

Abstract
CXCR3 and its ligands are important for the trafficking of activated CD4+ TH1 T cells, CD8+ T cells, and natural killer cells during inflammation. Recent functional studies demonstrate a more diverse role of CXCR3 in inflammatory diseases of the central nervous system (CNS). We examined the impact of CXCR3 on a less complex interferon-γ–dependent, type 1 cell–mediated immune response in the CNS, induced in mice by the transgenic production of glial fibrillary acidic protein IL-12 (GF-IL12) by astrocytes and retinal Müller cells. GF-IL12 mice develop ataxia because of severe cerebellar inflammation but have little overt ocular disease. Surprisingly, CXCR3-deficient GF-IL12 mice (GF-IL12/CXCR3KO) have drastically reduced ataxia but developed cataracts, severe ocular inflammation, and eye atrophy. Most GF-IL12/CXCR3KO mice had minimal cerebellar inflammation but severe retinal disorganization, loss of photoreceptors, and lens destruction in the eye. The number of CD3+, CD11b+, and natural killer 1.1+ cells were reduced in the CNS but highly increased in the eyes of GF-IL12/CXCR3KO compared with GF-IL12 mice. High levels of interferon-γ, IL-1, tumor necrosis factor α, CXCL9, CXCL10, and CCL5 were found in GF-IL12 cerebelli and GF-IL12/CXCR3KO eyes. Our findings demonstrate key but paradoxical functions for CXCR3 in IL-12–induced immune disease in the CNS, promoting inflammation in the brain yet restricting it in the eye. We conclude that the function of CXCR3 in cellular immune disease is driven by a common trigger and is controlled by tissue-specific factors.
Innate and adaptive immune responses play a crucial role in protecting the nervous system from dangerous pathogens. However, this is a two-edged sword because an excessive host immune response can cause more harm than good.1 The understanding of the pathogenetic mechanisms leading to immune-mediated tissue damage during pathogen-driven diseases, such as bacterial meningitis and herpes encephalitis,2 or autoimmune disease, such as multiple sclerosis,3 is important to develop novel treatment strategies to prevent detrimental effects of the immune response.
Chemokines, which are chemotactic cytokines, are key molecules in orchestrating innate and adaptive immune responses.4 The expression of chemokines is greatly increased in nearly all neuroinflammatory disorders examined; however, the precise functional properties of these multifunctional and promiscuous molecules remain somewhat unclear.5 The chemokines CXCL9, CXCL11, and in particular CXCL10 are highly induced in various neuroinflammatory disorders.6 They share a common chemokine receptor, which is CXCR3. CXCR3 is mainly found on activated CD4+ and CD8+ T cells but also on natural killer (NK) cells, monocytes, and dendritic cells.7, 8, 9, 10 CXCR3 is differentially activated by CXCL9, CXCL10, and CXCL11.11, 12 The ligands and the receptor itself are mainly induced and regulated by interferon (IFN)-γ.13, 14, 15 TH1 but not TH2 cells express CXCR3 in great amounts16; therefore, CXCR3 is a typical marker of TH1 cells.17
Descriptive studies initially pointed toward a key role of CXCR3 and its ligands in promoting the influx of activated T cells into the central nervous system (CNS).18, 19, 20 However, functional studies using gene-deficient mice or blocking antibodies revealed a much more complex functional profile of this chemokine system,21, 22, 23 which strongly depends on the pathogenesis of the disease model examined. In experimental autoimmune encephalomyelitis (EAE), CXCR3 surprisingly has a protective effect, which is demonstrated by a more severe and chronic disease in CXCR3-deficient mice.24
However, EAE has a complex pathogenesis, which is not, as previously assumed, a mainly TH1-driven autoimmune reaction. Different T-cell subsets—TH1, TH17, and Tregs—have an impact during different stages of the disease, and to understand these functions is currently an important issue in neuroimmunology.
To further clarify the functional relevance of CXCR3 in neuroinflammation, we examined the type 1 cell–mediated CNS inflammation in mice with CNS-restricted transgenic expression of the IL-12 gene.25 IL-12 is an important regulator of cellular immunity in both innate and adaptive immune responses. IL-12 activates NK cells, and in CD8+ and CD4+ T cells, IL-12 induces differentiation from a TH0 to a TH1 phenotype.19, 20 Furthermore, IL-12 increases the proliferation of T cells and NK cells and stimulates the production of numerous immune effector molecules, in particular IFN-γ. As IFN-γ induces the production of IL-12, the IL-12/IFN-γ cytokine system serves as a positive feedback mechanism, which initiates and maintains immune responses.
Glial fibrillary acidic protein (GFAP) IL-12 (GF-IL12) mice express the IL-12 gene under the control of an astrocyte-specific (GFAP) promoter and develop a severe neuroinflammatory response via induction of IFN-γ. In this transgenic mouse model, activated T cells and their production of IFN-γ are critical to the development IL-12–driven CNS inflammation,26 and the high expression of IFN-γ–inducible chemokines, such as CXCL9 or CXCL10, suggests an additional important role for these molecules. In this study, we examined whether CXCR3 is a critical molecule for the induction and course of IL-12–induced neuroinflammation. To address this question, we studied the impact of CXCR3 deficiency in transgenic mice with chronic CNS-restricted production of IL-12.
Materials and Methods
Animals
CXCR3-deficient (CXCR3KO) mice (originally provided by Drs. Bao Lu and Craig Gerard, Children's Hospital and Harvard Medical School, Boston, MA) have been described previously.27 The mice were backcrossed at least eight generations onto the C57BL/6 strain. CXCR3-deficient mice displayed no clinical or histologic abnormalities when compared with wild-type (WT) mice.
Transgenic mice expressing both subunits of the IL-12 heterodimer (p35/p40) under the transcriptional control of the astrocyte-specific GFAP promoter (GF-IL12 mice) were described previously.25 To obtain CXCR3-deficient GF-IL12 mice (GF-IL12/CXCR3KO), heterozygous GF-IL12 mice on a C57BL/6 background were successively crossed with CXCR3KO mice. Animals were kept under pathogen-free conditions in the Zentrale Tierexperimentelle Einrichtung animal facility of the University Hospital Münster (Münster, Germany) and in the Blackburn facility of the University of Sydney (Sydney, Australia). All procedures were approved by the veterinary office of the Bezirksregierung Münster (Münster, Germany) and of the Sydney Animal Care and Ethics Committee (Sydney, Australia).
Clinical Assessment of Mice
The GF-IL12/CXCR3KO, GF-IL12, CXCR3KO, and WT mice were clinically evaluated for 36 weeks at least twice a week. The clinical scores for ataxia were assessed for each animal according to a previously established protocol.28 Ataxia signs were scored using a cumulative scale of four points, giving one point to each of these four physical signs: splayed legs, dragging weight on the trunk rather than on the legs, wobbling, and falling from side to side. A second score was applied to assess the severity of the ocular phenotype: 1 indicating monocular cataract; 2, binocular cataract; 3, monocular cataract and monocular phthisis bulbi; and 4, binocular phthisis bulbi.
Tissue Processing for Histologic Analysis
The tissue for analysis (histology, immunohistochemistry, and molecular biology) was obtained from 8- and 24-week-old mice of each genotype (WT, CXCR3KO, GF-IL12/CXCR3KO, and GF-IL12). Immediately after euthanasia, the brain and eyes were removed and half of the brain (cut along the sagittal midline) and an eye were fixed overnight in PBS-buffered 4% paraformaldehyde at 4°C, washed in PBS, and subsequently embedded in paraffin. Sections (8 μm) were prepared from paraffin-embedded tissue.
For immunohistochemical analysis of cryosections, tissue was embedded with Tissue Tek (Sakura Finetek, Staufen, Germany). Sections (8 μm) were prepared and five cerebelli and eyes of each genotype were analyzed.
Routine Histologic Analysis and (Fluorescence) Immunohistochemistry
Paraffin-embedded sections were stained with H&E and Luxol fast blue (LFB) for routine histologic analysis and myelin evaluation. For immunohistochemistry, sections were rehydrated in graded ethanol series after deparaffination in xylene and some were pretreated with proteinase K digestion. Slides were then incubated for 1 hour at room temperature with primary antibodies (primary antibodies and corresponding protocols for immunohistochemistry are summarized in Table 1). After washing in PBS, a biotinylated secondary antibody (Axxora, Lörrach, Germany; 1/200) and horseradish peroxidase–coupled streptavidin (Axxora; 1/200) were used. The signal was visualized by NovaRED color reagent (Axxora), according to the manufacturer's instructions. Conventional and immunofluorescence-stained sections were examined under a DM4000B bright field and fluorescence microscope (Leica, Wetzlar, Germany). Bright field images and monochrome fluorescent images were acquired using a Leica DFC480 camera and Leica Firecam 1.7.1 software (Leica). The acquired monochrome fluorescence signals were merged using SPOT Advanced 4.5 software (Diagnostic Instruments, Sterling, MI) or “cell̂ P” imaging software (Olympus Soft Imaging Solutions, Münster, Germany).
Table 1.
Antibody and Lectin Reagents Used for Immunohistochemistry
| Antibody or lectin (source) | Specificity | Paraffin sections (dilution) | Cryosections (dilution) |
|---|---|---|---|
| Polyclonal rabbit anti-human CD3 (Dako, Hamburg, Germany) | T cell | 1:200 | 1:200 |
| Biotin-conjugated tomato lectin, Lycopersicon esculentum (Axxora, Lörrach, Germany) | Microglia and macrophages, endothelial cells | 1:50 | |
| Monoclonal mouse antihuman GFAP (Dako) | GFAP | 1:200 | |
| Polyclonal rabbit antilaminin reactive with human and mouse laminin (Sigma-Aldrich, Munich, Germany) | Basal lamina | 1:50 | |
| Polyclonal rabbit anti-Iba1 reactive with human, mouse, and rat Iba1 (Wako Chemicals, Neuss, Germany) | Microglia and macrophages | 1:500 |
To allow clear discrimination of IHC signal and the pigment layer of the eye and to detect epitopes that are not preserved in paraffin-embedded tissue, fluorescent immunohistochemistry on cryoembedded sections was performed (Table 1). After washing in PBS, OD594 and OD488 fluorescence-conjugated secondary antibody (Invitrogen, Darmstadt, Germany; 1/200) was used to visualize the primary antibody. Sections were mounted and counterstained with DAPI (Sigma-Aldrich, Munich, Germany).
