on the central Namibian shelf. Prog Oceanogr. 2009;83:169–179. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.