

Abstract
In this Commentary, Voss and colleagues discuss the work of Vana et al. who show that tau phosphorlyated at S422 in the cholinergic basal forebrain is the earliest marker of tau pathogenesis. The ramifications of these findings for future drug development specifically for tauopathies and Alzheimer's disease as discussed.
See related article on page 2533
Despite being first described in 1907, the causative mechanism driving progression of Alzheimer's disease (AD) from normal healthy neurons to a diseased state remains to be elucidated. There are two major pathological markers that are associated with AD diagnosis: amyloid β (Aβ) plaques and neurofibrillary tangles (NFTs). These extracellular plaques are composed of Aβ42, a proteolytically cleaved product from the larger amyloid precursor protein. The intracellular NFTs are primarily composed of the hyperphosphorylated microtubule-associated tau protein. Despite both markers being required for an AD diagnosis, only NFT formation has a positive correlation with the onset and severity of dementia.1, 2 The progression of tau pathological characteristics in AD is known to originate from the transentorhinal cortex and progress through the hippocampus and into the neocortex.2, 3 Tau pathological characteristics have also been observed in cholinergic basal forebrain (CBF) neurons early in AD (Braak stages I, II, and III).4, 5, 6 These cholinergic neurons innervate the areas of the originating tau pathological characteristics. The progression of NFT formation during AD in these cholinergic neurons is unknown. The precise mechanism of NFT formation still remains unknown; however, it is believed to begin with hyperphosphorylation of tau, resulting in a reduced affinity for microtubules. This free tau forms oligomeric structures that elongate into filaments and eventually coalesce into the larger aggregates, such as NFTs.
pS422 Provides a Clue
In this issue of The American Journal of Pathology, Vana et al7 sought to determine how progression of tau pathological characteristics is changed from normal aged brains to pathological AD brains in the CBF. They showed that pretangle formation in the CBF is associated with cognitive changes before NFT formation. This was achieved by analyzing brain sections of patients diagnosed as having no cognitive impairment, mild cognitive impairment, or AD, stained for biomarkers of AD progression. One of these biomarkers was tau phosphorylated on S422 (pS422). This post-translational modification is recognized by a site-specific antibody that correlates with early disease events.8 Other biomarkers were used to examine events associated with later-stage AD, such as caspase-mediated proteolytic cleavage of tau at aspartic acid 421, by using tau antibody TauC3.9, 10 To localize and monitor degeneration of the cholinergic neurons, Vana et al,7 used a marker for nerve growth factor receptor (p75NTR). p75NTR binds nerve growth factor, a vital substance for CBF survival; in the absence of the p75NTR, the CBF neurons undergo degeneration.11
S422 has recently gained notoriety owing to its involvement in NFT formation on phosphorylation in both in vitro and in vivo experiments.12, 13, 14, 15 Other results indicate that, although Aβ may not be correlative with degeneration, there is a link between fibrillization of Aβ42 and the induction of phosphorylation at S422 in the amygdala.12 Similarly, in a pro-aggregation (P301L) model of SY5Y cells, Aβ42 induction increased insoluble tau and paired helical filament–like filaments with P301L and P301L/S422E tau, a pseudophosphorylated form, but not P301L/S422A tau, an unphosphorylated form, of tau at S422.16
Intriguingly, recent work17 suggests that tau S422 is phosphorylated by a member of the mitogen-activated protein kinase (MAPK) superfamily, MAPK kinase 4. In addition to directly phosphorylating tau, MAPK kinase 4 also activates other MAPKs, and the downstream consequences of these other MAPKs can be vast. Other MAPKs can directly phosphorylate tau at distinct sites,18, 19, 20, 21 and they can also regulate synaptic plasticity, excitotoxicity, cell-cycle re-entry, and oxidative stress.22 Each of these events has been linked to NFT formation and Aβ deposition.23 Thus, phosphorylation of tau at S422 could be one of the earliest signs that the MAPK pathways are becoming dysregulated in AD, an outcome that could lead to a host of devastating problems for the brain.