RNase Protection Assays
The brain and eyes were collected as described above and snap frozen in liquid nitrogen. Total RNA was isolated using Trizol (Sigma-Aldrich). RNase protection assays (RPAs) were performed as described previously.29 Five micrograms of total RNA were used for each sample and hybridized with the following probes: CXCL9, transforming growth factor (TGF)-β, IL-1α, IFN- γ, IL-12p40, CCL5, and the RPL32–4A gene30 that served as an internal loading control. For autoradiography, Biomax films (Eastman-Kodak, Rochester, NY) were exposed for various periods and scanned using a ScanJet 4C (Hewlett-Packard Co., Palo Alto, CA).
Cytokine and Chemokine mRNA Determination by Real-Time Quantitative PCR
Total RNA (3 μg) was reverse transcribed into cDNA using SuperScript III Reverse Transcriptase (Invitrogen). Real-time quantitative PCR assays were performed using SYBR Green. The composition of the reaction mixture was as follows: 1 μL of cDNA corresponding to 300 ng of total RNA, 100 nmol/L of each primer, and 2× SYBR Green PCR Master Mix (Applied Biosystems, Darmstadt, Germany) in a total volume of 25 μL. Samples were analyzed simultaneously for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA as the internal control. The mRNA levels for each target were normalized to mRNA levels of GAPDH and expressed relative to that of nontransgenic C57BL/6J mice. Each sample was assayed in duplicate. Primer sequences used to amplify the GAPDH, IFN-γ, IL-17, CXCL10, tumor necrosis factor (TNF)-α, vascular endothelial growth factor (VEGF)-A, lymphatic vessel endothelial hyaluronan receptor 1 (LYVE1), and IL-10 cDNA were as follows: GAPDH sense 5′-TCACCAGGGCTGCCATTTGC-3′ and GAPDH antisense 5′-GACTCCACGACATACTCAGC-3′, IFN-γ sense 5′-CAGCAACAGCAAGGCGAAA-3′ and IFN-γ antisense 5′-GCTGGATTCCGGCAACAG-3′, IL-17 sense 5′-AAGGCAGCAGCGATCATCC-3′ and IL-17 antisense 5′-GGAACGGTTGAGGTAGTCTGAG-3′, CXCL10 sense 5′-GACGGTCCGCTGTTCT-3′ and CXCL10 antisense 5′-GCTTCCCTATGGCCCTCATT-3′, TNF-α sense 5′-ATGAGAAGTTCCCAAATGGCC-3′ and TNF-α antisense 5′-ACGTGGGCTACAGGCTTGTC-3′, VEGF-A sense 5′-TTACTGCTGTACCTCCACC-3′ and VEGF-A antisense 5′-ACAGGACGGCTTGAAGATG-3′, LYVE1 sense 5′-TCCAACACGGGGTAAAATGT-3′ and LYVE1 antisense 5′-CCTCCAGCCAAAAGTTCAAA-3′, and IL-10 sense 5′-TGTCAAATTCATTCATGGCCT-3′ and IL-10 antisense 5′-ATCGATTTCTCCCCTGTGAA-3′.
Flow Cytometry Analyses of Cerebellar and Ocular Leukocytes
Cerebelli and eyes from WT, CXCR3KO, GF-IL12, and GF-IL12/CXCR3KO (n = 3) animals were excised and placed into ice-cold PBS buffer solution. The tissue was cut into small pieces and digested for 30 minutes in PBS with collagenase I (0.05 g/mL; Roche Diagnostics, Mannheim, Germany) first and after DNase I (100 μg/mL; Sigma-Aldrich) digestion for 30 minutes at 37°C in a humidified atmosphere of 5% CO2. The eye homogenates were differentially digested by a combination of collagenase D (1 mg/mL; Roche) and DNase I (100 μg/mL, Sigma-Aldrich) for 30 minutes. Digestion was stopped with 10% fetal calf serum. A pellet was obtained after 10-minute centrifugation at 340 × g. Digested samples were resuspended in PBS and disrupted or homogenized using a needle (0.6 × 25) and a syringe (5 mL) before passing through a 70-μm cell strainer (BD Biosciences, Heidelberg, Germany). After pelleting, homogenates were dissolved in 30% Percoll (Amersham Pharmacia Biotech, Braunschweig, Germany). Subsequently, the 30% Percoll homogenate mix was layered over 70% Percoll. Leukocytes were collected from the 30%/70% interface after a 800 × g centrifugation step for 25 minutes at room temperature. The collected cells were washed in PBS and blocked with CD16/CD32 (Fc block; BD Biosciences) antibody. Isolated leukocytes were incubated with fluorochrome-conjugated antibodies (eBioscience, Frankfurt/Main, Germany) to detect CD3e (clone 145-2C11, PerCP-Cy5.5), CD4 (clone RM4-5, fluorescein isothiocyanate), CD8a (clone 53–6.7, APC-eFluor 780), CD11b (clone M1/70, APC), CD11c (clone N418, PE-Cy7), CD25 (clone PC61.5, APC), CD45 (clone 30-F11, fluorescein isothiocyanate), Ly6G (clone RB6-8C5, PerCP-Cy5.5), B220 (clone RA3-6B2, APC-eFluor 780) and NK-1/1 (clone PK136, PE-Cy7). Intracellular staining for FoxP3 (phycoerythrin) was performed using the Mouse Regulatory T Cell Staining Kit (with phycoerythrin Foxp3, clone FJK-16s, fluorescein isothiocyanate CD4, APC CD25; eBioscience) according to the manufacturer's instructions. After washing, bound antibody was detected with fluorescence activated cell sorting (FACS) (BD FACSCanto II; BD Biosciences), and the acquired data were analyzed using flow cytometry software (FlowJo; TreeStar, San Carlos, CA).
Protein Lysates and Western Blot
Tissue was homogenized using a Precellys 24 tissue homogenizer (Bertin Technologies, Saint-Quentin-en-Yvelines Cedex, France) in lysis buffer [25 mmol/L Tris HCl (pH 7.4), 150 mmol/L NaCl, 1% NP-40, 1 mmol/L EDTA, 1 mmol/L EGTA, 1% Na Deoxycholate 0.1% SDS, 2 mmol/L ortho-vanadate, 30 mmol/L NaF, 50 mmol/L sodium pyrophosphate, and 2 mmol/L phenylmethylsulfonyl fluoride] modified with freshly supplemented protease inhibitor mix (Sigma-Aldrich). Samples were centrifuged at 14,000 × g for 15 minutes and supernatants were taken. The protein concentrations were determined using the BCA Protein Assay Kit (Pierce, Rockford, IL). Protein lysates (50 μg) were separated by 10% SDS-PAGE gel using NuPage MES SDS running buffer (Invitrogen) at 150 V. PageRuler Prestained Protein Ladder (Fermentas, St. Leon-Rot, Germany) was used as standard. Proteins were transferred to 0.2-μm nitrocellulose membranes (Whatman, Dassel, Germany). Membranes were blocked for 30 minutes in Tris-buffered saline with Tween 30 containing 5% skim milk. Immunoblotting was performed using antiphosphorylated (Y693) STAT4 antibody (Invitrogen), anti-STAT4 (Santa Cruz, Heidelberg, Germany) antibody, and antibody CP06 (Oncogene Science, Cambridge, MA) detecting α-tubulin followed by incubation with the appropriate horseradish peroxidase–conjugated secondary antibodies (Jackson ImmunoResearch, Newmarket, UK). Immunoreactivity was detected by chemiluminescence reaction (Millipore, Schwalbach, Germany), and luminescence intensities were analyzed using the Chemidoc XRS imaging system (BioRad, Munich, Germany). With the Quantity One (BioRad) program, band density was determined for each lane and the intensity ratio for the detected proteins was calculated to α-tubulin.
Results
CXCR3-Deficient GF-IL12 Mice Only Rarely Develop Ataxia but Always Develop a Destructive Ocular Phenotype
To determine whether the clinical course of IL-12–driven CNS inflammation was altered in the absence of CXCR3 signaling, the physical status of a cohort of 13 GF-IL12 and 23 GF-IL12/CXCR3KO mice was observed for 36 weeks (Figure 1A). As described previously,25, 26 GF-IL12 mice developed a progressive ataxia at the age of 9 to 12 weeks. First clinical signs at that time point were belly dragging and the splaying of legs, resulting in 85% of GF-IL12 mice with an ataxia score of 1. At the same time point, only 9% of GF-IL12/CXCR3KO mice displayed any signs of ataxia. Progression of the ataxia in GF-IL12 mice could be observed, resulting in an ataxia score of 3 or 4 in 92% of the animals at 36 weeks. In contrast, only the GF-IL12/CXCR3KO mice (9%, 2/23) that displayed signs of ataxia at 12 weeks further progressed and developed severe ataxia at 36 weeks (GF-IL12/CXCR3KOsick). The remaining 91% of GF-IL12/CXCR3KO mice developed no signs of ataxia, demonstrating a strongly reduced incidence of the atactic phenotype in GF-IL12/CXCR3KO mice compared with GF-IL12 mice (P < 0.005).
Figure 1.
