

Abstract
Four-and-a-half LIM domain protein 1 isoform A (FHL1A) is predominantly expressed in skeletal and cardiac muscle. Mutations in the FHL1 gene are causative for several types of hereditary myopathies including X-linked myopathy with postural muscle atrophy (XMPMA). We here studied myoblasts from XMPMA patients. We found that functional FHL1A protein is completely absent in patient myoblasts. In parallel, expression of FHL1C is either unaffected or increased. Furthermore, a decreased proliferation rate of XMPMA myoblasts compared to controls was observed but an increased number of XMPMA myoblasts was found in the G0/G1 phase. Furthermore, low expression of Kv1.5, a voltage-gated potassium channel known to alter myoblast proliferation during the G1 phase and to control repolarization of action potential, was detected. In order to substantiate a possible relation between Kv1.5 and FHL1C, a pull-down assay was performed. A physical and direct interaction of both proteins was observed in vitro. In addition, confocal microscopy revealed substantial colocalization of FHL1C and Kv1.5 within atrial cells, supporting a possible interaction between both proteins in vivo. Two-electrode voltage clamp experiments demonstrated that coexpression of Kv1.5 with FHL1C in Xenopus laevis oocytes markedly reduced K+ currents when compared to oocytes expressing Kv1.5 only. We here present the first evidence on a biological relevance of FHL1C.
Introduction
LIM, an acronym of three homeodomain-containing transcription factors (Lin-11, Isl-1, and Mec-3), contains a 50–60 amino acid stretch and a highly conserved, double cysteine-rich Zink-finger domain. LIM domain-containing proteins play important roles in various cellular processes, such as cytoskeleton organization, signal transduction, gene expression and cell differentiation [1], [2]. Four and a half LIM protein 1 (FHL1) is a main representative of the LIM-only protein family that is characterized by a half LIM domain (located in the N-terminal portion) and four additional complete LIM domains stretching towards the C-terminal portion. FHL1 is suggested to play a role in sarcomere synthesis and assembly [3]. Overexpression of FHL1 in mouse skeletal muscle promotes myocardial fusion and hypertrophy, observations with potential implications for human myopathy [4]. In humans, FHL1 is predominantly expressed in skeletal muscle and heart, but also in other tissues e.g. brain, placenta, lung, liver and kidney, at a lower abundance.
In addition to the prevalently expressed FHL1A protein (280 amino acids) two minor splice variants, originally identified from murine studies, exist. FHL1B (323 amino acids, the homolog of murine KyoT3 and specifically expressed in brain [5]) comprises the N-terminal three and a half LIM domains and a unique C-terminus containing three nuclear localization signals, a nuclear export sequence, and a C-terminal binding site for RBP-Jk. FHL1C (194 amino acids, the homolog of murine KyoT2) contains the N-terminal two and a half LIM domains and the C-terminal binding site for RBP-Jk (Figure 1).
Figure 1. Wild-type FHL1 protein isoforms and mutated FHL1A proteins.

Top: FHL1A, FHL1B and FHL1C protein structures (resulting from alternative splicing of the wild-type FHL1 gene), including the LIM domain architecture and functional domains (RBP-Jk, recombination signal-binding protein 1 for J-kappa; NLS, nuclear localization signals; NES, nuclear export sequence) are shown for the corresponding variants. Bottom: Protein structures resulting from alternative splicing of the two different mutant FHL1 genes. An arrow in the fourth LIM domain of mutated FHL1A at position 224 (termed FHL1Ap.C224W) indicates the position of the amino acid exchange. The dotted line suggests alterations in the LIM-like architecture (termed MUT1 here). FHL1Ap.G168fs is identical to FHL1C protein (termed MUT2 here) (G = glycine, fs = frame shift).
Several mutations in the human FHL1 gene are associated with severe or less severe types of hereditary muscular dystrophies and myopathies [2], [6]–[11]. The majority of these FHL1 mutations are located in the second LIM domain of FHL1A. A specific mutation in the fourth LIM domain in the FHL1A gene as occurring is associated with X-linked myopathy with postural muscle atrophy (XMPMA, MIM ID #300696). This disease is characterized by atrophy of postural muscles with rigid spine syndrome, pseudo athleticism or hypertrophy and hypertrophic cardiomyopathy [6].
The present study aimed at investigating myoblast proliferation from two XMPMA patients with different mutations in FHL1A. The classical missense mutation c.672C>G (on protein level p.C224W) leads to disruption of the fourth LIM domain in FHL1A and subsequent degradation of the protein due to the loss of one cysteine-rich Zink-finger domain; in parallel expression of endogenous FHL1C remains unaffected [6]. The second, a splice site mutation c.688+1G>A (on protein level p.G168fs), results in the deletion of two C-terminal LIM domains in FHL1A; the resulting truncated FHL1-like protein is identical to FHL1C (Figure 1); as a consequence, concentrations of FHL1C (the sum of endogenous and truncated FHL1-like protein) should increase [9]. FHL1C is specifically expressed in testis, skeletal muscle, and heart at a relatively low level compared to FHL1A [12]; however, to date FHL1C is still considered a protein of unknown function.
Recent evidence suggested that the voltage-gated potassium channel Kv1.5 could act as a binding partner for LIM-domain containing proteins [13]. In myoblasts, cell cycle-dependent expression of Kv1.5 has been reported [14]. Kv1.5 plays a role in myocardial repolarisation and may regulate skeletal muscle proliferation [14], [15]. Furthermore, Kv1.5 forms the major basis of an atrial-specific ultra-rapid delayed rectifier potassium ion current I Kur [16] and most importantly, a missense mutation in Kv1.5 can result in atrial fibrillation [17]. Therefore, the second aim of this study was to clarify whether Kv1.5 is expressed in XMPMA myoblasts and whether this K+ channel could act as a specific binding partner for FHL1C. We further investigated whether FHL1C and Kv1.5 colocalize in cellular compartments. Using a pull-down assay we then tried to reveal a direct interaction between both proteins. Finally, electrophysiological studies were performed, aiming to test whether FHL1C functionally interacts with Kv1.5.
Materials and Methods
Cell culture
(i) Human myoblast cultures were established from muscle biopsies of two male unaffected controls (termed WT1 and WT2) and two male XMPMA patients with either the missense mutation p.C224W (termed MUT1) or the splice mutation p.G168fs (termed MUT2) (Figure 1). Clinical characteristics and histological examinations in addition to molecular genetics of MUT1 and MUT2 patients are described in detail in reference [9]. The ethic board of the Ludwig-Maximilians University, Munich, Germany, gave ethical approval. All patients gave their written informed consent to scientific analyses of their blood samples and myoblasts.