Proteolytic Cleavage of Tau May Spare Neurons
Vana et al7 also observed a significant correlation between the progression of disease from no cognitive impairment to mild cognitive impairment and AD, with increases in pS422 levels and decreases in p75NTR+ cholinergic neurons; however, the number of neurons immunoreactive for both pS422 and p75NTR progressively increased with Braak staging. More important, pS422 levels in CBF neurons significantly correlated with AD progression, but TauC3 levels, at least initially, did not, consistent with previous findings8 showing that phosphorylation at S422 can block caspase cleavage of tau. This finding also correlates well with data showing that neurons with caspase-mediated proteolytically cleaved tau (as indicated by TauC3 staining) actually live longer than those without TauC3 immunoreactivity.24 In this way, caspase-mediated cleavage of tau may actually be a neuroprotective response to the aberrant accumulation of tau.24 These results are also in agreement with previous results in a P301L pR5 mouse model, in which the phosphorylation of tau during pretangle formation (not the NFT number) had a greater impact on cognitive decline and neurodegeneration.14
The Two Pools of Tau: Accumulation Impairs Clearance
Perhaps the most intriguing aspect of this study is that as AD progressed, TauC3 immunoreactivity eventually emerged in the CBF neurons. At first, this may seem contradictory to previous data8 showing that phosphorylation at the S422 site prevents cleavage of tau by caspase 3 at D421. Recent data25 may shed light, however, on this seemingly surprising result. Indeed, caspase-mediated cleavage of tau must occur for its sequestration to the autophagy pathway. Without this proteolytic cleavage event, tau is not cleared by the workhorse pathway of the cell and may overburden the more specialized proteasome pathway, or even actively suppress it, facilitating accumulation of otherwise normal tau species (Figure 1). Thus, MAPK kinase 4 may precipitate tau dysfunction by producing a toxic seed incapable of clearance by autophagy: Phosphorylation at S422 prevents normal autophagic clearance of tau, which, in turn, leads to its accumulation and subsequent suppression of other clearance pathways. As a result, normal tau may start to accumulate secondarily. Thus, two distinct pools of tau may accumulate: one that is phosphorylated at S422 and cannot be proteolytically cleaved by caspases and another that is not phosphorylated at S422 but still cannot be cleared because of inadequacy of the clearance machinery. This sequence of events would explain the observation of TauC3+ aggregates that appear after the initial appearance of pS422 tau (Figure 1). In this way, the findings presented herein provide extremely compelling rationale for further mechanistic studies focused on the phosphorylation of tau at S422 and its role in the initiation of tau pathogenesis.
Figure 1.
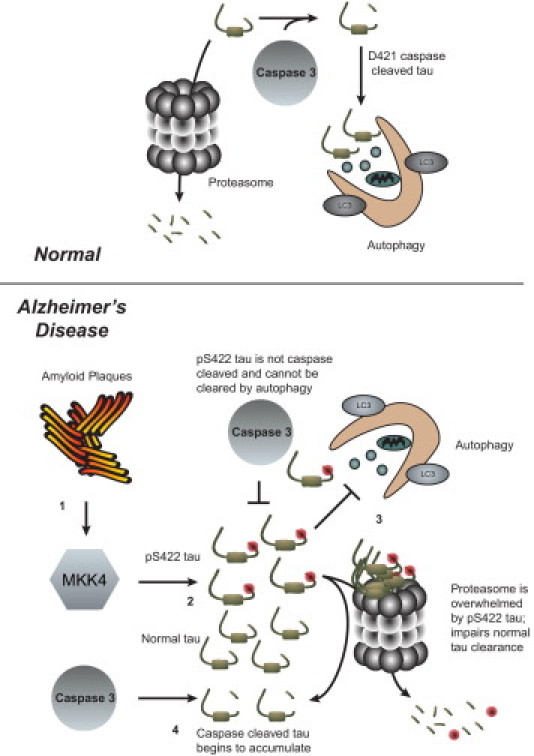
Proposed mechanism to explain data found by Vana et al.7 Under normal conditions, tau undergoes degradation by the proteasome or can be cleaved by caspase 3 and cleared via autophagy. In AD: 1) the presence of Aβ plaques activates MAPK kinase 4, which 2) phosphorylates tau at S422,12, 15, 17 preventing caspase 3 from proteolytically cleaving tau at aspartic acid 421.8 3) This pool of pS422 increases in size and cannot be cleared by autophagy, possibly actively inhibiting autophagy. The increasing amount of pS422 tau overwhelms the proteasome and leads to accumulation of soluble tau because the two major pathways of clearance are impaired. 4) Caspase 3 can cleave this new pool of soluble tau at D421, but because the two major clearance pathways are blocked, TauC3+ immunoreactivity is increased.