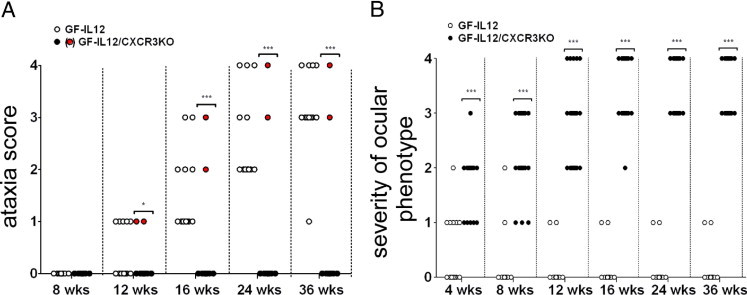
Progression of clinical ataxia (A) and the severity of ocular phenotype (B) in GF-IL12 (n = 13) and GF-IL12/CXCR3KO (n = 23) mice during 36 weeks. Ataxic signs were scored using an accumulative scale of four points, giving one point to each of these four physical signs: splayed legs, dragging weight on the trunk rather than on the legs, wobbling, and falling from side to side. The ocular score was applied to assess the severity of the ocular phenotype: 1 indicating monocular cataract; 2, binocular cataract; 3, monocular cataract and monocular phthisis bulbi; and 4, binocular phthisis bulbi. Although GF-IL12 mice developed severe ataxia during 24 weeks (black dots), most of the GF-IL12/CXCR3KO mice did not display signs of ataxia (circled dots). A few of the GF-IL12/CXCR3KO animals (GF-IL12/CXCR3KOsick; red dots) developed scores of ataxia comparable to GF-IL12 mice until 24 weeks. Surprisingly, during the first 4 weeks all GF-IL12/CXCR3KO mice developed a progressive disease of the eyes, ultimately leading to binocular bulbar atrophy after 36 weeks. The ocular phenotype of GF-IL12 mice at that age reached a maximum ocular score of 1. *P < 0.05; ***P < 0.005, mean ± SEM.
Unexpectedly, all GF-IL12/CXCR3KO mice (n = 23) developed mono-ocular or binocular cataracts during the first 4 weeks of life (ocular scores of 1 to 2, Figure 1B). In contrast, only 15% (2/13) of the GF-IL12 mice developed a very mild opacity of the lens (ocular score 1), which we did not observe in either WT or CXCR3KO controls. After 24 weeks all GF-IL12/CXCR3KO mice developed a bulbar atrophy (phthisis bulbi) and severe cataracts (ocular score of 3 to 4, Figure 1B), ultimately leading to blindness in these mice. None of the GF-IL12 mice developed these severe symptoms. The early lens opacity observed in some GF-IL12 animals (6/13) resolved in 66% of these animals, and in the remaining animals the ocular phenotype did not progress any further.
Less Severe Cerebellar Histopathologic Findings Correlate with the Attenuated Clinical Phenotype of CXCR3-Deficient GF-IL12 Mice
To correlate the clinical differences observed in GF-IL12 versus GF-IL12/CXCR3KO mice with histopathologic findings, we used routine histologic analysis and immunohistochemistry to examine WT, CXCR3KO, GF-IL12, nonatactic GF-IL12/CXCR3KO, and atactic GF-IL12/CXCR3KOsick mice at the age of 24 weeks. None of the WT or CXCR3KO animals showed any histopathologic changes in routine histologic findings. However, extensive tissue destruction, calcifications, and mononuclear cell accumulation in the parenchyma and meninges were observed in the brain of GF-IL12 mice and were consistent with previous reports.25, 26 Furthermore, widespread demyelination and spongiosis predominantly of the cerebellar white matter were observed (Figure 2A; GF-IL12, LFB).25, 26 In contrast, nonatactic GF-IL12/CXCR3KO mice had only minimal histologic alterations without demyelination and calcifications (Figure 2A; GF-IL12/CXCR3KO, LFB). Only the few GF-IL12/CXCR3KOsick mice with signs of ataxia (9% of all GF-IL12/CXCR3KO mice) displayed perivascular cell clustering and demyelination, without calcifications. However, these pathologic changes did not reach the level of severity observed in CXCR3-competent GF-IL12 mice (Figure 2A; GF-IL12, LFB). To further characterize the histopathologic differences, we examined accumulation and distribution of T cells (CD3) and microglia and macrophages (lectin) by immunohistochemistry. In CXCR3-competent GF-IL12 mice, T cells were found in a perivascular position and throughout the parenchyma (Figure 2A; GF-IL12, CD3). Activated microglia and macrophages were found in the parenchyma and correlated well with tissue destruction and demyelination after 24 weeks (Figure 2A; GF-IL12, lectin). In contrast, at this time point 91% of GF-IL12/CXCR3KO mice had only slightly increased numbers of CD3+ T cells, which were most often localized in a parenchymal perivascular position and not in the subarachnoidal space (Figure 2A; GF-IL12/CXCR3KO, CD3). T-cell–accompanying activated microglia and macrophages were drastically reduced in nonatactic CXCR3-deficient GF-IL12 mice (Figure 2A; GF-IL12/CXCR3KO, lectin). Only clinically affected GF-IL12/CXCR3KOsick mice showed perivascular infiltrates consisting of T cells surrounded by activated microglia and macrophages localized to the white matter of the cerebellum (Figure 2A; GF-IL12/CXCR3KOsick, CD3, lectin). Overall, these findings demonstrate that the less severe phenotype in GF-IL12/CXCR3KO mice is associated with a marked decrease in cerebellar inflammation.
Figure 2.
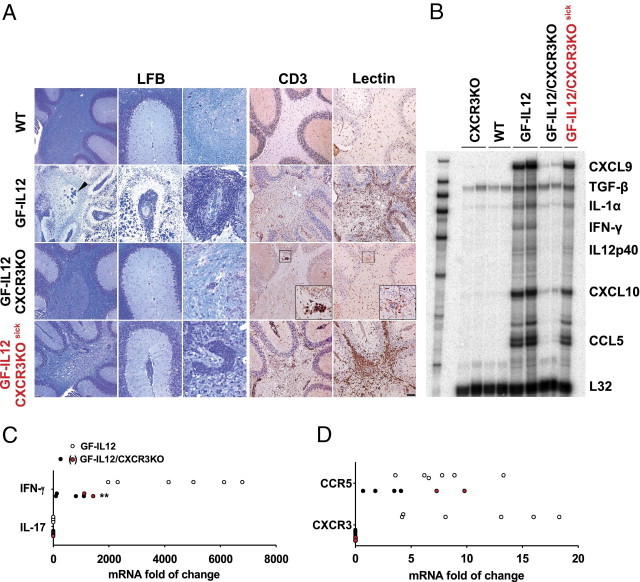
Histologic and molecular phenotype of CXCR3-deficient and CXCR3-competent cerebelli of GF-IL12 mice. A: Histologic alterations in the cerebelli of GF-IL12 and GF-IL12/CXCR3KO mice at 24 weeks of age. Colocalization of LFB routine histologic analysis, T cells (CD3), and microglia and macrophages (tomato lectin). Extensive cell loss in the granule layer (GF-IL12, LFB), white matter calcifications (GF-IL12, LFB, arrowhead), and demyelination were detectable in all GF-IL12 animals. In contrast, most GF-IL12/CXCR3KO mice had none or only minimal histopathologic alterations (GF-IL12/CXCR3KO, LFB). A few GF-IL12/CXCR3KO mice with moderate signs of ataxia displayed histopathologic alterations (GF-IL12/CXCR3KOsick, LFB), which did not reach the level of tissue disruption in GF-IL12 mice. Meningeal and perivascular (GF-IL12, GF-IL12/CXCR3KO, GF-IL12/CXCR3KOsick, LFB) infiltrates were drastically reduced in GF-IL12/CXCR3KO mice compared with GF-IL12 mice. Widespread distribution and perivascular accumulation of T cells (GF-IL12, GF-IL12/CXCR3KOsick, CD3) are accompanied by diffuse microglia and macrophage activation in the white matter (GF-IL12, GF-IL12/CXCR3KOsick, lectin) of GF-IL12 mice. Most GF-IL12/CXCR3KO displayed only minor T-cell infiltration and less activated microglia and macrophages (GF-IL12/CXCR3KO, CD3). Original magnification, ×20. Scale bars: 250 μm (overview of cerebellum) and 100 μm (meningeal/vascular focus). B: The mRNA expression pattern of selected cytokine and chemokines in the cerebellum of GF-IL12, GF-IL12/CXCR3KO, and control mice detected by RPA. Total RNA was isolated from the cerebelli of mice and 5 μg was used for analysis by RPA. The level of L32 was used as the loading control. An overall increase of proinflammatory cytokine and chemokine transcripts was found in GF-IL12 and GF-IL12/CXCR3KOsick mice with the exception of a low IFN-γ level in GF-IL12/CXCR3KOsick mice. C: Real-time quantitative PCR was performed to further determine the level of IFN-γ mRNA in the cerebelli of aged GF-IL12 (n = 6) and GF-IL12/CXCR3KO (n = 6) mice (left). Compared with GF-IL12 mice, the GF-IL12/CXCR3KO mice had significantly lower levels of IFN-γ RNA transcripts at 24 weeks of age. D: Further RNA analysis revealed high levels of cerebellar CXCR3 and CCR5 transcripts in GF-IL12 animals after 24 weeks (right). In contrast only the diseased GF-IL12/CXCR3KOsick mice (red dots) revealed elevated levels of CCR5 receptor transcripts in the cerebellum. For statistical significance, **P < 0.01, mean ± SEM.
Cerebellar Cytokine Profile Correlates with the Histopathologic Findings and the Clinical Outcome in GF-IL12 and GF-IL12/CXCR3KO Mice
We wanted to determine whether the different cerebral disease phenotypes in GF-IL12/CXCR3KO and GF-IL12 were linked with a specific cytokine RNA pattern. Therefore, the mRNA transcripts of key inflammatory cytokine and chemokine genes in the cerebellum of GF-IL12 versus GF-IL12/CXCR3KO mice were determined by RPA (Figure 2B).