Tissue sources for myoblast isolation were tibialial anterior muscle biopsy (WT1 and MUT1) and a biceps brachii muscle biopsy (WT2 and MUT2). Myoblasts were grown in skeletal muscle cell growth medium (PromoCell GmbH, Heidelberg, Germany) including supplement mix containing 10% (v/v) FCS (Invitrogen, Lofer, Austria), 1.5% (v/v) 100× Glutamax, 50 µl/ml gentamycin. Myoblasts isolated from controls and XMPMA patients consisted of a homogenous cell population and were confirmed to be of satellite origin (using a specific antibody raised human neuronal cell adhesion molecule, clone 123C3, Monosan, Uden, The Netherlands).
(ii) HL-1, established from the AT-1 mouse atrial cardiomyocyte cell line, was used for cotransfection experiments [18]. Cells were grown in Claycomb medium (Fluka, Sigma Aldrich, Munich, Germany) including 10% (v/v) FCS at 37°C (5% CO2, 95% air, relative humidity of 95%).
Myoblast proliferation assay
Myoblasts (2×105 cells, 40% confluence) were seeded in six-well plates at a density of 2.7×104/cm2 cultured up to 96 h in skeletal muscle cell growth medium (including a supplement mix containing 10% [v/v] FCS, 1.5% [v/v] 100× Glutamax, 50 µl/ml gentamycin). Cells were harvested after 24, 48, 72 and 96 h using 0.25% (v/v) trypsin. Cell proliferation was determined by measuring the number of viable cells at the indicated time periods using a cell counter and analyser system CASY®Model TT (Roche Innovatis AG, Bielefeld, Germany) as described [19].
Cell cycle analysis by flow cytometry
Myoblasts were seeded in six-well plates at a density of 2.7×104 cells/cm2 and cultured for 48 h, collected in PBS and centrifuged 5 min at 200 g. Cells were resuspended in 0.5 ml PBS and fixed in ice-cold 70% ethanol overnight. Thereafter, 0.25% (w/v) pepsin was added followed by CyStain DNA/protein staining solution. Samples were kept for 30 min at 4°C in the dark. Flow cytometric measurements were carried out using CyFlow® flow cytometer (Partec) according to the manufacturer's suggestions. For data analysis MultiCycleAV DNA cell cycle analysis software (Phoenix, San Diego, CA, USA) was used.
RNA isolation and reverse transcription PCR
Myoblasts (2×105 cells) were seeded in six-well plates, cultured for 48 h and harvested by treatment with 0.25% (v/v) trypsin. TRI Reagent (Invitrogen) was used for total RNA isolation according to the manufacturer's suggestions.
Final RNA concentrations were measured by Nanodrop technique (Thermo Scientific). Reverse transcription was performed from two µg of total RNA with random hexamer primers and SuperScript III Reverse Transcriptase (RT) (Invitrogen) [20], [21]. PCR primers (Table S1) were used to amplify the corresponding PCR products for FHL1 and Kv1.5. To ensure equal RNA loading, RT-PCR for human hypoxanthine phosphoribosyltransferase was performed for each experiment. Two control reactions (RNA template without primers and a water template only) were included in the absence of cDNA.
Protein isolation and Western blotting
Myoblasts were cultured and trypsinized as described above. The cells were washed with HBSS buffer, mixed with lysis buffer (0.1 M Tris pH 7.4, 10% [v/v] SDS, 0.1 M Na3VO4, 0.5 M NaF, 0.1 M sodium pyrophosphate buffer) and heated at 95°C for 10 min. Total cell lysate was frozen and stored at −20°C until use. Protein concentrations were measured with the Bradford Protein Assay Kit (Bio-Rad, Vienna, Austria). Aliquots of proteins were supplemented with NuPage® LDS sample buffer and NuPage® sample reducing agent (5%, v/v). After boiling for 5 min at 95°C, protein samples were subjected to electrophoresis at 4–12% NuPage® gradient SDS-PAGE gels [22] and transferred (90 min, 200 mA) to nitrocellulose membranes (0.45 µm, Schleicher and Schuell, Dassel, Germany, [23]). The membranes were blocked (30 min) with TBST (10 mM Tris-HCl [pH 7.4], 140 mM NaCl, 0.1% [v/v] Tween-20) containing 3% (w/v) non-fat dry milk followed by incubation overnight at 4°C with polyclonal rabbit anti-human FHL1 antibody (1∶1000, cross-reacting with the fourth LIM domain [coded for by exon 4–6] of FHL1A, AVIVA Systems Biology, San Diego, CA, USA) or polyclonal goat anti-human Kv1.5 (1∶200, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) as primary antibodies (diluted in 3% (w/v) BSA). After washing, the membranes were incubated (2 h, 25°C) with swine anti-rabbit IgG (1∶750) or donkey anti-goat IgG (1∶2000) as secondary antibodies (diluted in TBST containing 3% [w/v] non-fat dry milk). Immunoreactive bands were visualised with Immobilon Western Chemiluminescent HRP substrate (Millipore, Billerica, MA, USA). For normalisation, membranes were stripped with Restore™ Western Blot Stripping buffer (Pierce) and reprobed with anti-β-human actin antibody (1∶1000, in 3% [w/v] BSA, Santa Cruz) and goat anti-mouse IgG (1∶2500, 1 h, 25°C, Rockland, Gilbertsville, PA, USA) as secondary antibodies.
HL-1 cells transfection and confocal microscopy
FHL1C-peYFP-N1 (plasmid encoding for human FHL1C with the enhanced yellow fluorescence protein fused to its C-terminus) and Kv1.5-peYFP-N1 clones were raised using PCR technique and suitable primers (Table S2; see below). The peCFP-C1+human Kv1.5 construct (plasmid encoding for human Kv1.5 with the enhanced cyan fluorescence protein fused to its N-terminus) was kindly provided by Dr. Antonio Felipe (Barcelona, Spain). Plasmids encoding for eCFP-labelled ß-subunit of the signal recognition particle receptor Srß (SrßeCFP) and the eCFP-labelled glycosylphosphatidylinositol anchor domain (GPIeCFP) were kindly provided by Dr. Koret Hirschberg (Tel Aviv, Israel) and prepared as described [24], [25]. SrßeCFP was used as an endoplasmic reticulum marker while GPIeCFP was used to label lipid raft domains within the plasma membrane.
