

Summary
The inhibition of phosphatases by adenosine 5′-phosphorothioate (AMPS) was first reported in the late 1960s; however, the structural basis for the inhibition has remained unknown. Here, it is shown that AMPS is a submicromolar inhibitor of class C acid phosphatases, a group of bacterial outer membrane enzymes belonging to the haloacid dehalogenase structural superfamily. Furthermore, the 1.35 Å resolution crystal structure of the inhibited Haemophilus influenzae class C enzyme (aka rP4) was determined, which is the first structure of a phosphatase complexed with AMPS. The conformation of AMPS is identical to that of the substrate 5′-AMP, except that steric factors force a rotation of the thiophosphoryl out of the normal phosphoryl binding pocket. This conformation is catalytically nonproductive, because the P atom is not positioned optimally for nucleophilic attack by Asp64, and the O atom of the scissile O-P bond is too far from the Asp residue (Asp66) that protonates the leaving group. The structure of 5′-AMP complexed with the Asp64Asn mutant enzyme was also determined at 1.35 Å resolution. This mutation induces the substrate to adopt the same nonproductive binding mode that is observed in the AMPS complex. In this case, electrostatic considerations, rather than steric factors, underlie the movement of the phosphoryl. The structures not only provide an explanation for the inhibition by AMPS, but also highlight the precise steric and electrostatic requirements of phosphoryl recognition by class C acid phosphatases. Moreover, the Asp64Asn structure illustrates how a seemingly innocuous mutation can cause an unexpected structural artifact.
Keywords: X-ray crystallography, class C acid phosphatase, enzyme inhibition, adenosine 5′-phosphorothioate, substrate recognition
Introduction
Class C acid phosphatases (CCAPs) constitute a large group of bacterial enzymes related by several shared characteristics. Conserved features of CCAPs include a 25-residue bipartite sequence motif featuring four essential Asp residues [1], subunit molecular weight of approximately 30 kDa, and subcellular localization to the bacterial outer membrane. Structural and biophysical studies have further shown that CCAPs exhibit the haloacid dehalogenase (HAD) fold and are dimeric in solution [2–4]. Kinetic studies of several CCAPs indicate a substrate preference for nucleoside monophosphates, which has led to the classification of CCAPs as broad specificity 5′-,3′-nucleotidases [3, 5–10].
Although the defining characteristics of the family are established, the biological functions of CCAPs are less certain. The Haemophilus influenzae enzyme (aka P4) is the best studied member of the CCAP family. Kemmer et al. showed that one function of P4 is to dephosphorylate nicotinamide mononucleotide to nicotinamide riboside (NR) as part of a vestigial NAD+ utilization pathway [11]. Whether other CCAPs also function in NAD+ utilization is unknown.
High resolution crystal structures of recombinant P4 (rP4) complexed with substrates have provided insight into the nucleotidase activity of CCAPs [12]. Structures of a substrate-trapping mutant rP4 complexed with NMN, 5′-AMP, 3′-AMP, and 2′-AMP were determined. The structures revealed that the base binds in an aromatic box such that the hydrogen bonding groups do not contact the protein. This result is consistent with the general lack of base specificity of rP4 and other CCAPs. The region around the ribose is relatively open, which allows recognition of both 5′- and 3-′ substrates. The phosphoryl is tightly surrounded by numerous charged and polar groups. This crowded environment is necessary for positioning the phosphoryl group for nucleophilic attack by Asp64 and protonation of the leaving group by Asp66. Importantly, the span between the phosphoryl pocket and the aromatic box is optimal for binding nucleoside monophosphates, which is consistent with the classification of CCAPs as 5′-,3′-nucleotidases. Given the high sequence identity of the active sites of CCAPs, these features of substrate recognition are expected to be valid for the entire family.
The observation that the rP4 active site is well suited for binding nucleoside 5′-monophosphate substrates motivated us to investigate the inhibition of CCAPs by the nucleotide analog adenosine 5′-phosphorothioate (AMPS, Scheme 1, also called adenosine 5′-O-thiomonophosphate). AMPS differs from 5′-AMP by the substitution of an S atom for one of the O atoms of the phosphoryl and is thus isostructural with 5′-AMP. AMPS has been shown to be a competitive inhibitor or very poor substrate of 5′-nucleotidases [13, 14], but the structural basis of the inhibition is not known. We show here that AMPS is also an inhibitor of CCAPs and furthermore provide a structural explanation for the observed inhibition.