In the cerebellum of WT and CXCR3KO mice, only TGF-β could be detected unequivocally. Consistent with the clinical ataxia and the described histopathologic features, induction of all examined cytokine and chemokine genes was prominent in GF-IL12 mice at 24 weeks (Figure 2B; GF-IL12). In GF-IL12/CXCR3KO mice with a clinical score of 0 to 1 only and without prominent histologic damage, only low levels of CXCL9, IL-1, CXCL10, and CCL5 could be detected. In the GF-IL12/CXCR3KOsick mice, we observed a similar pattern to GF-IL12 mice with an induction of CXCL9, IL-1, CXCL10, and CCL5. However, the RNA level for IFN-γ and for CXCL9 and CXCL10 were considerably lower. Because IFN-γ is a key cytokine in this model, we additionally measured RNA levels of IFN-γ by quantitative PCR and found significantly higher levels in GF-IL12 mice (P = 0.0015, 4397-fold ± 804-fold increase) compared with GF-IL12/CXCR3KO mice (782-fold ± 227-fold increase) (Figure 2C). Within the GF-IL12/CXCR3KO group, the animals with clinical symptoms (GF-IL12/CXCR3KOsick, red dots) had the highest levels of IFN-γ RNA. To evaluate a possible induction of TH17 subset effector cells in parallel to the predominant TH1-driven immune response observed in this model, we examined the level of IL-17 transcripts in both GF-IL12 and GF-IL12/CXCR3KO cerebelli (Figure 2C). The IL-17 transcript levels in GF-IL12, GF-IL12/CXCR3KO, and CXCR3 in IL-12-Mediated CNS Immunity IL12/CXCR3KOsick mice were unaltered compared with WT animals. In summary, the RNA levels of various inflammatory cytokines correlated well with the clinical and histopathologic observations. The IFN-γ RNA level was markedly reduced, even in GF-IL12/CXCR3KOsick mice with clinical symptoms.
CXCR3 and CCR5 mRNA Expression in GF-IL12–Mediated Cerebellar Inflammation
CCL5 and CCR5 are, like CXCR3 and its ligands, involved in the trafficking of type 1 immune cells.17, 31, 32 The RPA results revealed a prominent up-regulation of CCL5 in clinically affected GF-IL12/CXCR3KOsick but not in unaffected GF-IL12/CXCR3KO mice. To determine whether CCL5 might compensate for the lack of CXCR3, we further examined and compared cerebellar CXCR3 and CCR5 RNA levels by real-time quantitative PCR in GF-IL12, unaffected GF-IL12/CXCR3KO, and clinically affected GF-IL12/CXCR3KOsick mice. The mRNA levels for each transcript were normalized to the mRNA levels of GAPDH and correlated with the level of control mice. We found increased levels of CXCR3 (10.7-fold ± 2.5-fold increase) and CCR5 (7.7-fold ± 1.3-fold increase) transcripts, suggesting a functional role of both CXCR3+ and CCR5+ cells. Significantly lower levels of CCR5 transcripts were found in unaffected GF-IL12/CXCR3KO animals (P < 0.01, 2.5-fold ± 0.8-fold increase). In clinically affected GF-IL12/CXCR3KOsick mice, we found CCR5 RNA at a level (8.6-fold ± 1.3-fold increase) comparable to the findings in GF-IL12 mice (Figure 2D, right). The high level of CCL5 and CCR5 in GF-IL12/CXCR3KOsick mice but not in clinically unaffected GF-IL12/CXCR3KO mice could argue for a compensation of the CXCR3 deficiency by CCR5 in GF-IL12/CXCR3KOsick mice.
Cerebellar Leukocyte Recruitment in GF-IL12/CXCR3KO Mice Is Reduced Early and Is Associated with Lower Levels of IFN-γ mRNA
To further examine the clinical and histopathologic differences observed at a late time point, we performed flow cytofluorometric analysis from cerebellar cell suspensions at the age of 8 weeks when both GF-IL12 versus GF-IL12/CXCR3KO mice did not show any clinical symptoms (Figure 3A). At that time point, the total number of CD45+ leukocytes were already increased in GF-IL12 mice compared with GF-IL12/CXCR3KO animals (243,250 ± 62,268 cells in GF-IL12 mice versus 102,067 ± 20,972 cells in GF-IL12/CXCR3KO mice) (Figure 3A). However, the relative proportion of T-cell subsets (CD4+ versus CD8+ T cells) remained similar between the two strains of transgenic mice (66.4% ± 13.3% versus 21.7% ± 0.4% in GF-IL12 mice and 59.1% ± 9.2% versus 29.2% ± 6.5% in GF-IL12/CXCR3KO mice). Further characterization of the CD45+ cells revealed an increase of CD45+/NK-1.1+ cells in GF-IL12 compared with GF-IL12/CXCR3KO mice (15.2% ± 4.9% in GF-IL12 mice versus 6.7% ± 4.0% in GF-IL12/CXCR3KO mice), resulting in a more than fivefold increase in the absolute cell counts in GF-IL12 cerebelli. An approximate fourfold increase in CD45+/CD11b+ macrophages isolated from GF-IL12 transgenic animals could be observed over that found in GF-IL12/CXCR3KO mice but with similar frequency of CD45+/CD11b+ cells (21.8% ± 12.5% in GF-IL12 versus 13.4% ± 2.5% in GF-IL12/CXCR3KO). Furthermore, real-time quantitative PCR revealed significantly higher IFN-γ transcript levels in the cerebelli of GF-IL12 mice than in GF-IL12/CXCR3KO animals (Figure 3B). In summary, cellular infiltrates and IFN-γ RNA level are increased early in GF-IL12 mice compared with GF-IL12/CXCR3KO animals, arguing for an early role of CXCR3 in cerebellar leukocyte accumulation and IFN-γ induction in the GF-IL12 mice.
Figure 3.
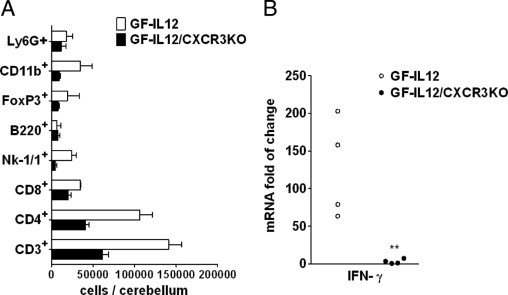
Specific leukocyte subsets and IFN-γ RNA are reduced in the cerebelli of GF-IL12/CXCR3KO mice at an early time point. A: Flow cytometric analysis of leukocyte subsets in the cerebelli of GF-IL12 and GF-IL12/CXCR3KO mice at the age of 8 weeks. Cerebellar tissue leukocytes were isolated and analyzed as described in Materials and Methods. A strong decrease of CD4+, Nk-1/1+, and CD45+/CD11b+ cells was found in GF-IL12/CXCR3KO mice compared with GF-IL12 animals. B: Low levels of IFN-γ transcripts (real-time quantitative PCR detection) at this early time point correlated with FACS analysis findings of diminished counts of NK and T cells found in cerebellar tissue of GF-IL12/CXCR3KO compared with GF-IL12 animals.
CXCR3KO Mice Develop a Progressive and Destructive Inflammation of the Eyes, Ultimately Leading to Bulbar Atrophy and Blindness
Macroscopic examination of the eyes of GF-IL12/CXCR3KO mice revealed a severe ocular atrophy (phthisis bulbi) at 24 weeks (Figure 4M). None of the GF-IL12 mice developed this ocular phenotype (Figure 4I); eyes from GF-IL12 mice were indistinguishable from WT or CXCR3KO mice at that time point (Figure 4, A and E). Next, we performed routine histologic staining on mice at 4, 8, and 24 weeks of age and immunohistochemistry at 8 weeks to further examine the histopathologic findings of the eye phenotype. The eyes of all genotypes were examined (each n = 6). Routine H&E staining at 4 weeks revealed that none of the genotypes displayed any pathological alterations, which ruled out developmental abnormalities (Figure 4, B, F, J, and N). At the age of 8 weeks, lens damage and changes in the retinal architecture with injury or elimination of photoreceptors inner and outer segments were found in GF-IL12/CXCR3KO mice (Figure 4O). At 24 weeks, GF-IL12/CXCR3KO mice displayed a severe retinopathy, a disrupted pigment epithelium, and destruction of the lens architecture (Figure 4P). In contrast, GF-IL12 animals did not display any major histopathologic alterations of the eye at 8 or 24 weeks (Figure 4, K and L) and neither did the WT or CXCR3 control mice (Figure 4, C, D, G, and H).
Figure 4.
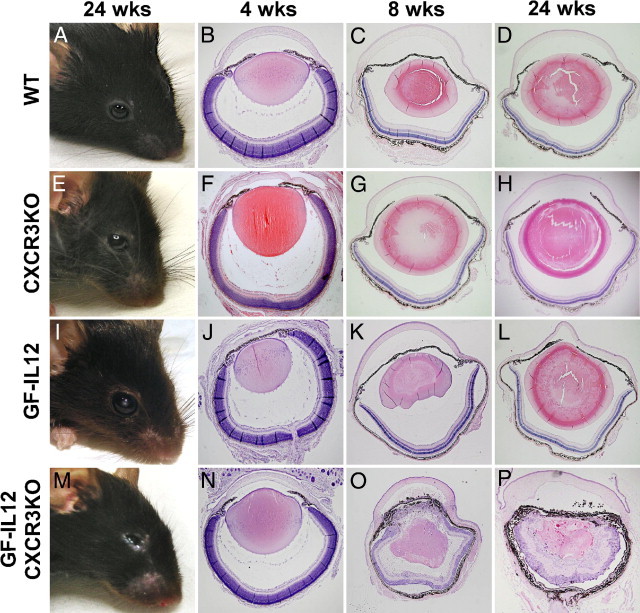
Progressive postnatal ocular inflammation of GF-IL12/CXCR3KO mice, ultimately leading to bulbar atrophy and destruction. Macroscopic ocular phenotype from WT, CXCR3KO, GF-IL12, and GF-IL12/CXCR3KO mice at the age 24 weeks (A, E, I, and M) and H&E-stained ocular sections at the age of 4, 8, and 24 weeks (B—D, F–H, J–L, and N–P). Sections of the eyes at the age of 4 weeks appeared normal (B, F, J, and N) in all genotypes, thereby ruling out developmental abnormalities in the mutant strains. At 8 weeks of age disorganization and loss of photoreceptors in the retina of CXCR3-deficient GF-IL12 mice and with cellular infiltrates in the peripheral cornea, iris, and ciliary body and pigment loaded cells in the anterior chamber (O). After 24 weeks of age the whole structure of the retinal layers and the lens were disrupted or destroyed in the eyes of GF-IL12/CXCR3KO mice (P). WT, CXCR3KO, and GF-IL12 mouse eyes or retinas without alterations observable in the overview of all ages (B–D, F–H, and J–L).