Transfection of HL-1 cells with the plasmids mentioned above was performed with Lipofectamine according to the manufacturer's suggestions. Cells were screened for signal 24 to 48 h after transfection using confocal imaging using a Leica inverted microscope with a laser-scanning module attached (DMIRE2 and TCS SL2; Leica Microsystems, Heidelberg, Germany). Using the 63×water immersion objective (NA: 1.20), confocal sections (1024×1024 pixels) were obtained at 12-bit resolution. Filter settings were: eYFP: excitation (514 nm); excitation beam splitter: DD 458/514; emission (540–570 nm). eCFP: excitation (458 nm); excitation beam splitter: DD 458/514; emission (477–500 nm). Interference between eCFP and eYFP channels was negligible. Analysis of confocal images was done using the ImageJ software (ImageJ 1.42 h by Wayne Rasband, NIH, and Bethesda, MD) supported with deconvolution plug-in (Bob Dougherty; http://www.optinav.com/imagej.html).
Pull-Down assay
A DNA fragment encoding amino acid residues 518 to 613 (numbering according to human Kv1.5, NP_002225.2) was synthesized by the PCR using a cDNA clone encoding full length Kv1.5 (kindly provided by Dr. Joachim Ehrlich, Frankfurt am Main, Germany) as a template and cloned into the vector pGEX-4T-1 using suitable primers (Table S2). The recombinant protein was heterologously expressed in E.coli and purified as described [26]. Four hundred fifty ng cRNA encoding FHL1C (or Gβ1 and Gγ2, respectively) were translated and radiolabelled in vitro using 9.4 µl rabbit reticulocyte lysate (Promega, Mannheim, Germany) and 0.95 µl 35S-methionine (Amersham GE Healthcare) by incubation at 37°C for 2 h. Purified GST-Kv1.5 fusion protein (10–20 µg) was used as bait and incubated with 10 µl of lysate containing 35S-labeled FHL1C in 300 µl buffer (25 mM HEPES-Na, 5 mM MgCl2, 5 mM EGTA, 0.05% Tween-20 [v/v, pH 7.0]), for 2 h at 25°C, with gentle rocking. GST protein alone was used as a negative control (i.e. no binding to FHL1C was expected). G-protein activated inwardly rectifying potassium channel (GIRK1) C-terminus (G1-CT) was used both as a positive control (in combination with 35S-labeled G-protein β subunits (Gβγ)) as well as a negative control (in combination with 35S-labeled FHL1C lysate). Control proteins were prepared as described [26]. Then, 30 µl of previously washed Glutathione Sepharose beads were added, the mixture was incubated (30 min, 25°C) and then washed three times in 1 ml of the same buffer. After washing, GST-fusion proteins were eluted with 30 µl of 15 mmol/l reduced glutathione in elution buffer (120 mM NaCl, 100 mM Tris, 0.05% Tween-20 [v/v, pH 8]) and subjected to 12% linear SDS-PAGE. Radioactivity was quantified by autoradiography of the dried gels, using a PhosphorImager Storm™ (GE Healthcare, Vienna, Austria). The amount of radioactivity was normalized to the amount of protein of a given band, as measured by staining of bands with Coomassie Brilliant Blue ( = relative specific radioactivity).
DNA constructs
For protein expression in Xenopus oocytes, FHL1C and Kv1.5 inserts were synthesized by PCR using suitable primers (Table S2) and cloned into the pBSmxt oocyte expression vector. A full-length human cDNA library (human fetal brain marathon cDNA) and Kv1.5-pcDNA3.1 (kindly provided by Joachim Ehrlich, Frankfurt, Germany) were used as PCR templates. The resulting pBSmxt constructs were linearized, cRNA was synthesized as described [27], aliquots were shock frozen in liquid nitrogen and stored at −70°C until use. Integrity of all DNA constructs was verified by DNA-sequencing (3130xl DNA Analyzer, Applied Biosystems, Vienna, Austria). DNA was prepared by Qiagen midiprep kit and concentration and purity was checked with Nanodrop (Thermo Scientific, Waltham, MA, USA).
Xenopus oocytes expression and electrophysiology
Xenopus oocytes, prepared as described [28], were injected with 50 nl of the following RNAs (either alone or in combination): Kv1.5 (2 ng RNA/nl), FHL1C (50 or 500 ng RNA/nl). Oocytes were incubated in ND96 solution (96 mM NaCl, 2 mM KCl, 1 mM CaCl2, 1 mM MgCl2, 5 mM HEPES/NaOH [pH 7.6]) supplemented with gentamycin (50 µg/ml) and sodium pyruvate (2.5 mM) at 19°C. Three to seven days after injection of oocytes, whole cell currents were recorded using agarose cushion electrodes [29]. Kv1.5 channels were characterized by the following voltage jump protocols: voltage-dependent activation was assessed by keeping the oocytes at a resting potential of −80 mV and K+ currents were elicited by consecutive voltage jumps repeated every 7 sec (from −35 mV to +45 mV in 5 mV increments). Inactivation of Kv1.5 was quantified by the ratio of the magnitude of K+ current 1.8 sec after a suprathreshold pulse to +15 mV divided by the peak current at this potential. Current traces were sampled at 300 msec sampling rate and low pass filtered at 500 kHz using a 4-pole Bessel filter. Parameters were normalized to the ones obtained from control oocytes (injected with cRNA encoding Kv1.5 alone) for a given experimental day and batch of oocytes. Data acquisition and analysis was performed using the digidata 1322A interface and the pClamp 9.2 software (both Molecular Devices, Sunnyvale, CA, USA).
Statistics
Each experiment was performed at least three times. Data of representative experiments are expressed as mean ± SD or ± SEM. Experimental parameters were analyzed between groups using Student's t-test (Sigma plot for Windows, v11, Systat Software). Means were checked for statistically significant differences (* p<0.05, ** p<0.01, and *** p<0.001). Sequence analysis and alignments were performed using Chromas Lite (v2.01, Technelysium Pty Ltd).
Results
Myoblast proliferation and cell cycle distribution
A significant reduction in the number of viable myoblasts isolated from both XMPMA patients when compared to controls was observed in between 24 h and 72 h (Figure 2A). Although the differences between WT and mutant myoblasts were statistically significant up to 72 h, the differences between WT2 and MUT2 were less pronounced when compared to WT1 and MUT1. To detect possible cell cycle alterations, FACS analyses were performed. A significantly higher number of patient myoblasts was found in the G0/G1 phase when compared to controls (Figure 2B). Statistical evaluation revealed that the increase of patient myoblasts in the G0/G1 phase was 28% in MUT1 (p<0.001) but only 8% in MUT2 (p<0.001).