The diversity of tau aggregate species, from dimers to NFTs, and the inability to precisely control oligomer size make it extremely challenging to identify which species of tau is toxic to neurons.26 The CBF human patient data from Vana et al7 show that pretangle tau species that occur early in disease progression and correlate with cognitive decline are more predictive of toxicity than NFT formation. These data corroborate previous studies using transgenic mouse models to elucidate the toxic species of tau and suggest that the oligomeric or pretangle tau aggregates are more toxic than NFTs. For example, a triple-transgenic (3×Tg) mouse model (PS1m146v, APPswe, and tauP301L) showed long-term potentiation deficits in an age-dependent manner, before either Aβ or tau pathological characteristics.27 The clearance of tau by injection of Aβ antibody in 3×Tg mice reduced Aβ and cleared early nonhyperphosphorylated tau but failed to clear later-stage hyperphosphorylated tau.28 When soluble amounts of Aβ and tau were reduced together (not Aβ alone), 3×Tg mice with both pathological conditions recovered cognitive deficits, indicating that soluble amounts of tau affected the cognition of the mice.29 In another model, regulable (r)Tg4510 mice exhibit tetracycline-controlled expression of mutant P301L tau and NFT formation in neurons leads to neuronal loss and cognitive impairment. On suppression of tau expression, cognition was recovered, but NFT formation continued.30 In a region-specific manner, neuronal loss was evident before NFT formation in the dentate gyrus, a region where tau pathological features originate.31 Also, tau multimers of 140 and 170 kDa that correlated with memory loss accumulated early in rTg4510 mice. These same multimers were found in a second tau transgenic line and in AD and FTDP-17 human brain lysates.32
Soluble pS422 Tau as the Earliest Marker for Disease
Another example of the contribution of soluble tau species to neurotoxicity was shown in rTg4510 mice treated with the phenothiazine, methylene blue (MB). Although all tested doses of MB were neuroprotective, only those mice receiving doses of MB in excess of approximately 400 mg/kg showed cognitive improvement. This cognitive improvement was inversely correlated with soluble tau reductions; however, NFT formation was completely unaffected by MB treatment.33 Thus, reducing neither NFT load nor neuronal loss was sufficient to improve cognition; only when neuroprotection was accompanied by reduced soluble tau levels were cognitive deficits reversed. Thus, identifying pS422 tau as the earliest soluble tau species, as indicated by Vana et al,7 may be a first step toward designing effective tau-based therapies that can halt tau pathogenesis and improve cognitive function.
The Importance of Additional Markers: TauC3 Reactivity and Absence of p75NTR
The progression of NFT formation in the medial temporal lobe neurons progressed such that the number of pS422+ neurons is high, and TauC3+ neuronal numbers are low, but this ratio equalizes as the disease worsens.8 Herein, Vana et al7 show that, in CBF neurons, the ratio of pS422+/TauC3+ neurons does not dramatically change between patients with no cognitive impairment and patients with AD. This is interesting because deposition of tau into NFTs can occur at different rates, depending on the environment in which NFT formation is occurring. This idea has been previously proposed: changes to the local environment of in vitro tau reactions can dramatically change the way tau assembles into filaments.34, 35 Vana et al7 also showed an increase in TauC3+ neurons, a decrease in p75NTR+ neurons, and a lack of TauC3+/p75NTR+ CBF neurons with the progression of AD. Caspase-mediated cleavage of tau is predictive of NFT formation, preceding even hyperphosphorylation.24, 36, 37 Because NFT+ neurons are correlated with Braak staging of AD and TauC3+ reactivity predicts NFT formation, the presence of TauC3 reactivity and the absence of p75NTR+ in these CBF neurons could represent a good marker for neurons that are on the cusp of degeneration. Combined with pS422 tau, this may be an excellent way to track the earliest stages of AD.
Improving Early Diagnosis Is Essential for Effective Intervention
AD research is limited by an inability to properly resolve indicators of disease onset. Although our ability to diagnose prodromal AD has certainly improved during the past several decades, it still is not possible to predict AD with 100% accuracy. This limitation has greatly weakened the chances for clinical trial success. The samples used in this study highlight how poor our ability to resolve patient “sameness” is. The postmortem brain samples from various patients were grouped based on current standards of pathological and clinical diagnoses, and the results from the study show just how different these seemingly similar patients are. Perhaps using tools, such as those described herein, will improve the resolution for prodromal diagnosis, assist in postmortem analyses to reduce variability, and allow for a finer level of disease staging than the current mild cognitive impairment and five Braak stages.