Scheme 1.
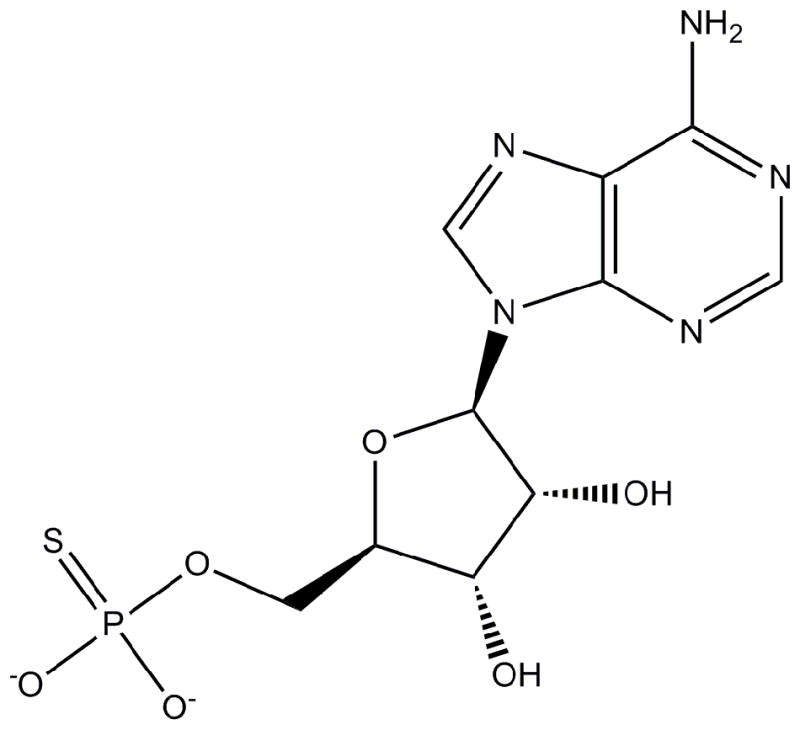
Results
Inhibition kinetics
AMPS was tested as an inhibitor of CCAPs using rP4 (UniProtKB C9MJZ6) and the CCAP from Francisella tularensis (FtCCAP, UniProtKB Q5NH51). The two enzymes share only 20 % global (26 % local) amino acid sequence identity and thus represent two distant branches of the CCAP family. Double reciprocal plots suggested that the inhibition mechanism for both enzymes is competitive, which is expected for an AMP analog. The inhibition constant (Ki) was estimated using global fitting to the competitive model (equation 1 in Experimental procedures). The fits were satisfactory (R2 of 0.97 for rP4 and 0.99 for FtCCAP) and yielded Ki values of 0.03 μM for rP4 and 0.3 μM for FtCCAP (Table 1). Thus, AMPS is a submicromolar inhibitor of these two CCCAPs.
Table 1.
Inhibition of CCAPs by AMPS using pNPP as the substrate
| CCAP | Km of (mM) | kcat (s−1) | Ki (μM) |
|---|---|---|---|
| rP4 | 0.12 ± 0.02 | 3.3 ± 0.1 | 0.035 ± 0.006 |
| FtCCAP | 0.56 ± 0.04 | 0.17 ± 0.01 | 0.30 ± 0.03 |
Structure of rP4 inhibited by AMPS
The crystal structure of rP4 complexed with AMPS was determined at 1.35 Å resolution (Table 2). AMPS binds in the previously identified substrate binding site, which is located at the junction between the core and cap domains (Fig. 1). The adenine base interacts exclusively with the cap domain, while the thiophosphoryl interacts with the core domain. The binding of AMPS to the substrate site is consistent with AMPS being a competitive inhibitor.
Table 2.