Figure 2. Proliferation rate of human myoblasts.

Myoblasts from controls (WT1, WT2) and patients (MUT1, MUT2) were cultured for the indicated times (A) or for 48 h (B). Myoblast samples WT1 and MUT1 originated from tibialial anterior muscle biopsy, samples WT2 and MUT2 from biceps brachii muscle biopsy. (A) Cell proliferation was determined by measuring the number of viable cells using Casy cell counter. Values represent mean ± SD of three experiments (six wells per one experiment). **p<0.01 and *** p<0.001. (B) Flow cytometric measurements (see Methods) were performed to estimate cell cycle, i.e. G0/G1 phase, S phase, and M phase, respectively. One representative experiment (performed in triplicate) out of three is shown.
Expression of Kv1.5 and FHL1 in myoblasts of XMPMA patients and controls
Expression of Kv1.5 on mRNA (Figure 3A) and protein level (Figure 3B) was almost absent (MUT1) or decreased (MUT2) in patient myoblasts, when compared to controls. Expression of total FHL1 mRNA transcripts (coding for all known FHL1 isoforms (FHL1A–C) is comparable between control and XMPMA myoblasts (Figure 3A). While expression of FHL1A mRNA is similar in control myoblasts (WT1 and WT2), it is remarkably decreased (MUT1) or almost absent (MUT2) in XMPMA cells (Figure 3A). In parallel, expression of mRNA encoding FHL1C was found to be low (MUT1) or high (MUT2). This observation is in line with the assumption that the production of mRNA encoding for FHL1C is unaffected in MUT1 but apparently increased in MUT2; this splice site mutation generates additional mRNA encoding a truncated FHL1-like protein that is identical to FHL1C.
Figure 3. RT-PCR and Western blot for FHL1 and Kv1.5 in human myoblasts.

(A) RNA was isolated from myoblasts from controls (WT1, WT2) and XMPMA patients (MUT1, MUT2) and the corresponding Kv1.5 and FHL1 regions were amplified by RT-PCR. Primers, spanning from exon 1 to 2 (termed FHL1) amplify the mRNA stretch coding for the common N-terminus of FHL1 in all three FHL1 isoforms (FHL1A, FHL1B and FHL1C), while primers termed FHL1A or FHL1C specifically amplify mRNAs encoding FHL1A or FHL1C, respectively. (P = positive control: human fetal brain marathon cDNA). To ensure equal gel loading, RT-PCR for human HPRT1 was performed. One representative experiment out of three is shown. See Table S1 for the primer sequences used. (B) Protein lysates from myoblasts from controls (WT1, WT2) and XMPMA patients (MUT1, MUT2) were subjected to SDS-PAGE. Proteins were transferred to nitrocellulose membranes and immunoreactive bands were detected with anti-Kv1.5 or anti-FHL1A as primary antibodies. After stripping, the membranes were incubated with anti-β-actin antibody. (Kv1.5 = 68 kDa; FHL1A = 32 kDa; β-actin = 45 kDa). One representative experiment out of three is shown.
Subsequently, FHL1 expression on the protein level was investigated. Due to the lack of commercially available antibodies against FHL1C only expression of FHL1A could be followed. Western blot experiments demonstrated the presence of FHL1A in myoblasts from controls while only trace amounts were present in XMPMA patient myoblasts (Figure 3). Densitometric evaluation of immunoreactive Kv1.5 and FHL1A bands is shown in Figure S1.
Subcellular localisation of FHL1C and Kv1.5
Since FHL1A (a protein containing four and the half LIM domains) has recently been shown to interact with Kv1.5 [13], we were interested whether FHL1C (lacking the two C-terminal LIM domains being present in FHL1A) could behave in a similar manner. First, we checked whether FHL1C colocalized with Kv1.5 in vivo. Fluorescently labelled Kv1.5 and FHL1C chimeras were generated by PCR technique and the corresponding constructs were expressed in atrial cells. Figure 3A shows pronounced expression and predominant nuclear localization of FHL1CeYFP and, to a lesser extent, in the cytoplasm; these results partly parallel previous findings when FHL1C (tagged with the green fluorescence protein) was expressed in a hepatoma cell line [12] or embryonal fibroblasts [30]. On the other hand Kv1.5 eCFP was predominantly localized within the cytoplasm with low fluorescence intensity in the nucleus.
The precise subcellular distribution of Kv1.5 and FHL1C was studied by coexpression of both proteins with marker proteins that localize within specific cellular compartments. Coexpression of Kv1.5 eYFP with SrßeCFP revealed predominant localization of the channel in the endoplasmic reticulum (Figure 4B). Partial colocalization of Kv1.5 eYFP with GPIeCFP is further indicative for its additional presence in the plasma membrane including lipid rafts (Figure 4C). Distribution of FHL1CeYFP showed considerable overlap with SrßeCFP (Figure 4B) and partial overlap with GPIeCFP (Figure 4C), indicating that substantial amounts of FHL1C localize intimately close to both subcellular compartments. Intensity histograms (Figure 4A–C) show the number of pixels in the images at each different intensity value. During our experiments we have obtained individual histograms for each signal (CFP and YFP, presented in red or green) and the combination of both is shown (Figure 4A–C). Although FHL1C and Kv1.5 exerted quite different subcellular localization patterns as a whole, there was substantial overlap within both the plasma membrane and the endoplasmic reticulum, indicating that interaction of these proteins is likely within a living cell.
Figure 4. Subcellular localization of heterologous expressed chimaeric FHL1C and Kv1.5 constructs in the atrial HL-1 cell line.

Cells were cultured as described and after transfection, fluorescence of either eYFP- and eCFP-labelled proteins was detected as described in the Methods section. Each horizontal sequence of images was taken from the same cell and vertical section. For better visualization of colocalization, the eYFP and eCFP channels are shown in green and red, respectively, hence yellow in the overlay image indicates colocalization (merge). Intensity histograms are shown to quantify colocalization of both markers (right). Histogram shows the pixel-by-pixel analysis of the section indicated by the yellow line in the merged channel (from left to right). (A) Staining for FHL1CeYFP and Kv1.5 eCFP and colocalization. (B) Staining for Kv1.5 eYFP (upper) and FHL1CeYFP (lower) and colocalization with SrßeCFP (a marker for the endoplasmic reticulum). (C) Staining for Kv1.5 eYFP (upper) and FHL1CeYFP (lower) and colocalization with GPIeCFP (a marker for lipid raft domains of the plasma membrane). One representative series of images out of three independent series of experiments is shown.