Another possible utility for these findings is in the area of noninvasive imaging. Much work has been devoted to noninvasive tracking of AD pathological characteristics. These types of studies provide a mechanism for longitudinal analyses of AD pathological features in individual patients. By using labeled tracers, such as Pittsburgh compound B, in conjunction with positron emission tomographic scanning, amyloid plaque deposition can be tracked in patients who develop AD.38 However, few tau tracers exist to be used in conjunction with Pittsburgh compound B and related probes. Recently, the compound 18F-Thk523 was developed to track the formation of NFTs; unlike Pittsburgh compound B and other amyloid tracers, this tracer shows specificity for NFTs compared with Aβ plaques.39 The insights provided by Vana et al7 may allow for the identification of a more specific tau epitope-specific tracer, such as one that recognized pS422, that could show pretangle tau pathogenesis. As we develop better technology, a beneficial compound that recognizes specific tau phosphorylation states or conformations would allow researchers to monitor these earlier changes.
In conclusion, Vana et al7 show that pretangle events in CBF neurons are associated with declines in cognitive functions. This is in opposition to NFT formation being the correlative factor. Future work investigating other changes to tau that cause the CBF neurons to degenerate would provide further insight into the mechanism of how tau causes toxicity. The results showing that NFT formation progresses at different rates, depending on the location of the neurons, are also of significance, suggesting that the microenvironment within discreet brain regions could be essential for disease susceptibility. Phosphorylation of tau at S422 within the CBF may be the earliest malfunction of a protein that is critical to the pathogenesis of many neurodegenerative diseases. These insights may lead to new therapeutic strategies targeting this toxic moiety.
Footnotes
Supported by NIH (R01NS073899 and R00AG031291), Alzheimer’s Association (IIRG-09-130689 and NIRG-10-174517), Rosalinde and Arthur Gilbert New Investigator Awards in Alzheimer’s disease/American Federation for Aging Research, and Irene and Abe Pollin Fund for Corticobasal Degeneration Research/Cure PSP.
CME Disclosure: None of the authors disclosed any relevant financial relationships.
References
- 1.Wilcock G.K., Esiri M.M. Plaques, tangles and dementia: a quantitative study. J Neurol Sci. 1982;56:343–356. doi: 10.1016/0022-510x(82)90155-1. [DOI] [PubMed] [Google Scholar]
- 2.Arriagada P.V., Growdon J.H., Hedley-Whyte E.T., Hyman B.T. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 3.Braak H., Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 4.Sassin I., Schultz C., Thal D.R., Rub U., Arai K., Braak E., Braak H. Evolution of Alzheimer's disease-related cytoskeletal changes in the basal nucleus of Meynert. Acta Neuropathol. 2000;100:259–269. doi: 10.1007/s004019900178. [DOI] [PubMed] [Google Scholar]
- 5.Mesulam M., Shaw P., Mash D., Weintraub S. Cholinergic nucleus basalis tauopathy emerges early in the aging-MCI-AD continuum. Ann Neurol. 2004;55:815–828. doi: 10.1002/ana.20100. [DOI] [PubMed] [Google Scholar]
- 6.Wu C.K., Thal L., Pizzo D., Hansen L., Masliah E., Geula C. Apoptotic signals within the basal forebrain cholinergic neurons in Alzheimer's disease. Exp Neurol. 2005;195:484–496. doi: 10.1016/j.expneurol.2005.06.020. [DOI] [PubMed] [Google Scholar]
- 7.Vana L., Kanaan N.M., Ugwu I.C., Wuu J., Mufson E.J., Binder L.I. Progression of tau pathology in cholinergic basal forebrain neurons in MCI and AD. Am J Pathol. 2011;179:2535–2552. doi: 10.1016/j.ajpath.2011.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guillozet-Bongaarts A.L., Cahill M.E., Cryns V.L., Reynolds M.R., Berry R.W., Binder L.I. Pseudophosphorylation of tau at serine 422 inhibits caspase cleavage: in vitro evidence and implications for tangle formation in vivo. J Neurochem. 2006;97:1005–1014. doi: 10.1111/j.1471-4159.2006.03784.x. [DOI] [PubMed] [Google Scholar]