Data collection and refinement statisticsa
| Complex | rP4/AMPS | D64N/5′-AMP |
|---|---|---|
| Wavelength | 0.9792 | 0.9795 |
| Space group | P6522 | P6522 |
| Unit cell (Å) | a = 98.2, c = 107.0 | a = 97.9, c = 107.0 |
| Data collection resolution (Å) | 50 – 1.35 (1.40 – 1.35) | 50 – 1.35 (1.40 – 1.35) |
| No. of observations | 547582 | 614437 |
| No. of unique reflections | 67160 | 66328 |
| Rmerge(I)b | 0.072 (0.607) | 0.063 (0.537) |
| Rmeas(I)b | 0.071 (0.697) | 0.060 (0.528) |
| Rp.i.m.(I)b | 0.025 (0.247) | 0.019 (0.179) |
| Average I/σ(I) | 31.8 (3.2) | 39.2 (3.4) |
| Completeness (%) | 100 (99.9) | 99.8 (99.8) |
| Redundancy | 8.2 (7.9) | 9.3 (9.1) |
| Mosaicity (deg) | 0.2 | 0.3 |
| Refinement resolution (Å) | 50 – 1.35 (1.37 – 1.35) | 50 – 1.35 (1.40 – 1.35) |
| Rcryst | 0.138 (0.224) | 0.142 (0.214) |
| Rfreec | 0.159 (0.252) | 0.159 (0.257) |
| No. of protein residues | 247 | 247 |
| No. of protein atoms | 1938 | 1928 |
| No. of water molecules | 305 | 251 |
| Average B-factor (Å2) | ||
| Protein | 13.2 | 15.1 |
| Water | 24.6 | 25.1 |
| Ligand | 13.3 | 13.9 |
| Mg+2 | 7.4 | 8.6 |
| rmsdd | ||
| Bonds (Å) | 0.006 | 0.006 |
| Angles (deg) | 1.08 | 1.13 |
| Ramachandran plote | ||
| Favored (%) | 98.0 | 97.6 |
| Allowed (%) | 2.0 | 2.4 |
| Outliers (%) | 0.0 | 0.0 |
| Coordinate error (Å)f | 0.13 | 0.14 |
| PDB code | 3OCZ | 3SF0 |
Values for the outer resolution shell of data are given in parenthesis.
Rmerge was calculated using HKL2000; Rmeas and Rp.i.m. were calculated using SCALA [28] after integration with XDS [29].
A common set of test reflections (5 %) was used for refinement of all structures.
Compared to the parameters of Engh and Huber [30].
The Ramachandran plot was generated with RAMPAGE [31].
Maximum likelihood-based coordinate error estimate.
Fig. 1.

Ribbon representation of rP4 complexed with AMPS. The core and cap domains are colored blue and pink, respectively. AMPS is colored gray; the S atom of AMPS is colored magenta. The yellow sphere represents Mg2+. This figure and others were created with PyMOL [32].
The electron density at 1.35 Å resolution allowed for unequivocal determination of the inhibitor conformation, as well as the conformations of surrounding side chains and positions of active site water molecules (Fig. 2). The conformation of AMPS is identical to that of 5′-AMP bound to the D66N substrate trapping mutant of rP4 [12], except that the thiophosphoryl is shifted by 1.2 Å from the phosphoryl of 5′-AMP (Fig. 3). In both structures, the adenine base binds in an aromatic box formed by residues Phe86, Trp91 and Tyr221, and the ribose interacts with Glu131 via a water-mediated hydrogen bond. The shift of the thiophosphoryl reflects an apparent rotation of about 20° around the C4′-C5′ bond.
Fig. 2.

Electron density maps showing the presence of AMPS in the active site (relaxed stereographic view). The cage (cyan for the ligand, silver for protein side chains) represents a simulated annealing σA-weighted Fo - Fc omit map contoured at 3.0 σ. Prior to map calculation, the ligand and surrounding residues and water molecules were removed, and simulated annealing refinement was performed using PHENIX. The magenta surface represents an anomalous difference Fourier map contoured at 3.0 σ. Note that this map exhibits peaks corresponding to the P and S atoms of the thiophosphoryl.
Fig. 3.

Active site of rP4 inhibited by AMPS (relaxed stereographic view). AMPS is colored white; the S atom of AMPS is colored magenta. For reference, 5′-AMP from the D66N/5′-AMP structure (PDB code 3OCV) is also shown (yellow). Secondary structural elements of the core and cap domains are colored blue and pink, respectively. The yellow sphere represents Mg2+. The dashed lines denote electrostatic interactions in the AMPS complex.