Physical interaction between FHL1C and Kv1.5
Subsequently, it was of interest to investigate whether FHL1C physically interacts with Kv1.5. As a positive control for protein/protein interaction, the pull-down assay was performed using a GST fusion of the cytosolic C-terminus of GIRK1 (isoform 1 of the G-protein activated inwardly rectifying K+ channel; a well known direct G-protein effector) as bait and the G-protein ß1/γ2 dimer (Gß/γ) as binding partner [26]. As a negative control, i.e. no specific protein/protein interaction, pull-down between GST alone and Gß/γ or FHL1C was performed. Although minimal non-specific interaction of FHL1C with GST could be seen, when compared to Gß/γ (Figure 5, lane 2/3 vs. lane 1) considerable physical interaction of FHL1C with the C-terminal portion of Kv1.5 was clearly detected (Figure 5A/B, lane 4/5).
Figure 5. Physical interaction of FHL1C with the Kv1.5 cytosolic C-terminus.

(A) Autoradiography of the dried gel, showing the amount of radioactive protein associated with the bait. As radioactive protein either FHL1C or the ß/γ subunits of heterotrimeric G-protein (Gß/γ; control) was used as indicated at the bottom of the lanes. (B) Commassie Brilliant Blue staining of the SDS gel shown in panel A. Bait proteins used are indicated at the bottom of the lanes: GST protein alone (GST); GST-Kv1.5 C-terminus fusion protein (Kv1.5); C-terminus of G-protein activated inwardly rectifying potassium channel (GIRK1) fused with GST (G1-CT) as positive control. (C) Statistics of the bound radioactivity normalized to the amount of bait protein (relative specific radioactivity) for FHL1C using Kv1.5 and GST as a bait (left) and Gß/γ using G1-CT and GST as a bait (right). Values represent the mean values from different experiments (N is given in parenthesis above each bar). Significant difference (**p<0.01) between GST and Kv1.5 /G1-CT.
Functional effect of FHL1 coexpression on heterologously expressed Kv1.5 channels
In order to test whether FHL1C exerts functional effects on Kv1.5, both proteins were heterologously coexpressed in the Xenopus laevis oocyte system. Functional characterization of Kv1.5 was performed by assessing its voltage-dependent activation. Most remarkably, coexpression of FHL1C reduced Kv1.5 currents at all potentials tested, when compared to oocytes expressing Kv1.5 alone (Figure 6A and B). Such reduction in current magnitude could in principle be brought about by either (i) effects of FHL1C association on Kv1.5 kinetics (i.e. voltage-dependent activation or channel inactivation) or (ii) interference of FHL1C with channel biosynthesis and/or trafficking (resulting in reduced insertion of Kv1.5 complexes into the plasma membrane). Parameters of voltage-dependant activation and channel inactivation are shown in Table 1. Steady-state activation (Figure 6B and C) was analyzed by fitting the measured peak current (Ipeak) as a function of membrane potential (Em) to a Boltzmann isotherm resulting in corresponding values for reversal potential (Erev), potential for half-maximal activation (E0.5), slope of Boltzmann distribution (k) and the conductivity for K+ ions at maximal activation (Gmax). Analysis of voltage-dependent activation parameters revealed that neither the potential of half-maximal activation nor the gating charge, i.e. the steepness of the Boltzmann isotherm, was affected by FHL1C (Figure 6C). Also Kv1.5 inactivation was apparently unchanged upon coexpression with FHL1C (Figure 6D). In conclusion, all analyzed parameters of channel kinetics did not alter when Kv1.5 was coexpressed with either low or high cRNA levels of FHL1C (Table 1). Interestingly, the maximal conductance (Gmax) of the oocytes for K+ ions produced by Kv1.5 complexes was reduced considerably and statistically significant upon FHL1C coexpression. This reduction was observed during the entire time-span tested after cRNA injections (Figure 6E) and clearly correlated with the amount of coinjected cRNA encoding FHL1C (Figure 6F). Coexpression of oocytes with Kv1.5 and FHL1C reduced Gmax by approx. 33% (50 ng FHL1C cRNA/nl) or approx. 75% (500 ng FHL1C cRNA/nl) (Figure 6F).
Figure 6. Coexpession of FHL1C reduces currents through Kv1.5 channels in Xenopus laevis oocytes.

(A) Representative original current traces recorded from two oocytes expressing either Kv1.5 alone (upper panel) or Kv1.5 in combination with FHL1C (lower panel). Each family of current traces originated from four different suprathreshold voltage pulses (−35, −10, +10 and +40 mV). Recordings were taken five days after cRNA injection. (B) Ipeak/Em relation for the oocytes shown in (A). Original data (circles) and fit through the data according to a Boltzmann isotherm (solid lines) are shown. (C) Representative statistics of steady-state activation (n∞) five days after cRNA injection (with and w/o 500 ng cRNA encoding FHL1C coexpressed). Circles represent mean values ± SEM from six batches of oocytes (five oocytes per batch and experimental condition) and the dotted lines fits trough the data according to Boltzmann isotherms, respectively. (D) Kv1.5 inactivation kinetics, same data set as in C. Mean values of the current inactivation ratio (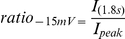 ) ± SEM are shown. (E) Time course of the effect of FHL1C coexpression on maximal K+ conductance. Data were normalized to Gmax of oocytes expressing Kv1.5 alone for five days (mean values ± SEM of six different batches of oocytes are shown); at least five oocytes were recorded from every batch and experimental group. (F) Dose dependent effect of coexpression of FHL1C on Gmax; five days after cRNA injection. The amount of cRNA coinjected, encoding FHL1C, is shown at the bottom. Mean values ± SEM from six different experiments (five oocytes per batch and experimental group) is shown (ns: the difference is statistically not significant, *: p<0.05 and ***: p<0.001, respectively).
) ± SEM are shown. (E) Time course of the effect of FHL1C coexpression on maximal K+ conductance. Data were normalized to Gmax of oocytes expressing Kv1.5 alone for five days (mean values ± SEM of six different batches of oocytes are shown); at least five oocytes were recorded from every batch and experimental group. (F) Dose dependent effect of coexpression of FHL1C on Gmax; five days after cRNA injection. The amount of cRNA coinjected, encoding FHL1C, is shown at the bottom. Mean values ± SEM from six different experiments (five oocytes per batch and experimental group) is shown (ns: the difference is statistically not significant, *: p<0.05 and ***: p<0.001, respectively).