Using immunofluorescence staining of sagittal horizontal sections of eyes, we further characterized the histopathologic features of the ocular destruction in GF-IL12/CXCR3KO mice at 8 weeks of age. We found high counts of CD3+ T cells throughout the eye (204 ± 23 per section of the eye; n = 5 sections) but in particular an accumulation in the ganglion cell layer of the retina (Figure 5M). At this stage T-cell accumulation was closely associated with Iba1+ microglial and macrophage infiltration of the retinal ganglion cell layer, widely found in proximity to the retinal pigment epithelium (Figure 5O) and anterior chamber (data not shown). GF-IL12 mice had only minor ocular CD3+ T-cell accumulation (Figure 5I; <10 ± 6 per section of the eye; n = 8 sections). At 8 weeks we found Iba1+ microglia and macrophages in the GF-IL12 ganglion cell layer and between the inner and outer nuclear layer of retina (Figure 5K). No Iba1+ cells were observable in the intact layer, where one can find the inner and outer segments of rod and cone photoreceptors. Staining of the vasculature with laminin revealed an increased amount of vessels in the inner plexiform and the granule layers of GF-IL12/CXCR3KO mice but not GF-IL12 mice (Figure 5, L and P). In addition, Müller cells, the retinal astrocytes, were found highly activated in terms of morphology and GFAP levels in GF-IL12/CXCR3KO mice (Figure 5N). None of the examined WT and CXCR3KO eyes showed any histopathologic features in respect to CD3 (Figure 5, A and E), GFAP (Figure 5, B and F), Iba (Figure 5, C and G), or laminin (Figure 5, D and H) immunohistochemistry.
Figure 5.
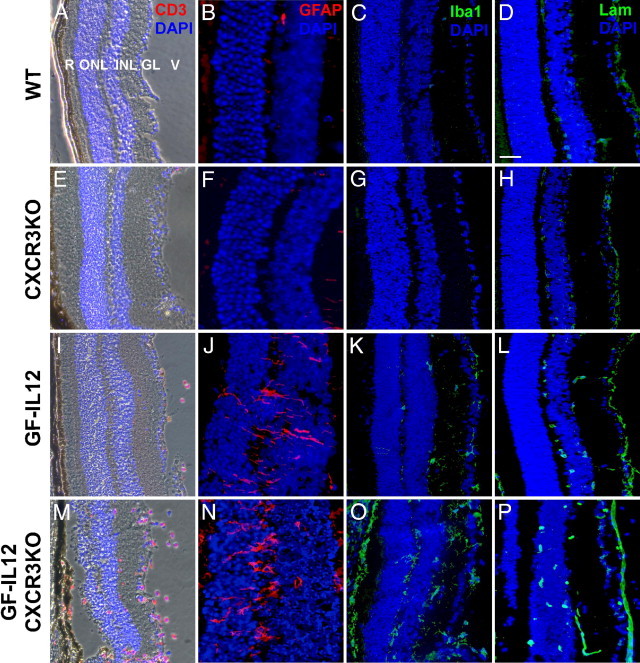
T-cell infiltration, glial activation, and vessel formation characterize the early ocular histopathologic findings in GF-IL12/CXCR3KO mice. Combined immunofluorescence staining for T cells (CD3)/phase contrast microscopy (A, E, I, M) and single immunofluorescence staining of the retina of examined genotypes (B–D, F–H, J–L, N–P); GFAP to detect Müller cells; Iba-1 to detect microglia and macrophages; and laminin to detect blood vessels on serial sections. GF-IL12/CXCR3KO but not GF-IL12 mice at 8 weeks revealed massive CD3+ T-cell accumulation and destruction of the retinal architecture (V, vitreous; GC, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; R, rod & cones outer segments) with loss of the photoreceptors inner and outer segments (I and M). In contrast to only minor changes in the eyes from GF-IL12 mice, pathological neovascularization in the inner plexiform layer and the granule layer (L and P; Lam) was observed in the retina of GF-IL12/CXCR3KO mice. Retinal microglia and macrophages were detectable in GF-IL12 and GF-IL12/CXCR3KO genotypes, but Iba-1 positive cells within the photoreceptor layer were only detected in the GF-IL12/CXCR3KO genotype (K and O; Iba-1). In addition, Müller cells in GF-IL12/CXCR3KO mice were highly activated in terms of morphology and GFAP level (J and N). Nuclear counterstaining with DAPI (blue signal). Scale bars: 50 μm (A, C–E, G–I, K–M, O, and P) and 25 μm (B, F, J, and N).
Increase of Specific Leukocyte Subsets in the Eyes of GF-IL12/CXCR3KO Mice
To characterize the composition and numbers of immune cells infiltrating the eyes of GF-IL12/CXCR3KO and GF-IL12 animals at 8 weeks, flow cytometric analysis was performed (Figure 6A). Corresponding with the histologic findings of high numbers of CD3+ cells in the eyes of GF-IL12/CXCR3KO mice, an increase in CD3+CD4+ and CD3+CD8+ T cells was observed by FACS compared with the GF-IL12 genotype (5299 ± 93 CD4+ T cells and 1606 ± 319 CD8+ T cells in GF-IL12/CXCR3KO versus 1224 ± 232 CD4+ T cells and 257 ± 176 CD8+ T cells in GF-IL12). Furthermore, the number and relative proportion of Ly6G+ neutrophil granulocytes were found to be markedly elevated in the GF-IL12/CXR3KO compared with the GF-IL12 eyes (4.6% ± 0.3% in GF-IL12 mice versus 20.7% ± 3.2% in GF-IL12/CXCR3KO mice). However, the percentage of CD4+ (79.0% ± 15.0% in GF-IL12 mice versus 74.5% ± 1.3% in GF-IL12/CXCR3KO mice), CD8+ (16.6% ± 11.3% in GF-IL12 mice versus 22.6% ± 4.5% in GF-IL12/CXCR3KO mice), NK-1.1+ (6.6% ± 3.0% in GF-IL12 mice versus 9.2% ± 0.8% in GF-IL12/CXCR3KO mice), and CD11b+ (13.6% ± 0.0% in GF-IL12 mice versus 14.2% ± 4.7% in GF-Il12/CXCR3KO mice) cells remained similar between the two strains. These findings further demonstrate the inflammatory response in the ocular phenotype observed in GF-IL12/CXCR3KO mice. When comparing the cerebellar leukocyte subsets (Figure 3A) with the ocular leukocyte subsets (Figure 6A) of individual mice, we could not detect any relation between the amount of specific inflammatory cell population in these organs (data not shown).
Figure 6.
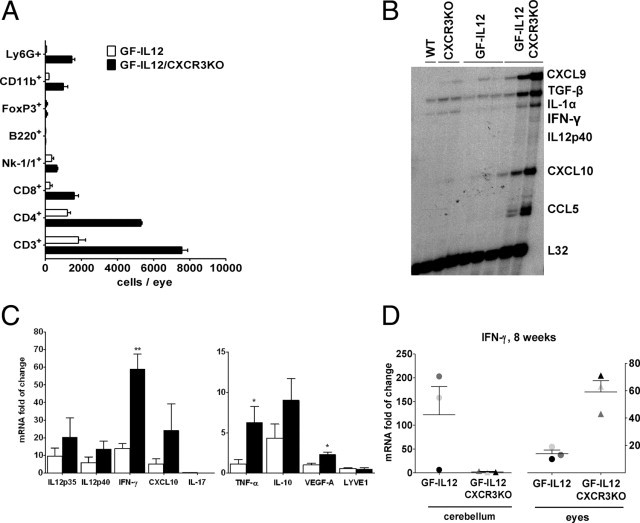
Ocular leukocyte infiltration is related to the induction of various inflammatory gene transcripts in GF-IL12/CXCR3KO mice. A: Markedly increased accumulation of various leukocyte subsets in the eyes of GF-IL12/CXCRKO mice at the age of 8 weeks. Ocular leukocytes were isolated and analyzed by cytometry as described in Materials and Methods. Ocular cell counts of CD4+, CD8+, CD45+ CD11b+, CD45+ Ly6G+, CD4+ FoxP3+, and CD19+ B220+ cell populations in GF-IL12 and GF-IL12/CXCR3KO mice. B: Increased levels of cytokine and chemokine gene transcripts in the eyes of GF-IL12/CXCR3KO compared with GF-IL12 mice. C: Real-time quantitative PCR was performed to determine the ocular gene expression level of various proinflammatory transcripts in GF-IL12 and GF-IL12/CXCR3KO mice. D: IFN-γ transcript analysis in the eyes and cerebelli of individual animals (each symbol represents eyes or cerebelli of one analyzed animal). For statistical significance, *P < 0.05; **P < 0.01, mean ± SEM.
Induction of Inflammation-Related Genes in the Eyes of GF-IL12/CXCR3KO Mice
To examine the RNA levels of key inflammatory cytokines in the eye of GF-IL12 vs GF-IL12/CXCR3KO mice, we performed RPA with total RNA from all analyzed genotypes (Figure 6B). Correlating with the histopathologic changes and inflammatory cells accumulated in the eye at 8 weeks, we found highly increased RNA levels of all examined cytokines, including IFN-γ, CXCL9, CXCL10, and CCL5 (Figure 6B).
To further characterize the ocular inflammatory response in GF-IL12/CXCR3KO mice, we used real-time quantitative PCR to confirm the RPA results and to compare RNA levels from selected key cytokines, chemokines (IL-12p40/35, IFN-γ, CXCL10, IL-17, TNF-α, and IL-10), or angiogenesis and lymphangiogenesis markers (VEGF-A and LYVE1) from the eyes of 8-week-old animals (Figure 6C). Fold increase was calculated with respect to expression of the respective target in the CXCR3KO and WT mice. No significant differences were detected between CXCR3KO and WT mice.