The 20° rotation around the C4′-C5′ bond of AMPS is significant, because it moves the thiophosphoryl away from two essential catalytic residues, Asp64 and Asp66. The former residue functions as the nucleophile that attacks the substrate P atom, and the latter residue is the general acid that protonates the substrate leaving group. Both residues are conserved in HAD superfamily enzymes [15]. The distance between the nucleophile and the P atom of AMPS is 3.3 Å, and the angle formed by the nucleophile, P, and O5′ is 137°. The corresponding values for 5′-AMP are 3.0 Å and 173°, which are more consistent with backside nucleophilic attack. The rotation also displaces the O atom of the scissile O-P bond beyond the reach of Asp66. The O5′-Asp66 distances are 3.4 Å and 2.8 Å for AMPS and 5′-AMP, respectively. These subtle changes in the positioning of thiophosphoryl group lock the inhibitor in a nonproductive binding mode.
The identity of the S atom of AMPS was deduced from anomalous difference Fourier analysis in order to better understand the basis for the displaced thiophosphoryl group. Although the anomalous signal for sulfur is low at the wavelength of the data collection (f ″ is 0.18 e− for P and 0.23 e− for S), the anomalous difference Fourier map was unambiguous. Peaks 1 and 5 of the map correspond to the P and S atom of AMPS, respectively (magenta surface in Fig. 2). We note that peaks 2 – 4 and 6 correspond to S atoms of Met residues. The P-S bond points 180° away from the back wall of the active site. Consequently, the S atom sits in an open solvent-filled region of the active site and makes no direct contacts with the enzyme.
With the S atom identified, the interactions involving the O atoms of the thiophosphoryl are evident (Fig. 3). One of the O atoms interacts with the Mg2+ ion, while the other one interacts with Lys161. Unlike the phosphoryl of 5′-AMP, the thiophosphoryl does not interact directly with conserved Thr124 or the backbone of Asp66. Instead, a water molecule mediates the interactions with these groups (denoted wat1 in Fig. 3).
Binding of AMPS does not disrupt the octahedral coordination geometry of the Mg2+ ion. In ligand-free rP4 (PDB code 3ET4), the metal ion binds to Asp64, Asp181, the backbone carbonyl of Asp66, and three water molecules. One of the coordinating water molecules dissociates upon substrate binding, and an O atom of the phosphoryl takes its place. An analogous exchange occurs during the binding of AMPS.
Mutation of the Asp nucleophile induces the nonproductive conformation in the substrate
The conformation of AMPS is reminiscent of substrates bound to human mitochondrial deoxyribonucleotidase (mdN) [16]. This enzyme also belongs to the HAD structural superfamily, but the cap domain fold and quaternary structure are substantially different from those of rP4. In order to trap nucleoside monophosphate substrates bound to mdN, Walldén et al. mutated the essential nucleophile (Asp41) to Asn. The substrates bound with the phosphoryl in one or both of two alternative conformations, which were dubbed Mode A and Mode B. Mode A is the active conformation and is identical to the phosphoryl position of 5′-AMP bound to D66N. Mode B is a nonproductive conformation and resembles AMPS. Presumably, the adoption of the Mode B conformation results from the alteration of the electrostatic environment caused by the mutation of the negatively charged Asp41 to Asn.
Mutation of the Asp64 nucleophile in rP4 also induces the substrate to adopt the nonproductive Mode B conformation. The structure of D64N complexed with 5′-AMP was determined at 1.35 Å resolution (Table 2). The electron density map definitively indicates just a single conformation, which is very similar to that of AMPS (Fig. 4). The only substantial difference between the two structures involves Lys161. In the AMPS complex, Lys161 interacts with Asp64, Asp185, and a thiophosphoryl O atom (Fig. 5, black dashes). In the D64N/5′-AMP complex, however, Lys161 has rotated 90° around the Cδ-Cε bond to avoid an electrostatic clash with the side chain amine group of Asn64. This new conformation of Lys161 maintains the ion pair with Asp185 and also allows a new interaction with a different phosphoryl oxygen atom (the O atom corresponding to the S of the thiophosphoryl, see Fig. 5, cyan dashes).
Fig. 4.

Electron density map showing the presence of 5′-AMP in the active site of D64N (relaxed stereographic view). The cage (cyan for the ligand, silver for protein side chains) represents a simulated annealing σA-weighted Fo - Fc omit map contoured at 3.0 σ. Prior to map calculation, the ligand and surrounding residues and water molecules were removed, and simulated annealing refinement was performed using PHENIX.
Fig. 5.