Table 1. Effect of FHL1C coexpression on Kv1.5 kinetics: reversal potential (Erev), potential of half-maximal activation (E0.5), slope of Boltzmann isotherm (k) and channel inactivation (ratio −15 mV).
| Kv1.5 | Kv1.5+FHL1C(50 ng cRNA) | Kv1.5+FHL1C(500 ng cRNA) | |
| Erev (mV) | −33.58±5.71 | −36.40±6.25 | −36.23±6.20 |
| E0.5 (mV) | −1.37±1.94 | −1.11±1.53 | −1.43±2.34 |
| k (mV−1) | 6.14±0.12 | 6.51±0.16 | 5.95±0.17 |
| ratio −15 mV | 0.76±0.05 | 0.74±0.05 | 0.71±0.04 |
Values are mean ± SEM of six independent experiments (≥5 oocytes were analysed per experimental day).
Summarizing all data from Figure 6, we conclude that FHL1C coexpression of Kv1.5 with FHL1C resulted in a reduced membrane insertion of Kv1.5 complexes into the plasma membrane, without influencing kinetics.
Discussion
Mutations in FHL1 (previously called skeletal muscle LIM protein 1) cause distinct types of muscular dystrophies e.g. XMPMA [2], which may be associated with cell cycle alterations of myoblasts. We here show a reduced proliferation rate of myoblasts from XMPMA patients; furthermore, an increased number of myoblasts in the G0/G1 phase is paralleled by low expression of Kv1.5.
Villalonga and coworkers [14] previously reported that Kv1.5 is involved in skeletal muscle proliferation. In their studies [14] Kv1.5 was operative by controlling G1 phase progression through a mechanism that involves the CDKIs p21cip-1 and p27kip1 as well as cyclins A and D1. Our study provides additional evidence for a possible involvement of Kv1.5 in myotube growth; however, the aim of our study was to link myoblast proliferation, expression of Kv1.5 and a possible interaction with FHL1C, a LIM-domain-containing protein that is abundantly expressed in XMPMA. Although a panel of different interacting proteins as well as putative binding partners have been identified for FHL1 [31], [32], no data are available for FHL1C specifically. We here show for the first time both, a physical and a functional interaction of Kv1.5 with FHL1C.
A recent paper [33] reported that FHL1 knockdown significantly inhibits proliferation of rat aortic smooth muscle cells. Traffic and subcellular localization regulate ion channel activities [34] and these conditions may affect both skeletal and cardiac muscle tissues [6]–[9]. We here found that FHL1C is localized in the nucleus and the cytoplasm, in the latter colocalizing with Kv1.5 that can be found mostly in the endoplasmic reticulum and the plasma membrane. Our studies were performed in HL-1 cells, a cellular model that retains important atrial features like contractility and excitability [18]. Our data is the first report on Kv1.5 expression in myoblasts from patients with a myopathy associated with mutations in FHL1. The presence of intact FHL1C as occurring in XMPMA patients might contribute to the relative mild phenotype when compared to other FHL1 mutation-associated myopathies [2]. We therefore could speculate that FHL1C might compensate either absent or reduced levels of other FHL1 isoforms.
Our in vitro data (pull-down assay) confirm a physical interaction of Kv1.5 with FHL1C. From our observations, we may assume that the two and a half N-terminal LIM domains in FHL1 are responsible for effective interaction with Kv1.5. Previous genetic analyses of XMPMA patients [6], [9] suggested that FHL1C could play a specific role in cardiomyophathy. In the present study we show that in myoblasts from XMPMA patients (with specific mutations in FHL1) FHL1C is expressed at mRNA levels comparable to controls. However, due to degradation of FHL1A (MUT1) or expression of a truncated FHL1C-like protein (MUT2), FHL1C is likely to increase on a percentage level when compared to total FHL1 expression. Further studies performed in the Xenopus oocyte system (Figure 5) demonstrate reduced conductivity of Kv1.5 (Gmax) indicative for a reduced density of the K+ channel within the plasma membrane. This in turn suggests that FHL1C could act as a scaffold protein that modulates Kv1.5 trafficking to the plasma membrane. We therefore may speculate that increased levels of FHL1C could be the cause for a reduced current in the human atrium leading to enhanced atrial fibrillation in further consequence. Kv1.5 has been reported to interact with FHL1A [13]. The lack of this FHL1 isoform in XMPMA patients could affect stability of Kv1.5. Unfortunately, to date, no studies are available on atrial fibrillation and subsequent heart failure in XMPMA patients.
Yang and coworkers [13] have recently reported that coexpression of Kv1.5 with FHL1A in Chinese hamster ovary cells resulted in increased currents compared to expression of Kv1.5 alone, findings that are in apparent contrast to our observations coexpressing Kv1.5 and FHL1C in oocytes. This difference could be due to the fact that we were expressing FHL1C (a protein that lacks two LIM domains in the C-terminal portion) in oocytes where a different processing/trafficking mechanism of FHL1 splice variants may occur. Such alternative processing/trafficking has been reported already for FHL1C but translocation to the nucleus may be mediated by particular protein modifications and/or protein interactions [12], [31]. Another explanation might be the lack of a functional domain, RBP-Jk, in the C-terminal portion of FHL1A (Figure 1). The RBP-Jk moiety, when complexed with the signal-transducing Notch intracellular domain, may act as a transcriptional activator [35], a process recently addressed to occur in myocyte differentiation [36]. Most importantly, KyoT2, the murine homolog of FHL1C, and Notch1 competes with each other for binding to RBP-Jk [30]. Taken these structural elements into account, different effects of the three main FHL1 isoforms on channel activities may not be surprising at all.
In conclusion, we have described the interaction of FHL1C, a specific FHL1 isoform, with Kv1.5. Since Kv1.5 expression and functionality are altered in XMPMA patients we assume that FHL1 might regulate channel in both cardiac and skeletal muscle. Absence of FHL1 could result in lack of regulation of the channel and explain at least a part of the (patho)physiology found in XMPMA patients.
Supporting Information
Densitometric evaluation of FHL1A and Kv1.5 expression in human myoblasts. Immunoreactive bands (shown in Figure 3B) were scanned and their intensity was determined using ImageJ 1.40 g software from Wayne Rasband of the National Institutes of Health (Bethesda, MD, USA). The relative intensity of each band was calculated by dividing the absolute intensity of the respective band by the absolute intensity of the respective loading control band (β-actin). (A) Kv1.5 expression in control (WT1, WT2) and XMPMA patient (MUT1, MUT2) myoblasts. Values for control samples are set to 1 and for XMPMA patients normalized to corresponding controls. Experiments were performed in triplicates (WT1 and MUT1) and quadruplicates (WT2 and MUT2). (B) FHL1A expression in myoblasts using the same samples as listed in (A). Experiments were performed in triplicates.
