

Abstract
Vascular endothelial growth factor receptor–1 (VEGFR-1) is a member of the VEGFR family, and binds to VEGF-A, VEGF-B, and placental growth factor. VEGFR-1 contributes to tumor growth and metastasis, but its role in the pathological formation of blood vessels is still poorly understood. Herein, we used infantile hemangioma (IH), the most common tumor of infancy, as a means to study VEGFR-1 activation in pathological vasculogenesis. IH arises from stem cells (HemSCs) that can form the three most prominent cell types in the tumor: endothelial cells, pericytes, and adipocytes. HemSCs can recapitulate the IH life cycle when injected in immuncompromised mice, and are targeted by corticosteroids, the traditional treatment for IH. We report here that VEGF-A or VEGF-B induces VEGFR-1–mediated ERK1/2 phosphorylation in HemSCs and promotes differentiation of HemSCs to endothelial cells. Studies of VEGFR-2 phosphorylation status and down-regulation of neuropilin-1 in the HemSCs demonstrate that VEGFR-2 and NRP1 are not needed for VEGF-A– or VEGF-B–induced ERK1/2 activation. U0216-mediated blockade of ERK1/2 phosphorylation or shRNA-mediated suppression of VEGFR-1 prevents HemSC-to-EC differentiation. Furthermore, the down-regulation of VEGFR-1 in the HemSCs results in decreased formation of blood vessels in vivo and reduced ERK1/2 activation. Thus, our study reveals a critical role for VEGFR-1 in the HemSC-to-EC differentiation that underpins pathological vasculogenesis in IH.
The human vascular endothelial growth factor receptor–1 [VEGFR-1, or fms-like tyrosine kinase-1 (Flt-1)] has been implicated in tumor activation, metastatasis, and vascular development.1, 2, 3 VEGFR-1 binds with high affinity to the ligands VEGF-A, VEGF-B, and placental growth factor (PlGF). VEGF-A binds to both VEGFR-1 and VEGFR-2, whereas VEGF-B and PlGF are specific ligands for VEGFR-1.4 VEGF-A is well known as a central mediator of angiogenesis and vasculogenesis.5 Less well studied, VEGF-B has been correlated with vascular invasion in hepatocellular cancer and with increased microvascular density in oral squamous cancer6,7 and has been reported as a coronary growth factor in rat.8 PlGF is upregulated in advanced colorectal and breast cancer as well in melanoma.9, 10, 11
VEGFR-1 is considered a negative regulator of angiogenesis and vasculogenesis during development. It has been shown to act as a decoy receptor for VEGF-A, thereby reducing VEGF-A ligand availability for VEGFR-2.12, 13, 14 Indeed, a reduction in this decoy mechanism has been implicated in the proliferation of infantile hemangioma (IH)-derived endothelial cells (HemECs).15 Despite its weak tyrosine kinase activity, VEGFR-1 has an active role in inflammation and metastasis but its specific function in tumor angiogenesis is not fully understood. However, a positive role for VEGFR-1 in angiogenesis has been demonstrated in a variety of settings. VEGFR-1 activation induced by PlGF increases tumor angiogenesis in the Lewis lung carcinoma murine model.2 Furthermore, VEGFR-1–blocking antibodies showed reduced tumor angiogenesis in epidermoid and colorectal cancer in mice,16, 17, 18 and reduced neovascularization of the eye.19
VEGFR-1−/− mice show disorganized blood vessels and overgrowth of immature endothelial cells,1, 13, 14 features reminiscent of IH. This common neonatal tumor is characterized by immature endothelial cells (ECs) that assemble into a disorganized vascular network. IH typically appears around the second week of life, grows over the course of 6 to 9 months and involutes during early childhood.20, 21 We previously reported that VEGFR-1 is relatively underexpressed in IH compared with placenta, infantile skin, and congenital hemangiomas.22 HemECs isolated from IH were shown to express reduced levels of VEGFR-1 compared with mature human dermal ECs (HDMECs).15 The mechanism was shown to involve sequestration of β1-integrin into a complex with VEGFR-2 and tumor endothelial marker–8 (TEM8), thereby inhibiting NFAT-driven VEGFR-1 expression. Operationally, the diminished VEGFR-1 levels in HemECs lead to increased VEGF-A availability that induces VEGFR-2 activation and subsequent increased cellular proliferation.15
Based on results using our in vivo model in which IH-derived stem cells (HemSCs) differentiate into hemangioma-like EC-lined blood vessels, we propose that IH arises from a vasculogenic process initiated by HemSC-to-EC differentiation. The central function of HemSCs in IH is supported by our recent study in which we showed corticosteroids, the most common treatment for IH, act specifically on the HemSCs by down-regulating VEGF-A expression.23 We report here that VEGFR-1, through VEGF-A or VEGF-B ligand activation, plays a fundamental role in the HemSC-to-EC differentiation and IH blood vessel formation. Furthermore we demonstrate that VEGFR-1 activity in this endothelial differentiation is mediated by ERK1/2 phosphorylation.