Comparison of rP4/AMPS (gray with magenta S atom) and D64N/5′-AMP (cyan) (relaxed stereographic view). The black and cyan dashes denote electrostatic interactions in rP4/AMPS and D64N/5′-AMP, respectively. For reference, the 5′-AMP bound to D66N is shown in yellow. The yellow sphere represents Mg2+.
Discussion
Murray and Atkinson reported in 1968 that AMPS is a competitive inhibitor (Ki value of 20 μM) of the type II 5′-nucleotidase from Crotalus adamanteus venom [13]. One plausible explanation for the inhibition is that the lower electronegativity of S compared to O causes the P atom of AMPS to be less electrophilic than that of 5′-AMP and therefore less susceptible to nucleophilic attack by the enzyme. In this scenario, one might expect the inhibitor to adopt the same conformation as the substrate.
The rP4/AMPS structure suggests a different mechanism of inhibition based on steric considerations. The larger size of S compared to O prevents the thiophosphoryl from occupying the catalytically active conformation. The phosphoryl binding site appears to be too congested, and apparently not flexible enough, to accommodate the larger S atom. This steric conundrum is solved by rotating the thiophosphoryl away from the back wall of the active site, which allows the S atom to protrude into an open solvent filled cavity. As a consequence of the rotation, the P atom is not positioned optimally for nucleophilic attack by Asp64. Furthermore, the O atom of the scissile O-P bond is too far from the Asp residue (Asp66) that protonates the leaving group. Thus, the AMPS is an inhibitor of rP4.
This nonproductive mode of binding is also adopted by substrates bound to the mutant enzyme D64N. In this case, electrostatic considerations underlie the rotation of the phosphoryl. Mutation of Asp to Asn decreases the negative charge of the active site and places an obligate hydrogen bond donor group next to Lys161. It appears that Lys161 rotates to avoid the electrostatic repulsion, which, in turn, induces rotation of the phosphoryl into the Mode B position. Taken together, the two rP4 structures reported here highlight the precise steric and electrostatic requirements of phosphoryl recognition by rP4.
We note that a similar rotation of the phosphoryl was not observed for D66N. Asp66 functions as the acid that protonates the leaving group, and is therefore protonated at the beginning of the catalytic cycle. Thus, mutation of Asp66 to Asn does not change the overall charge of the active site. For this reason, D66N is superior to D64N as a substrate-trapping enzyme.
The D64N structure also illustrates the potential pitfalls of the common practice of using active site mutants to understand enzyme structure and mechanism. Most biochemists consider the mutation of Asp to Asn to be a conservative change. Nevertheless, this mutation at residue 64 of rP4 induced the substrate to adopt a conformation that does not reflect the conformation of the substrate bound to the native enzyme. Interestingly, this artifact was avoided in rP4 by mutating Asp66 rather than Asp64. These results remind us that seemingly innocuous mutations can induce unexpected structural artifacts depending on the three-dimensional context of the mutated residue.
The data reported here could potentially aid inhibitor design efforts. For example, it has been suggested that targeting the NAD+ utilization pathway may be a possible route to the development of narrow spectrum antimicrobial agents against H. influenzae [17]. The NAD+ utilization pathway of H. influenzae includes an uptake system that imports NAD+, NMN, and NR into the periplasm. The periplasmic NAD+ nucleotidase NadN catalyzes the hydrolysis of NAD+ to generate NMN and AMP. NMN is dephosphorylated in the periplasm by P4 to NR, which is transported across the inner membrane into the cytosol by the NR-specific permease PnuC. Finally, the bifunctional NR kinase/NMN adenylyltransferase NadR converts NR into NAD+. All of these enzymes and transporters are potential targets for inhibitor design. The structural and kinetic data reported here provide a potential starting point for the design of specific inhibitors of P4.
The similarities between the nonproductive nucleotide conformations reported here for rP4 and observed previously for mdN may provide new insight into designing inhibitors of human 5′-nucleotidases. 5′-nucleotidases regulate the nucleotide pool by reversing the action of nucleoside kinases [18]. They have been implicated in decreasing the efficacy of antiviral and antiblastic nucleoside analogs. Phosphorylation in vivo activates these drugs. Since intracellular 5′-nucleotidases can reverse this activation step, they decrease drug efficacy. For this reason, designing inhibitors of 5′-nucleotidases for co-therapy is of interest. P4 and mdN share a similar core domain that includes the essential DDDD motif and a hydrophobic region for binding the nucleotide base. The occurrence of the Mode B substrate conformation in structures of rP4 D64N and mdN D41N attest to the similarities of their phosphoryl binding pockets. These observations raise the possibility that AMPS could be an inhibitor of mdN.