(TIF)
Description of primers for the RT-PCR assay, expected amplified fragment size, and annealing temperature.
(DOC)
Description of primers for DNA constructs, the expected amplified fragment size, and the annealing temperature.
(DOC)
Footnotes
Competing Interests: The authors have declared that no competing interests exist.
Funding: This work received financial supported by the Country of Styria (GZ: A3-16.R-10/2009-112). The PhD program Molecular Medicine of the Medical University of Graz, Austria funded IP. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Kadrmas JL, Beckerle MC. The LIM domain: from the cytoskeleton to the nucleus. Nat Rev Mol Cell Biol. 2004;5:920–931. doi: 10.1038/nrm1499. [DOI] [PubMed] [Google Scholar]
- 2.Cowling BS, Cottle DL, Wilding BR, D'Arcy CE, Mitchell CA, et al. Four and a half LIM protein 1 gene mutations cause four distinct human myopathies: A comprehensive review of the clinical, histological and pathological features. Neuromusc Disord. 2011;21:237–251. doi: 10.1016/j.nmd.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 3.McGrath MJ, Cottle DL, Nguyen MA, Dyson JM, Coghill ID, et al. Four and a half LIM protein 1 binds myosin-binding protein C and regulates myosin filament formation and sarcomere assembly. J Biol Chem. 2006;281:7666–7683. doi: 10.1074/jbc.M512552200. [DOI] [PubMed] [Google Scholar]
- 4.Cowling BS, McGrath MJ, Nguyen MA, Cottle DL, Kee AJ, et al. Identification of FHL1 as a regulator of skeletal muscle mass: implications for human myopathy. J Cell Biol. 2008;183:1033–1048. doi: 10.1083/jcb.200804077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee SM, Li HY, Ng EK, Or SM, Chan KK, et al. Characterization of a brain-specific nuclear LIM domain protein (FHL1B) which is an alternatively spliced variant of FHL1. Gene. 1999;237:253–263. doi: 10.1016/s0378-1119(99)00251-6. [DOI] [PubMed] [Google Scholar]
- 6.Windpassinger C, Schoser B, Straub V, Hochmeister S, Noor A, et al. An X-linked myopathy with postural muscle atrophy and generalized hypertrophy, termed XMPMA, is caused by mutations in FHL1. Am J Hum Genet. 2008;82:88–99. doi: 10.1016/j.ajhg.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gueneau L, Bertrand AT, Jais JP, Salih MA, Stojkovic T, et al. Mutations of the FHL1 gene cause Emery-Dreifuss muscular dystrophy. Am J Hum Genet. 2009;85:338–353. doi: 10.1016/j.ajhg.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schessl J, Taratuto AL, Sewry C, Battini R, Chin SS, et al. Clinical, histological and genetic characterization of reducing body myopathy caused by mutations in FHL1. Brain. 2009;132:452–464. doi: 10.1093/brain/awn325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schoser B, Goebel HH, Janisch I, Quasthoff S, Rother J, et al. Consequences of mutations within the C terminus of the FHL1 gene. Neurology. 2009;73:543–551. doi: 10.1212/WNL.0b013e3181b2a4b3. [DOI] [PubMed] [Google Scholar]
- 10.Chen DH, Raskind WH, Parson WW, Sonnen JA, Vu T, et al. A novel mutation in FHL1 in a family with X-linked scapuloperoneal myopathy: phenotypic spectrum and structural study of FHL1 mutations. J Neurol Sci. 2010;296:22–29. doi: 10.1016/j.jns.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Knoblauch H, Geier C, Adams S, Budde B, Rudolph A, et al. Contractures and hypertrophic cardiomyopathy in a novel FHL1 mutation. Ann Neurol. 2010;67:136–140. doi: 10.1002/ana.21839. [DOI] [PubMed] [Google Scholar]
- 12.Ng EK, Lee SM, Li HY, Ngai SM, Tsui SK, et al. Characterization of tissue-specific LIM domain protein (FHL1C) which is an alternatively spliced isoform of a human LIM-only protein (FHL1). J Cell Biochem. 2001;82:1–10. doi: 10.1002/jcb.1110. [DOI] [PubMed] [Google Scholar]
- 13.Yang Z, Browning CF, Hallaq H, Yermalitskaya L, Esker J, et al. Four and a half LIM protein 1: a partner for KCNA5 in human atrium. Cardiovasc Res. 2008;3:449–457. doi: 10.1093/cvr/cvn038. [DOI] [PubMed] [Google Scholar]
- 14.Villalonga N, Martínez-Mármol R, Roura-Ferrer M, David M, Valenzuela C, et al. Cell cycle-dependent expression of Kv1.5 is involved in myoblast proliferation. Biochem Biophys Acta. 2008;1783:728–736. doi: 10.1016/j.bbamcr.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 15.Wang J, Juhaszova M, Conte JV, Jr, Gaine SP, Rubin LJ, et al. Action of fenfluramine on voltage-gated K+ channels in human pulmonary artery smooth-muscle cells. Lancet. 1998;352:290. doi: 10.1016/s0140-6736(05)60264-4. [DOI] [PubMed] [Google Scholar]
- 16.Wang Z, Fermini B, Nattel S. Sustained depolarization-induced outward current in human atrial myocytes: Evidence for a novel delayed rectifier K+ current similar to Kv1.5 cloned channel currents. Circ Res. 1993;73:1061–1076. doi: 10.1161/01.res.73.6.1061. [DOI] [PubMed] [Google Scholar]
- 17.Olson TM, Alekseev AE, Liu XK, Park S, Zingman LV, et al. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum Mol Genet. 2006;15:2185–2191. doi: 10.1093/hmg/ddl143. [DOI] [PubMed] [Google Scholar]
- 18.Claycomb WC, Lanson NA, Jr, Stallworth BS, Egeland DB, Delcarpio JB, et al. HL-1 cells: A cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci USA. 1998;95:2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weiss U, Cervar M, Puerstner P, Schmut O, Haas J, et al. Hyperglycaemia in vitro alters the proliferation and mitochondrial activity of the choriocarcinoma cell lines BeWo, JAR and JEG-3 as models for human first-trimester trophoblast. Diabetologia. 2001;44:209–219. doi: 10.1007/s001250051601. [DOI] [PubMed] [Google Scholar]