We found significantly higher IFN-γ mRNA levels in the eyes of GF-IL12/CXCR3KO (P < 0.01; 59.0 ± 8.3) versus GF-IL12 (14.0 ± 2.7) mice (Figure 6C). However, analysis of the cerebelli and eyes of individual mice at 8 weeks revealed no correlation between cerebellar and ocular IFN-γ mRNA levels (Figure 6D). The IFN-γ–inducible chemokine CXCL10 mRNA was induced in eyes of GF-IL12/CXCR3KO (24.2 ± 14.9) versus GF-IL12 (5.0 ± 3.1) mice. However, similar to the cerebellum (see above) no significant induction of IL-17 mRNA was found in the eyes of any genotypes. Next, the mRNA level in the eyes for the proinflammatory cytokine TNF-α was found to be significantly increased in GF-IL12/CXCR3KO mice (P < 0.05; 6.3 ± 2.0) versus GF-IL12 (1.1 ± 0.6) mice. Because of the extensive vascularization observed in the eyes of GF-IL12/CXCR3KO mice, we examined the expression of the VEGFA gene. VEGF induces microvascular permeability and plays a central role in both angiogenesis and vasculogenesis.33 VEGF-A RNA was found at significantly higher levels in the eye of GF-IL12/CXCR3KO (P < 0.05; 2.3 ± 0.3) versus GF-IL12 (1.0 ± 0.2) (n = 3, mean ± SEM; Figure 6C) mice. To further evaluate whether increased vessel formation was also accompanied by an increase in lymphangiogenesis in the eye, we examined LYVE1 transcripts as a lymphatic endothelial cell marker but did not find any significant difference between GF-IL12/CXCR3KO and GF-IL12 mice. In summary, the expression of inflammatory genes is highly up-regulated in the eyes of GF-IL12/CXCR3KO mice compared with GF-IL12 mice, which further provides evidence for an inflammatory-driven pathogenesis, leading to the severe destruction of eyes in GF-IL12/CXCR3KO mice.
IL12rb1 mRNA Levels and STAT4 Phosphorylation Is Increased in the Cerebellum of GF-IL12 Mice and Eyes of GF-IL12/CXCR3KO Mice
To evaluate possible differences in the cerebellar and ocular tissue response to IL-12, we examined the RNA levels of IL12rb1 by quantitative PCR and STAT4 phosphorylation using immunoblot detection. Corresponding with the described inflammatory response, we observed higher RNA levels of IL12rb1 in cerebelli of GF-IL12 mice compared with GF-IL12/CXCR3KO mice. Conversely, ocular IL12rb1 was higher in GF-IL12/CXCR3KO mice compared with GF-IL12 mice (Figure 7, C and F). Densitometric analysis of immunoblots (Figure 7, A, B, D, and E) revealed significantly higher protein levels of total and phosphorylated STAT4 protein levels in the cerebelli of GF-IL12 mice versus GF-Il12/CXCR3KO mice (total STAT4, 1.65 ± 0.08 versus 0.96 ± 0.06; phosphorylated STAT4, 0.81 ± 0.07 versus 0.27 ± 0.02; Figure 7, A and B). The opposite was observed in the examined eyes: lower levels of total and phosphorylated STAT4 were found in the eyes of GF-Il12 mice compared with GF-IL12/CXCR3KO mice (total STAT4, 1.94 ± 0.14 versus 2.76 ± 0.06; phosphorylated STAT4, 0.91 ± 0.09 versus 1.97 ± 0.02; Figure 7, D and E).
Figure 7.
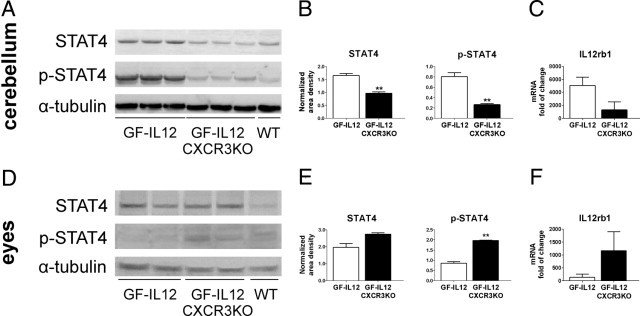
Increased levels of STAT4, phosphorylated STAT4, and IL12rb1 transcripts in GF-IL12GF cerebelli and GF-IL12/CXCR3KO eyes (n = 3). Immunoblot (A and D) and densitometric analysis (B and E) of the cerebellar and ocular STAT4 and phospho-STAT4 level in GF-IL12 and GF-IL12/CXCR3KO mice were performed as described in Materials and Methods. mRNA levels (C and F) of IL12rb1 in cerebellum and eyes of GF-IL12 mice compared with GF-IL12/CXCR3KO mice were assessed by quantitative PCR. For statistical significance, **P < 0.01, mean ± SEM.
Discussion
CXCR3 and its ligands CXCL9, CXCL10, and CXCL11 are implicated in the pathogenesis of many neuroinflammatory diseases.18, 34, 35 However, functional studies examining the role of CXCR3 and its ligands in neuroinflammatory disease models have led to conflicting results without clearly defining the role of the CXCR3 chemokine system in inflammatory CNS diseases.6, 36 In particular, studies of EAE did not reveal the expected disease-promoting effect of CXCR3 but rather protective and disease-limiting functions.24, 37 We examined the role of CXCR3 in a less complex TH1-mediated model of spontaneous CNS inflammation in transgenic mice with astrocyte-targeted production of IL-12. Within the CNS of these mice with CXCR3 deficiency, we observed a markedly attenuated inflammatory response, which corresponded with the well-characterized impact of the CXCR3 chemokine system on the migration and attraction of type 1 immune cells.38, 39 Unexpectedly, these same animals developed a very severe inflammatory disease of the eyes, demonstrating that the effect of CXCR3 in neuroinflammation is not only dependent on the underlying cause but also strongly influenced by the site of the inflammation.
Neuroinflammation in GF-IL12 transgenic mice is well characterized. Astrocytic IL-12 activates CD4+ and CD8+ T cells and NK cells, all of which produce proinflammatory cytokines, including IFN-γ and the CXCR3 ligands CXCL9 and CXCL10.40 An IFN-γ–dependent immune response further increases the local accumulation of activated immune cells and mediates the tissue damage of the CNS.41, 42, 43, 44 Phenotypically, these GF-IL12 transgenic mice develop a severe cerebellitis and, correspondingly, progressive ataxia.25 However, most GF-IL12/CXCR3KO mice did not develop a clinical phenotype and had only minor histopathologic alterations of the cerebellum. This finding argues for a strong disease-promoting function of the CXCR3 chemokine system in IL-12–induced neuroinflammation in the GFAP-IL12 model and is in contrast to previous findings in EAE, where CXCR3 has a disease-limiting function independent of the recruitment of effector T cells to the CNS.24, 37
The gross reduction of leukocytes in the parenchyma and subarachnoid space of GF-IL12/CXCR3KO mice provides evidence of a functional role of CXCR3 in mediating the leukocyte accumulation in the CNS as a result of IL-12 overproduction. However, CXCR3 deficiency did not completely prevent cerebellar inflammation in all GF-IL12/CXCR3KO mice. A low percentage of GF-IL12/CXCR3KO mice developed clinical signs of ataxia and histopathologic alterations, which still were less prominent than observed in CXCR3 competent GF-IL12 mice. Because CCL5 and CCR5 are highly up-regulated in GF-IL12/CXCR3KOsick mice, this chemokine system may possibly have compensated for the CXCR3 deficiency in GF-IL12/CXCR3KO mice that develop clinical signs.
To define early differences between GF-IL12 and GF-IL12/CXCR3KO mice in the inflammatory response, we examined animals of each genotype at the age of 8 weeks, where clinical signs were not yet observable. FACS analysis revealed substantial differences in the amount and composition of immune cells in cerebelli from GF-IL12 and GF-IL12/CXCR3KO mice at that early time point. We observed not only a difference in T-cell subsets but also a marked reduction of CD11b+ microglia and macrophages and NK cells in GF-IL12/CXCR3KO mice. This finding demonstrates that CXCR3 is involved not only in the accumulation of T cells in this model but also, directly or indirectly, in the accumulation of macrophages and NK cells at an early time point. The observation that CXCR3 and its ligands modulate the CNS accumulation of a variety of immune cells, and not only T cells, is further supported by previous observations in transgenic mice with long-term astrocyte-targeted production of the CXCR3 ligand CXCL10. These mice develop subarachnoidal cellular infiltrates consisting not only of T cells but also of macrophages and neutrophils.45
Surprisingly, all GF-IL12/CXCR3KO mice developed a severe and progressive ocular phenotype with severe chorioretinitis. In GF-IL12 mice, only mild lens opacities were observed in 50% of the animals. As judged from histologic analysis, prominent T-cell and neutrophil accumulation in the eyes of GF-IL12/CXCR3KO mice resulted in a severe disruption of the internal structures of the eye and the loss of the photoreceptor outer segments. FACS analysis revealed a substantial accumulation of both CD4+ and CD8+ T cells, in particular CD4+ T cells, NK cells, CD11b+, and neutrophils. Although the total number of T cells was drastically increased, we did not detect an increase of FoxP3+ regulatory T cells, suggesting a role for CXCR3 in directing and accumulating regulatory T cells into the eye. Proinflammatory cytokines, such as IFN-γ, TNF-α, and VEGF, were highly up-regulated in the eyes of GF-IL12/CXCR3KO mice, which further underlines the inflammatory nature of the ocular phenotype. A similar ocular condition was observed in a previous study examining a transgenic mouse line using the rhodopsin promoter to direct the expression of IFN-γ to photoreceptor cells (rhoγ mice).46 It is therefore likely that the ocular condition is driven by the production of IFN-γ from IL-12 activated T cells or NK cells. This assumption is supported by the increased levels of phosphorylated STAT4 in the eyes of GF-IL12/CXCR3KO mice. Because GF-IL12 animals do not develop a severe ocular phenotype and have only slightly increased levels of IFN-γ, it is likely that CXCR3 is, indirectly or directly, restricting the activation of immune cells by IL-12, which in turn prevents the inflammatory cascade, ultimately leading to the accumulation of leukocytes and the ocular destruction observed in GF-IL12/CXCR3KO mice.