Experimental procedures
Kinetics of inhibition
His-tagged rP4 and FtCCAP were expressed and purified as described previously [12, 19]. AMPS was purchased from Sigma-Aldrich, St. Louis, MO, USA (catalogue number A1640). Steady-state enzymatic activity was assessed at 25 °C with p-nitrophenyl phosphate (pNPP) as the substrate and using a discontinuous assay that measures the production of p-nitrophenolate [20, 21]. The assay buffer consisted of 0.2 M sodium acetate and 1 mM MgCl2 at pH 5.5. The substrate concentration was varied in the range of 0.1 – 5 mM at fixed AMPS concentrations of 0.25 – 1.0 μM. The reaction was stopped using 1.0 M glycine at pH 10 after reaction times of 15 s, 75 s, 135 s and 195 s. The p-nitrophenolate concentration was determined spectrophotometrically at 405 nm by reference to a standard curve constructed from solutions of known p-nitrophenolate concentration. The initial rate was estimated by fitting data from the four time points to a line. The kinetic parameters for pNPP (Vmax, Km) and Ki for AMPS were estimated by the global fitting using Origin 8.5 software to the competitive inhibition model,
| (1) |
Preparation of rP4/AMPS and D64N/5′-AMP crystals
His-tagged rP4 was expressed from a pET20b plasmid and purified as described previously [12]. A site-directed mutant of rP4 in which Asp64 is changed to Asn (D64N) was created using overlap extension PCR from the pET20b plasmid containing the rP4 coding sequence. The mutation was verified by DNA sequencing, and the mutated plasmid was transformed into BL21(AI) for large scale protein expression using Studier's auto-induction method [22]. His-tagged D64N was purified as described previously for rP4 [12].
Hexagonal crystals of rP4 and D64N were grown as described previously for rP4 and the D66N mutant of rP4 [12]. The crystals were grown in sitting drops (1 μL of protein and 1 μL of reservoir) at room temperature over reservoirs containing 0.05 – 0.2 M ammonium citrate, 0.05 – 0.15 mM MgCl2, and 18 – 23 % (w/v) polyethylene glycol (PEG) 3350 in the pH range of 6.8 – 7.2.
Crystals of enzyme/ligand complexes were obtained by soaking. Crystals of rP4 were first cryoprotected at room temperature in 23 – 28 % (w/v) PEG 3350, 0.1 M ammonium citrate buffer pH 7.0, and 20% PEG 200. The cryoprotected crystals were transferred to a solution of the cryobuffer supplemented with 200 mM MgCl2 and 10 – 15 mM AMPS. After 15 – 35 minutes, the crystals were picked up with Hampton loops and plunged into liquid nitrogen. Crystals of D64N complexed with 5′-AMP were prepared similarly, except that the substrate concentration was 10 – 23 mM, the MgCl2 concentration was 100 mM, and the soaking times were 30 – 75 minutes.
Structure determination
X-ray diffraction data were collected at the Advanced Photon Source beamline 24-ID-C and processed with HKL2000 [23] (Table 1). The crystals have space group P6522 with unit cell lengths of a = 98 Å and c = 107 Å, and there is one molecule in the asymmetric unit with 54% solvent content and Vm of 2.6 Å3/Da [24, 25]. We note that this crystal form is the same one used to determine structures of the D66N mutant of rP4 complexed with substrates [12]. PHENIX [26] and COOT [27] were used for structure refinement and model building, respectively. Refinement was initiated from a model derived from the coordinates of a 1.35 Å resolution structure of rP4 (PDB code 3OCU). The test set of reflections (5 %) was based on the one used for previous refinements of rP4 structures [12]. The B-factor model used during the initial rounds of refinement consisted of an isotropic B-factor for each non-hydrogen atom and TLS refinement (one TLS group for the entire protein chain). During the final few rounds of refinement, anisotropic B-factors were used, which decreased Rfree by 0.016 for the AMPS structure (0.175 to 0.159) and 0.023 for the D64N structure (0.185 to 0.162).