- 20.Kovacevic A, Hammer A, Sundl M, Pfister B, Hrzenjak A, et al. Expression of serum amyloid A transcripts in human trophoblast and fetal-derived trophoblast-like choriocarcinoma cells. FEBS Lett. 2006;580:161–167. doi: 10.1016/j.febslet.2005.11.067. [DOI] [PubMed] [Google Scholar]
- 21.Kitz K, Windischhofer W, Leis HJ, Huber E, Kollroser M, et al. 15-Deoxy-Δ12,14-prostaglandin J2 induces Cox-2 expression in human osteosarcoma cells through MAPK and EGFR activation involving reactive oxygen species. Free Radic Biol Med. 2011;50:854–865. doi: 10.1016/j.freeradbiomed.2010.12.039. [DOI] [PubMed] [Google Scholar]
- 22.Rauh A, Windischhofer W, Kovacevic A, DeVaney T, Huber E, et al. Endothelin (ET)-1 and ET-3 promote expression of c-fos and c-jun in human choriocarcinoma via ET(B) receptor-mediated G(i)- and G(q)-pathways and MAP kinase activation. Br J Pharmacol. 2008;154:13–24. doi: 10.1038/bjp.2008.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goti D, Hammer A, Galla HJ, Malle E, Sattler W. Uptake of lipoprotein-associated alpha-tocopherol by primary porcine brain capillary endothelial cells. J Neurochem. 2000;74:1374–1383. doi: 10.1046/j.1471-4159.2000.0741374.x. [DOI] [PubMed] [Google Scholar]
- 24.Glebov OO, Nichols BJ. Lipid raft proteins have a random distribution during localized activation of the T-cell receptor. Nat Cell Biol. 2004;6:238–243. doi: 10.1038/ncb1103. [DOI] [PubMed] [Google Scholar]
- 25.Ward TH, Brandizzi F. Dynamics of proteins in Golgi membranes: Comparisons between mammalian and plant cells highlighted by photobleaching techniques. Cell Mol Life Sci. 2004;61:172–185. doi: 10.1007/s00018-003-3355-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ivanina T, Rishal I, Varon D, Mullner C, Frohnwieser-Steinecke B, et al. Mapping the Gbetagamma-binding sites in GIRK1 and GIRK2 subunits of the G protein-activated K+ channel. J Biol Chem. 2003;278:29174–29183. doi: 10.1074/jbc.M304518200. [DOI] [PubMed] [Google Scholar]
- 27.Wagner V, Stadelmeyer E, Riederer M, Regitnig P, Gorischek A, et al. Cloning and characterisation of GIRK1 variants resulting from alternative RNA editing of the KCNJ3 gene transcript in a human breast cancer cell line. J Cell Biochem. 2010;110:598–608. doi: 10.1002/jcb.22564. [DOI] [PubMed] [Google Scholar]
- 28.Hofer D, Lohberger B, Steinecker B, Schmidt K, Quasthoff S, et al. A comparative study of the action of tolperisone on seven different voltage dependent sodium channel isoforms. Eur J Pharmacol. 2006;538:5–14. doi: 10.1016/j.ejphar.2006.03.034. [DOI] [PubMed] [Google Scholar]
- 29.Schreibmayer W, Lester HA, Dascal N. Voltage clamping of Xenopus laevis oocytes utilizing agarose-cushion electrodes. Pflugers Arch. 1994;426:453–458. doi: 10.1007/BF00388310. [DOI] [PubMed] [Google Scholar]
- 30.Taniguchi A, Furukawa T, Tun T, Han H, Honjo T. LIM Protein KyoT2 negatively regulates transcription by association with the RBP-J DNA-binding protein. Mol Cell Biol. 1994;18:644–654. doi: 10.1128/mcb.18.1.644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shathasivam T, Kislinger T, Gramolini AO. Genes, proteins and complexes: The multifaceted nature of FHL family proteins in diverse tissues. J Cell Mol Med. 2010;14:2702–2720. doi: 10.1111/j.1582-4934.2010.01176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharma P, Shathasivam T, Ignatchenko V, Kislinger T, Gramolini AO. Identification of an FHL1 protein complex containing ACTN1, ACTN4, and PDLIM1 using affinity purifications and MS-based protein-protein interaction analysis. Mol Biosyst. 2011;7:1185–1196. doi: 10.1039/c0mb00235f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weng J, Liao M, Zou S, Bao J, Zhou J, et al. Downregulation of FHL1 Expression in Thoracic Aortic Dissection: Implications in Aortic Wall Remodeling and Pathogenesis of Thoracic Aortic Dissection. Ann Vasc Surg. 2011;25:240–247. doi: 10.1016/j.avsg.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 34.Vicente R, Villalonga N, Calvo M, Escalada A, Solsona C, et al. Kv1.5 association modifies Kv1.3 traffic and membrane localization. J Biol Chem. 2008;283:8756–8764. doi: 10.1074/jbc.M708223200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iso T, Hamamori Y, Kedes L. Notch signaling in vascular development. Arterioscler Thromb Vasc Biol. 2003;23:543–553. doi: 10.1161/01.ATV.0000060892.81529.8F. [DOI] [PubMed] [Google Scholar]
- 36.Boni A, Urbanek K, Nascimbene A, Hosoda T, Zheng H, et al. Notch1 regulates the fate of cardiac progenitor cells. Proc Natl Acad Sci USA. 2008;105:15529–15534. doi: 10.1073/pnas.0808357105. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Densitometric evaluation of FHL1A and Kv1.5 expression in human myoblasts. Immunoreactive bands (shown in Figure 3B) were scanned and their intensity was determined using ImageJ 1.40 g software from Wayne Rasband of the National Institutes of Health (Bethesda, MD, USA). The relative intensity of each band was calculated by dividing the absolute intensity of the respective band by the absolute intensity of the respective loading control band (β-actin). (A) Kv1.5 expression in control (WT1, WT2) and XMPMA patient (MUT1, MUT2) myoblasts. Values for control samples are set to 1 and for XMPMA patients normalized to corresponding controls. Experiments were performed in triplicates (WT1 and MUT1) and quadruplicates (WT2 and MUT2). (B) FHL1A expression in myoblasts using the same samples as listed in (A). Experiments were performed in triplicates.
(TIF)
Description of primers for the RT-PCR assay, expected amplified fragment size, and annealing temperature.
(DOC)
Description of primers for DNA constructs, the expected amplified fragment size, and the annealing temperature.
(DOC)