This finding is contrary to other experimental studies, which suggest that CXCR3 increases the production of IFN-γ by T cells in the CNS.37, 47 An approach to explain the different impact of CXCR3 deficiency for the brain and the eye could be differences in the local immune milieu. It is known that the eye has an immune-suppressive milieu in which factors such as high constitutional levels of TGF-β and the induction of high levels of IL-10 during an immune response are able to prevent a potentially harmful TH1 response.48 Although the brain also displays properties of a so-called immune privileged micromilieu, milieu differences between the brain and eye could contribute to the different functional properties of CXCR3, leading to enhanced ocular but attenuated cerebellar inflammation in our model.49 Future in vitro studies examining the functional properties of CXCR3+ immune cells in the presence or absence of a specific cytokine milieu, including TGF-β and IL-10, could further help to clarify our observations.
The ocular inflammation in GF-IL12/CXCR3KO mice was accompanied by increased vessel formation in the retina. The finding of neovascularization in the retina points toward an angiostatic role of CXCR3 and its ligands in ocular inflammation, which has been described for CXCR3 in many other models of inflammation.50 Retinal neovascularization is not a feature commonly observed in animal models of ocular inflammation.46 We therefore suggest that the observed neovascularization is likely linked to the CXCR3 deficiency in our model.
Our data delineate that CXCR3 is important to initiate and maintain an IL-12–driven inflammation in the brain. CXCR3 deficiency drastically reduces the incidence of IL-12–induced inflammation of the cerebellum. However, the protective effect of CXCR3 deficiency in IL-12–driven inflammation is not as complete as previously observed for IFN-γ deficiency.26 Some GF-IL12 animals developed cerebral inflammation despite CXCR3 deficiency. This observation suggests that other molecules may compensate for the loss of CXCR3 and mediate the IL-12–induced inflammation even in the absence of CXCR3. Our data suggest that CCL5 and the corresponding receptor CCR5 might be candidates in this role because we found high levels of both CCL5 and CCR5 RNA in affected GF-IL12/CXCR3KO mice.
However, in contrast to the cerebellar effects, CXCR3 deficiency led to a destructive inflammatory phenotype of the eyes, arguing for a protective role of CXCR3 in IL-12–induced ocular inflammation and for micromilieu-dependent functional properties of CXCR3, which remain to be further defined by future studies. Our data support the perspective that CXCR3 can have both striking protective and harmful functions in CNS and ocular inflammation and that this effect does not only depend on the trigger as suggested by previous studies but likely also on the micromilieu of the affected organ. CXCR3 is a potential therapeutic target, but our data further underline the functional complexity of this chemokine system, which has to be better defined in future studies. Until then, our findings caution against the therapeutic targeting of CXCR3.6
Acknowledgments
We thank Jane Radford (University of Sydney, NSW, Australia) for expert technical assistance.
Footnotes
Supported by US Public Health Service NIH grant NS044905 and a startup grant from the University of Sydney to I.L.C. S.L.E. was supported by an Endeavor International Postgraduate Research Scholarship and International Postgraduate Award from the University of Sydney. M.M. was a postdoctoral fellow from the Deutsche Forschungsgemeinschaft (Mu17-07/3-1) and was also supported by the fund Innovative Medical Research of the University of Münster Medical School, Münster, Germany.
I.L.C. and M.M. contributed equally to this study.
References
- 1.Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol. 2009;9:429–439. doi: 10.1038/nri2565. [DOI] [PubMed] [Google Scholar]
- 2.Conrady C.D., Drevets D.A., Carr D.J.J. Herpes simplex type I (HSV-1) infection of the nervous system: is an immune response a good thing? J Neuroimmunol. 2010;220:1–9. doi: 10.1016/j.jneuroim.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gandhi R., Laroni A., Weiner H.L. Role of the innate immune system in the pathogenesis of multiple sclerosis. J Neuroimmunol. 2010;221:7–14. doi: 10.1016/j.jneuroim.2009.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Charo I.F., Ransohoff R.M. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 5.Cardona A.E., Li M., Liu L., Savarin C., Ransohoff R.M. Chemokines in and out of the central nervous system: much more than chemotaxis and inflammation. J Leukoc Biol. 2008;84:587–594. doi: 10.1189/jlb.1107763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Müller M., Carter S., Hofer M.J., Campbell I.L. Review: the chemokine receptor CXCR3 and its ligands CXCL9: CXCL10 and CXCL11 in neuroimmunity: a tale of conflict and conundrum. Neuropathol Appl Neurobiol. 2010;36:368–387. doi: 10.1111/j.1365-2990.2010.01089.x. [DOI] [PubMed] [Google Scholar]
- 7.Liu L., Callahan M.K., Huang D.R., Ransohoff R.M. Chemokine receptor CXCR3: an unexpected enigma. Curr Top Dev Biol. 2005;68:149. doi: 10.1016/S0070-2153(05)68006-4. [DOI] [PubMed] [Google Scholar]
- 8.Biber K., Dijkstra I., Trebst C., De Groot C.J.A., Ransohoff R.M., Boddeke H. Functional expression of CXCR3 in cultured mouse and human astrocytes and microglia. Neurosci. 2002;112:487–497. doi: 10.1016/s0306-4522(02)00114-8. [DOI] [PubMed] [Google Scholar]
- 9.Rappert A., Bechmann I., Pivneva T., Mahlo J., Biber K., Nolte C., Kovac A.D., Gerard C., Boddeke H.W., Nitsch R., Kettenmann H. CXCR3-dependent microglial recruitment is essential for dendrite loss after brain lesion. J Neurosci. 2004;24:8500. doi: 10.1523/JNEUROSCI.2451-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cella M., Jarrossay D., Facchetti F., Alebardi O., Nakajima H., Lanzavecchia A., Colonna M. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med. 1999;5:919–923. doi: 10.1038/11360. [DOI] [PubMed] [Google Scholar]
- 11.Weng Y., Siciliano S.J., Waldburger K.E., Sirotina-Meisher A., Staruch M.J., Daugherty B.L., Gould S.L., Springer M.S., DeMartino J.A. Binding and functional properties of recombinant and endogenous CXCR3 chemokine receptors. J Biol Chem. 1998;273:18288–18291. doi: 10.1074/jbc.273.29.18288. [DOI] [PubMed] [Google Scholar]
- 12.Loetscher M., Loetscher P., Brass N., Meese E., Moser B. Lymphocyte-specific chemokine receptor CXCR3: regulation, chemokine binding and gene localization. Eur J Immunol. 1998;28:3696–3705. doi: 10.1002/(SICI)1521-4141(199811)28:11<3696::AID-IMMU3696>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 13.Cole K.E., Strick C.A., Paradis T.J., Ogborne K.T., Loetscher M., Gladue R.P., Lin W., Boyd J.G., Moser B., Wood D.E., Sahagan B.G., Neote K. Interferon-inducible T cell alpha chemoattractant (I-TAC): a novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J Exp Med. 1998;187:2009–2021. doi: 10.1084/jem.187.12.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferber I.A., Brocke S., Taylor-Edwards C., Ridgway W., Dinisco C., Steinman L., Dalton D., Fathman C.G. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE) J Immunol. 1996;156:5–7. [PubMed] [Google Scholar]
- 15.Luster A.D., Unkeless J.C., Ravetch J.V. Gamma-interferon transcriptionally regulates an early-response gene containing homology to platelet proteins. Nature. 1985;315:672–676. doi: 10.1038/315672a0. [DOI] [PubMed] [Google Scholar]
- 16.Kim T.S., Kang B.Y., Lee M.H., Choe Y.K., Hwang S.Y. Inhibition of interleukin-12 production by auranofin, an anti-rheumatic gold compound, deviates CD4(+) T cells from the Th1 to the Th2 pathway. Br J Pharmacol. 2001;134:571–578. doi: 10.1038/sj.bjp.0704298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Loetscher M., Gerber B., Loetscher P., Jones S.A., Piali L., Clark-Lewis I., Baggiolini M., Moser B. Chemokine receptor specific for IP10 and mig: structure, function, and expression in activated T-lymphocytes. J Exp Med. 1996;184:963–969. doi: 10.1084/jem.184.3.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sørensen T.L., Trebst C., Kivisäkk P., Klaege K.L., Majmudar A., Ravid R., Lassmann H., Olsen D.B., Strieter R.M., Ransohoff R.M., Sellebjerg F. Multiple sclerosis: a study of CXCL10 and CXCR3 co-localization in the inflamed central nervous system. J Neuroimmunol. 2002;127:59–68. doi: 10.1016/s0165-5728(02)00097-8. [DOI] [PubMed] [Google Scholar]