Acknowledgments
H.S. was supported by a pre-doctoral fellowship from National Institutes of Health grant DK071510 and a Chancellor’s Dissertation Completion Fellowship from the University of Missouri-Columbia. We thank Dr. Jonathan Schuermann for help with X-ray data collection. Part of this work is based upon research conducted at the Northeastern Collaborative Access Team beam lines of the Advanced Photon Source, supported by award RR-15301 from the National Center for Research Resources at the National Institute of Health. Use of the Advanced Photon Source is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under contract No. W-31-109-ENG-38.
Abbreviations
- AMPS
adenosine 5′-phosphorothioate
- rP4
recombinant Haemophilus influenzae e (P4) class C acid phosphatase
- CCAP
class C acid phosphatase
- HAD
haloacid dehalogenase
- NR
nicotinamide riboside
- FtCCAP
Francisella tularensis class C acid phosphatase
- pNPP
p-nitrophenyl phosphate
- mdN
human mitochondrial deoxyribonucleotidase
- D64N
mutant of rP4 in which Asp64 is replaced by Asn
- PDB
Protein Data Bank
- PEG
polyethylene glycol
Footnotes
Database
Coordinates and structure factor amplitudes are available in the Protein Data Bank database under the accession numbers 3OCZ (rP4/AMPS) and 3SF0 (D64N/5′-AMP).
References
- 1.Thaller MC, Schippa S, Rossolini GM. Conserved sequence motifs among bacterial, eukaryotic, and archaeal phosphatases that define a new phosphohydrolase superfamily. Protein Sci. 1998;7:1647–52. doi: 10.1002/pro.5560070722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Felts RL, Ou Z, Reilly TJ, Tanner JJ. Structure of Recombinant Haemophilus Influenzae e (P4) Acid Phosphatase Reveals a New Member of the Haloacid Dehalogenase Superfamily. Biochemistry. 2007;46:11110–9. doi: 10.1021/bi701016m. [DOI] [PubMed] [Google Scholar]
- 3.Reilly TJ, Chance DL, Calcutt MJ, Tanner JJ, Felts RL, Waller SC, Henzl MT, Mawhinney TP, Ganjam IK, Fales WH. Characterization of a unique class C acid phosphatase from Clostridium perfringens. Appl Environ Microbiol. 2009;75:3745–54. doi: 10.1128/AEM.01599-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh H, Malinski TJ, Reilly TJ, Henzl MT, Tanner JJ. Crystal structure and immunogenicity of the class C acid phosphatase from Pasteurella multocida. Arch Biochem Biophys. 2011;509:76–81. doi: 10.1016/j.abb.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reilly TJ, Chance DL, Smith AL. Outer membrane lipoprotein e (P4) of Haemophilus influenzae is a novel phosphomonoesterase. J Bacteriol. 1999;181:6797–6805. doi: 10.1128/jb.181.21.6797-6805.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reilly TJ, Smith AL. Purification and characterization of a recombinant Haemophilus influenzae outer membrane phosphomonoesterase e (P4) Protein Expr Purif. 1999;17:401–409. doi: 10.1006/prep.1999.1157. [DOI] [PubMed] [Google Scholar]
- 7.Malke H. Cytoplasmic membrane lipoprotein LppC of Streptococcus equisimilis functions as an acid phosphatase. Appl Environ Microbiol. 1998;64:2439–42. doi: 10.1128/aem.64.7.2439-2442.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Passariello C, Schippa S, Iori P, Berlutti F, Thaller MC, Rossolini GM. The molecular class C acid phosphatase of Chryseobacterium meningosepticum (OlpA) is a broad-spectrum nucleotidase with preferential activity on 5′-nucleotides. Biochim Biophys Acta. 2003;1648:203–9. doi: 10.1016/s1570-9639(03)00147-x. [DOI] [PubMed] [Google Scholar]
- 9.Reilly TJ, Calcutt MJ. The class C acid phosphatase of Helicobacter pylori is a 5′ nucleotidase. Protein Expr Purif. 2004;33:48–56. doi: 10.1016/j.pep.2003.08.020. [DOI] [PubMed] [Google Scholar]
- 10.Wang R, Ohtani K, Wang Y, Yuan Y, Hassan S, Shimizu T. Genetic and biochemical analysis of a class C non-specific acid phosphatase (NSAP) of Clostridium perfringens. Microbiology. 2010;156:167–73. doi: 10.1099/mic.0.030395-0. [DOI] [PubMed] [Google Scholar]