- 9.Guillozet-Bongaarts A.L., Garcia-Sierra F., Reynolds M.R., Horowitz P.M., Fu Y., Wang T., Cahill M.E., Bigio E.H., Berry R.W., Binder L.I. Tau truncation during neurofibrillary tangle evolution in Alzheimer's disease. Neurobiol Aging. 2005;26:1015–1022. doi: 10.1016/j.neurobiolaging.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 10.Guillozet-Bongaarts A.L., Glajch K.E., Libson E.G., Cahill M.E., Bigio E., Berry R.W., Binder L.I. Phosphorylation and cleavage of tau in non-AD tauopathies. Acta Neuropathol. 2007;113:513–520. doi: 10.1007/s00401-007-0209-6. [DOI] [PubMed] [Google Scholar]
- 11.Hefti F. Is Alzheimer disease caused by lack of nerve growth factor? Ann Neurol. 1983;13:109–110. doi: 10.1002/ana.410130127. [DOI] [PubMed] [Google Scholar]
- 12.Gotz J., Chen F., van Dorpe J., Nitsch R.M. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Aβ 42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 13.Gotz J., Chen F., Barmettler R., Nitsch R.M. Tau filament formation in transgenic mice expressing P301L tau. J Biol Chem. 2001;276:529–534. doi: 10.1074/jbc.M006531200. [DOI] [PubMed] [Google Scholar]
- 14.Deters N., Ittner L.M., Gotz J. Divergent phosphorylation pattern of tau in P301L tau transgenic mice. Eur J Neurosci. 2008;28:137–147. doi: 10.1111/j.1460-9568.2008.06318.x. [DOI] [PubMed] [Google Scholar]
- 15.Grueninger F., Bohrmann B., Czech C., Ballard T.M., Frey J.R., Weidensteiner C., von Kienlin M., Ozmen L. Phosphorylation of Tau at S422 is enhanced by Aβ in TauPS2APP triple transgenic mice. Neurobiol Dis. 2010;37:294–306. doi: 10.1016/j.nbd.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 16.Ferrari A., Hoerndli F., Baechi T., Nitsch R.M., Gotz J. β-Amyloid induces paired helical filament-like tau filaments in tissue culture. J Biol Chem. 2003;278:40162–40168. doi: 10.1074/jbc.M308243200. [DOI] [PubMed] [Google Scholar]
- 17.Grueninger F., Bohrmann B., Christensen K., Graf M., Roth D., Czech C. Mol Cell Biochem. 2011. Novel screening cascade identifies MKK4 as key kinase regulating Tau phosphorylation at Ser422. [Epub ahead of press] [DOI] [PubMed] [Google Scholar]
- 18.Sheng J.G., Jones R.A., Zhou X.Q., McGinness J.M., Van Eldik L.J., Mrak R.E., Griffin W.S. Interleukin-1 promotion of MAPK-p38 overexpression in experimental animals and in Alzheimer's disease: potential significance for tau protein phosphorylation. Neurochem Int. 2001;39:341–348. doi: 10.1016/s0197-0186(01)00041-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feijoo C., Campbell D.G., Jakes R., Goedert M., Cuenda A. Evidence that phosphorylation of the microtubule-associated protein Tau by SAPK4/p38delta at Thr50 promotes microtubule assembly. J Cell Sci. 2005;118:397–408. doi: 10.1242/jcs.01655. [DOI] [PubMed] [Google Scholar]
- 20.Yoshida H., Goedert M. Sequential phosphorylation of tau protein by cAMP-dependent protein kinase and SAPK4/p38delta or JNK2 in the presence of heparin generates the AT100 epitope. J Neurochem. 2006;99:154–164. doi: 10.1111/j.1471-4159.2006.04052.x. [DOI] [PubMed] [Google Scholar]
- 21.Zhu X., Rottkamp C.A., Boux H., Takeda A., Perry G., Smith M.A. Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J Neuropathol Exp Neurol. 2000;59:880–888. doi: 10.1093/jnen/59.10.880. [DOI] [PubMed] [Google Scholar]
- 22.Munoz L., Ammit A.J. Targeting p38 MAPK pathway for the treatment of Alzheimer's disease. Neuropharmacology. 2010;58:561–568. doi: 10.1016/j.neuropharm.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 23.Moh C., Kubiak J.Z., Bajic V.P., Zhu X., Smith M.A., Lee H.G. Cell cycle deregulation in the neurons of Alzheimer's disease. Results Probl Cell Differ. 2011;53:565–576. doi: 10.1007/978-3-642-19065-0_23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Calignon A., Fox L.M., Pitstick R., Carlson G.A., Bacskai B.J., Spires-Jones T.L., Hyman B.T. Caspase activation precedes and leads to tangles. Nature. 2010;464:1201–1204. doi: 10.1038/nature08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dolan P.J., Johnson G.V. A caspase cleaved form of tau is preferentially degraded through the autophagy pathway. J Biol Chem. 2010;285:21978–21987. doi: 10.1074/jbc.M110.110940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rankin C.A., Gamblin T.C. Assessing the toxicity of tau aggregation. J Alzheimers Dis. 2008;14:411–416. doi: 10.3233/jad-2008-14408. [DOI] [PubMed] [Google Scholar]