- 19.Kivisäkk P., Trebst C., Liu Z., Tucky B.H., Sørensen T.L., Rudick R.A., Mack M., Ransohoff R.M. T-cells in the cerebrospinal fluid express a similar repertoire of inflammatory chemokine receptors in the absence or presence of CNS inflammation: implications for CNS trafficking. Clin Exp Immunol. 2002;129:510–518. doi: 10.1046/j.1365-2249.2002.01947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balashov K.E., Rottman J.B., Weiner H.L., Hancock W.W. CCR5(+) and CXCR3(+) T cells are increased in multiple sclerosis and their ligands MIP-1alpha and IP-10 are expressed in demyelinating brain lesions. Proc Natl Acad Sci U S A. 1999;96:6873–6878. doi: 10.1073/pnas.96.12.6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fife B.T., Kennedy K.J., Paniagua M.C., Lukacs N.W., Kunkel S.L., Luster A.D., Karpus W.J. CXCL10 (IFN-gamma-inducible protein-10) control of encephalitogenic CD4+ T cell accumulation in the central nervous system during experimental autoimmune encephalomyelitis. J Immunol. 2001;166:7617–7624. doi: 10.4049/jimmunol.166.12.7617. [DOI] [PubMed] [Google Scholar]
- 22.Narumi S., Kaburaki T., Yoneyama H., Iwamura H., Kobayashi Y., Matsushima K. Neutralization of IFN-inducible protein 10/CXCL10 exacerbates experimental autoimmune encephalomyelitis. Eur J Immunol. 2002;32:1784–1791. doi: 10.1002/1521-4141(200206)32:6<1784::AID-IMMU1784>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 23.Klein R.S., Izikson L., Means T., Gibson H.D., Lin E., Sobel R.A., Weiner H.L., Luster A.D. IFN-inducible protein 10/CXC chemokine ligand 10-independent induction of experimental autoimmune encephalomyelitis. J Immunol. 2004;172:550–559. doi: 10.4049/jimmunol.172.1.550. [DOI] [PubMed] [Google Scholar]
- 24.Müller M., Carter S.L., Hofer M.J., Manders P., Getts D.R., Getts M.T., Dreykluft A., Lu B., Gerard C., King N.J.C., Campbell I.L. CXCR3 signaling reduces the severity of experimental autoimmune encephalomyelitis by controlling the parenchymal distribution of effector and regulatory T cells in the central nervous system. J Immunol. 2007;179:2774–2786. doi: 10.4049/jimmunol.179.5.2774. [DOI] [PubMed] [Google Scholar]
- 25.Pagenstecher A., Lassmann S., Carson M.J., Kincaid C.L., Stalder A.K., Campbell I.L. Astrocyte-targeted expression of IL-12 induces active cellular immune responses in the central nervous system and modulates experimental allergic encephalomyelitis. J Immunol. 2000;164:4481–4492. doi: 10.4049/jimmunol.164.9.4481. [DOI] [PubMed] [Google Scholar]
- 26.Hofer M., Hausmann J., Staeheli P., Pagenstecher A. Cerebral expression of interleukin-12 induces neurological disease via differential pathways and recruits antigen-specific T cells in virus-infected mice. Am J Pathol. 2004;165:949–958. doi: 10.1016/S0002-9440(10)63356-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hancock W.W., Lu B., Gao W., Csizmadia V., Faia K., King J.A., Smiley S.T., Ling M., Gerard N.P., Gerard C. Requirement of the chemokine receptor CXCR3 for acute allograft rejection. J Exp Med. 2000;192:1515–1520. doi: 10.1084/jem.192.10.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quintana A., Muller M., Frausto R.F., Ramos R., Getts D.R., Sanz E., Hofer M.J., Krauthausen M., King N.J.C., Hidalgo J., Campbell I.L. Site-specific production of IL-6 in the central nervous system retargets and enhances the inflammatory response in experimental autoimmune encephalomyelitis. J Immunol. 2009;183:2079–2088. doi: 10.4049/jimmunol.0900242. [DOI] [PubMed] [Google Scholar]
- 29.Ousman S.S., Campbell I.L. Regulation of murine interferon regulatory factor gene expression in the central nervous system determined by multiprobe RNase protection assay. Methods Mol Med. 2005;116:115. doi: 10.1385/1-59259-939-7:115. [DOI] [PubMed] [Google Scholar]
- 30.Dudov K.P., Perry R.P. The gene family encoding the mouse ribosomal protein L32 contains a uniquely expressed intron-containing gene and an unmutated processed gene. Cell. 1984;37:457–468. doi: 10.1016/0092-8674(84)90376-3. [DOI] [PubMed] [Google Scholar]
- 31.Schrum S., Probst P., Fleischer B., Zipfel P.F. Synthesis of the CC-chemokines MIP-1alpha, MIP-1beta, and RANTES is associated with a type 1 immune response. J Immunol. 1996;157:3598–3604. [PubMed] [Google Scholar]
- 32.Sallusto F., Lanzavecchia A., Mackay C.R. Chemokines and chemokine receptors in T-cell priming and Th1/Th2-mediated responses. Immunol Today. 1998;19:568–574. doi: 10.1016/s0167-5699(98)01346-2. [DOI] [PubMed] [Google Scholar]
- 33.Malecaze F., Clamens S., Simorre-Pinatel V., Mathis A., Chollet P., Favard C., Bayard F., Plouet J. Detection of vascular endothelial growth factor messenger RNA and vascular endothelial growth factor-like activity in proliferative diabetic retinopathy. Arch Ophthalmol. 1994;112:1476–1482. doi: 10.1001/archopht.1994.01090230090028. [DOI] [PubMed] [Google Scholar]
- 34.Rupprecht T.A., Koedel U., Muhlberger B., Wilske B., Fontana A., Pfister H. CXCL11 is involved in leucocyte recruitment to the central nervous system in neuroborreliosis. J Neurol. 2005;252:820–823. doi: 10.1007/s00415-005-0752-9. [DOI] [PubMed] [Google Scholar]
- 35.Lepej S.Z., Rode O.D., Jeren T., Vince A., Remenar A., Barsić B. Increased expression of CXCR3 and CCR5 on memory CD4+ T-cells migrating into the cerebrospinal fluid of patients with neuroborreliosis: the role of CXCL10 and CXCL11. J Neuroimmunol. 2005;163:128–134. doi: 10.1016/j.jneuroim.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 36.Tsunoda I., Lane T.E., Blackett J., Fujinami R.S. Distinct roles for IP-10/CXCL10 in three animal models: Theiler's virus infection EAE, and MHV infection, for multiple sclerosis: implication of differing roles for IP-10. Mult Scler. 2004;10:26–34. doi: 10.1191/1352458504ms982oa. [DOI] [PubMed] [Google Scholar]
- 37.Liu L.P., Huang D.R., Matsui M., He T.T., Hu T., DeMartino J., Lu B., Gerard C., Ransohoff R.M. Severe disease, unaltered leukocyte migration, and reduced IFN-gamma production in CXCR3-/- mice with experimental autoimmune encephalomyelitis. J Immunol. 2006;176:4399–4409. doi: 10.4049/jimmunol.176.7.4399. [DOI] [PubMed] [Google Scholar]
- 38.Xie J.H., Nomura N., Lu M., Chen S., Koch G.E., Weng Y., Rosa R., Di Salvo J., Mudgett J., Peterson L.B., Wicker L.S., DeMartino J.A. Antibody-mediated blockade of the CXCR3 chemokine receptor results in diminished recruitment of T helper 1 cells into sites of inflammation. J Leukoc Biol. 2003;73:771–780. doi: 10.1189/jlb.1102573. [DOI] [PubMed] [Google Scholar]
- 39.Gao P., Zhou X., Yashiro-Ohtani Y., Yang Y., Sugimoto N., Ono S., Nakanishi T., Obika S., Imanishi T., Egawa T., Nagasawa T., Fujiwara H., Hamaoka T. The unique target specificity of a nonpeptide chemokine receptor antagonist: selective blockade of two Th1 chemokine receptors CCR5 and CXCR3. J Leukoc Biol. 2003;73:273–280. doi: 10.1189/jlb.0602269. [DOI] [PubMed] [Google Scholar]
- 40.Gately M.K., Renzetti L.M., Magram J., Stern A.S., Adorini L., Gubler U., Presky D.H. The interleukin-12/interleukin-12-receptor system: role in normal and pathologic immune responses. Annu Rev Immunol. 1998;16:495–521. doi: 10.1146/annurev.immunol.16.1.495. [DOI] [PubMed] [Google Scholar]
- 41.Wenner C., Guler M., Macatonia S., O'Garra A., Murphy K. Roles of IFN-gamma and IFN-alpha in IL-12-induced T helper cell-1 development. J Immunol. 1996;156:1442–1447. [PubMed] [Google Scholar]
- 42.Komatsu T., Bi Z., Reiss C.S. Interferon-gamma induced type I nitric oxide synthase activity inhibits viral replication in neurons. J Neuroimmunol. 1996;68:101–108. doi: 10.1016/0165-5728(96)00083-5. [DOI] [PubMed] [Google Scholar]
- 43.Binder G.K., Griffin D.E. Interferon-gamma-mediated site-specific clearance of alphavirus from CNS neurons. Science. 2001;293:303–306. doi: 10.1126/science.1059742. [DOI] [PubMed] [Google Scholar]
- 44.Chesler D.A., Reiss C.S. The role of IFN-[gamma] in immune responses to viral infections of the central nervous system. Cytokine Growth Factor Rev. 2002;13:441–454. doi: 10.1016/s1359-6101(02)00044-8. [DOI] [PubMed] [Google Scholar]
- 45.Boztug K., Carson M.J., Pham-Mitchell N., Asensio V.C., DeMartino J., Campbell I.L. Leukocyte infiltration, but not neurodegeneration, in the CNS of transgenic mice with astrocyte production of the CXC chemokine ligand 10. J Immunol. 2002;169:1505–1515. doi: 10.4049/jimmunol.169.3.1505. [DOI] [PubMed] [Google Scholar]
- 46.Geiger K.D., Sarvetnick N.E. Transgenic expression of IFN-g in the eye as a model for studying ocular inflammatory disease. Methods. 1996;10:392–405. doi: 10.1006/meth.1996.0117. [DOI] [PubMed] [Google Scholar]
- 47.Christensen J.E., Nansen A., Moos T., Lu B., Gerard C., Christensen J.P., Thomsen A.R. Efficient T-cell surveillance of the CNS requires expression of the CXC chemokine receptor 3. J Neurosci. 2004;24:4849–4858. doi: 10.1523/JNEUROSCI.0123-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.D'Orazio T.J., Niederkorn J.Y. A novel role for TGF-β and IL-10 in the induction of immune privilege. J Immunol. 1998;160:2089–2098. [PubMed] [Google Scholar]
- 49.Galea I., Bechmann I., Perry V.H. What is immune privilege (not) Trends Immunol. 2007;28:12–18. doi: 10.1016/j.it.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 50.Petrai I., Rombouts K., Lasagni L., Annunziato F., Cosmi L., Romanelli R.G., Sagrinati C., Mazzinghi B., Pinzani M., Romagnani S., Romagnani P., Marra F. Activation of p38 (MAPK) mediates the angiostatic effect of the chemokine receptor CXCR3-B. Int J Biochem Cell Biol. 2008;40:1764–1774. doi: 10.1016/j.biocel.2008.01.008. [DOI] [PubMed] [Google Scholar]