- 11.Kemmer G, Reilly TJ, Schmidt-Brauns J, Zlotnik GW, Green BA, Fiske MJ, Herbert M, Kraiss A, Schlor S, Smith A, Reidl J. NadN and e (P4) are essential for utilization of NAD and nicotinamide mononucleotide but not nicotinamide riboside in Haemophilus influenzae. J Bacteriol. 2001;183:3974–3981. doi: 10.1128/JB.183.13.3974-3981.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Singh H, Schuermann JP, Reilly TJ, Calcutt MJ, Tanner JJ. Recognition of Nucleoside Monophosphate Substrates by Haemophilus influenzae Class C Acid Phosphatase. J Mol Biol. 2010;404:639–649. doi: 10.1016/j.jmb.2010.09.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murray AW, Atkinson MR. Adenosine 5′-phosphorothioate. A nucleotide analog that is a substrate, competitive inhibitor, or regulator of some enzymes that interact with adenosine 5′-phosphate. Biochemistry. 1968;7:4023–9. doi: 10.1021/bi00851a032. [DOI] [PubMed] [Google Scholar]
- 14.Skladanowski AC, Hoffmann C, Krass J, Jastorff B, Makarewicz W. Structure-activity relationship of cytoplasmic 5′-nucleotidase substrate sites. Biochem J. 1996;314(Pt 3):1001–7. doi: 10.1042/bj3141001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allen KN, Dunaway-Mariano D. Phosphoryl group transfer: evolution of a catalytic scaffold. Trends Biochem Sci. 2004;29:495–503. doi: 10.1016/j.tibs.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 16.Wallden K, Ruzzenente B, Rinaldo-Matthis A, Bianchi V, Nordlund P. Structural basis for substrate specificity of the human mitochondrial deoxyribonucleotidase. Structure. 2005;13:1081–8. doi: 10.1016/j.str.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 17.Gerlach G, Reidl J. NAD+ utilization in Pasteurellaceae: simplification of a complex pathway. J Bacteriol. 2006;188:6719–27. doi: 10.1128/JB.00432-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bianchi V, Spychala J. Mammalian 5′-nucleotidases. J Biol Chem. 2003;278:46195–8. doi: 10.1074/jbc.R300032200. [DOI] [PubMed] [Google Scholar]
- 19.Singh H, Felts RL, Ma L, Malinski TJ, Calcutt MJ, Reilly TJ, Tanner JJ. Expression, purification and crystallization of class C acid phosphatases from Francisella tularensis and Pasteurella multocida. Acta Crystallogr. 2009;F65:226–31. doi: 10.1107/S1744309109001511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lanzetta PA, Alvarez LJ, Reinach PS, Candia OA. An improved assay for nanomole amounts of inorganic phosphate. Anal Biochem. 1979;100:95–97. doi: 10.1016/0003-2697(79)90115-5. [DOI] [PubMed] [Google Scholar]
- 21.Carter SG, Karl DW. Inorganic phosphate assay with malachite green: an improvement and evaluation. J Biochem Biophys Methods. 1982;7:7–13. doi: 10.1016/0165-022x(82)90031-8. [DOI] [PubMed] [Google Scholar]
- 22.Studier FW. Protein production by auto-induction in high density shaking cultures. Protein Expr Purif. 2005;41:207–34. doi: 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 23.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 24.Matthews BW. Solvent content of protein crystals. J Mol Biol. 1968;33:491–497. doi: 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- 25.Kantardjieff KA, Rupp B. Matthews coefficient probabilities: Improved estimates for unit cell contents of proteins, DNA, and protein-nucleic acid complex crystals. Protein Sci. 2003;12:1865–71. doi: 10.1110/ps.0350503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr, Sect D. 2010;66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Cryst. 2004;D60:2126–32. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 28.Evans P. Scaling and assessment of data quality. Acta Cryst. 2006;D62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 29.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr. 2010;66:125–32. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engh RA, Huber R. Accurate bond and angle parameters for x-ray protein structure refinement. Acta Cryst. 1991;A47:392–400. [Google Scholar]
- 31.Lovell SC, Davis IW, Arendall WB, 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins. 2003;50:437–50. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- 32.DeLano WL. The PyMOL User’s Manual. DeLano Scientific; Palo Alto, CA, USA: 2002. [Google Scholar]