- 27.Oddo S., Caccamo A., Shepherd J.D., Murphy M.P., Golde T.E., Kayed R., Metherate R., Mattson M.P., Akbari Y., LaFerla F.M. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 28.Oddo S., Billings L., Kesslak J.P., Cribbs D.H., LaFerla F.M. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 29.Oddo S., Vasilevko V., Caccamo A., Kitazawa M., Cribbs D.H., LaFerla F.M. Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. 2006;281:39413–39423. doi: 10.1074/jbc.M608485200. [DOI] [PubMed] [Google Scholar]
- 30.Santacruz K., Lewis J., Spires T., Paulson J., Kotilinek L., Ingelsson M., Guimaraes A., DeTure M., Ramsden M., McGowan E., Forster C., Yue M., Orne J., Janus C., Mariash A., Kuskowski M., Hyman B., Hutton M., Ashe K.H. Tau suppression in a neurodegenerative mouse model improves memory function. Science. 2005;309:476–481. doi: 10.1126/science.1113694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spires T.L., Orne J.D., SantaCruz K., Pitstick R., Carlson G.A., Ashe K.H., Hyman B.T. Region-specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am J Pathol. 2006;168:1598–1607. doi: 10.2353/ajpath.2006.050840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berger Z., Roder H., Hanna A., Carlson A., Rangachari V., Yue M., Wszolek Z., Ashe K., Knight J., Dickson D., Andorfer C., Rosenberry T.L., Lewis J., Hutton M., Janus C. Accumulation of pathological tau species and memory loss in a conditional model of tauopathy. J Neurosci. 2007;27:3650–3662. doi: 10.1523/JNEUROSCI.0587-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.O'Leary J.C., 3rd, Li Q., Marinec P., Blair L.J., Congdon E.E., Johnson A.G., Jinwal U.K., Koren J., 3rd, Jones J.R., Kraft C., Peters M., Abisambra J.F., Duff K.E., Weeber E.J., Gestwicki J.E., Dickey C.A. Phenothiazine-mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden. Mol Neurodegener. 2010;5:45. doi: 10.1186/1750-1326-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carlson S.W., Branden M., Voss K., Sun Q., Rankin C.A., Gamblin T.C. A complex mechanism for inducer mediated tau polymerization. Biochemistry. 2007;46:8838–8849. doi: 10.1021/bi700403a. [DOI] [PubMed] [Google Scholar]
- 35.Voss K., Gamblin T.C. GSK-3β phosphorylation of functionally distinct tau isoforms has differential, but mild effects. Mol Neurodegener. 2009;4:1–12. doi: 10.1186/1750-1326-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rissman R.A., Poon W.W., Blurton-Jones M., Oddo S., Torp R., Vitek M.P., LaFerla F.M., Rohn T.T., Cotman C.W. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J Clin Invest. 2004;114:121–130. doi: 10.1172/JCI20640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spires-Jones T.L., de Calignon A., Matsui T., Zehr C., Pitstick R., Wu H.Y., Osetek J.D., Jones P.B., Bacskai B.J., Feany M.B., Carlson G.A., Ashe K.H., Lewis J., Hyman B.T. In vivo imaging reveals dissociation between caspase activation and acute neuronal death in tangle-bearing neurons. J Neurosci. 2008;28:862–867. doi: 10.1523/JNEUROSCI.3072-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Herholz K., Ebmeier K. Clinical amyloid imaging in Alzheimer's disease. Lancet Neurol. 2011;10:667–670. doi: 10.1016/S1474-4422(11)70123-5. [DOI] [PubMed] [Google Scholar]
- 39.Fodero-Tavoletti M.T., Okamura N., Furumoto S., Mulligan R.S., Connor A.R., McLean C.A., Cao D., Rigopoulos A., Cartwright G.A., O'Keefe G., Gong S., Adlard P.A., Barnham K.J., Rowe C.C., Masters C.L., Kudo Y., Cappai R., Yanai K., Villemagne V.L. 18F-THK523: a novel in vivo tau imaging ligand for Alzheimer's disease. Brain. 2011;134:1089–1100. doi: 10.1093/brain/awr038. [DOI] [PubMed] [Google Scholar]