Materials and Methods
Cell Isolation and Culture
Specimens of IH were obtained under a human subject protocol approved by the Committee on Clinical Investigation, Children's Hospital Boston. The clinical diagnosis was confirmed in the Department of Pathology at Children's Hospital Boston. Informed consent was obtained for the specimens, according to the Declaration of Helsinki. Single cell suspensions were prepared from the proliferating phase specimens and HemSCs were selected and expanded as described.24 Human dermal microvascular endothelial cells (HDMECs) and hemangioma-derived endothelial cells (HemECs) were isolated as described,25 and bone marrow mesenchymal stem cells (bmMSCs) were purchased from Cambrex (East Rutherford, NJ).
Short interference (siRNA) silencing of neuropilin-1 (NRP-1) (NM_001024629) was performed on the HemSCs and HMDECs to test the effects of NRP-1 down-regulation. A pool of four different siRNA oligonucleotides (Dharmacon, Lafayette, CO) was used to transfect the cells at a concentration of 40 nmol/L. Efficiency was tested by Western blot for NRP-1 expression.
Short hairpin RNA (shRNA) lentiviral particles for VEGFR-1 targeting (NM_002019) and nontargeting shRNA (control) were used to infect HemSCs (Sigma-Mission, St. Louis, MO). HemSCs with stable expression of VEGFR-1 shRNA were subjected to puromycin selection, according to the manufacturer's instructions. The pool used for the in vivo experiments was selected after testing five different shRNA sequences for silencing efficacy.
Endothelial Differentiation Protocol
To induce endothelial differentiation, HemSCs were cultured on plates coated with fibronectin (10 ng/cm2) at a density of 2 × 104 cells/cm2 in regular growth medium (EBM2/20%FBS, plus EGM2 Single Quots). After 18 to 24 hours, the growth medium was replaced with endothelial differentiation medium [EBM2, 1× insulin–transferrin–selenium, 1:5000 linoleic acid–albumin, 1 μmol/L dexamethasone, 60 μmol/L ascorbic acid-2–phosphate, 10 ng/mL VEGF-B (R&D Systems, Minneapolis, MN)]. Thereafter, fresh endothelial differentiation medium was added every 2 days. A list of all of the IH specimens used to derive HemSCs that have been positively tested for the EC differentiation is shown in Supplemental Table S1 (available at http://ajp.amjpathol.org).
Assay for Tube Formation
Wells were precoated with Matrigel and incubated for 30 minutes at 37 °C. HemSCs were seeded at a density of 2.5 × 104 cells/cm2 in 500 μL of EBM2/0.1%BSA. After 18 hours, cultures were fixed with 10% formalin and pictures taken with an inverted microscope Nikon Eclipse TE300 (Nikon, Melville, NY) using SPOT Advanced 3.5.9 software (Diagnostic Instruments Inc., Sterling Heights, MI).
Assays for in Vitro Cellular Proliferation and Viability
Proliferation was assessed after seeding 104 cells/cm2 on fibronectin-coated, 48-well plates and culturing in regular growth medium. After attachment (24 hours), plating efficiency was determined (0 hours). Cell numbers at 24, 48, and 72 hours were determined using a Coulter Counter (Beckman, Brea, CA). Cell survival was assessed by seeding 4000 cells per well on 96-well plates. After 24 hours, cells were treated with AAC789 or U0126, and vitality was assessed at different time points by measuring absorbance at 570 to 690 nm 4 hours after the addition of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), 0.5 mg/mL.
Inhibitors
U0126 was purchased from Sigma, and dissolved in dimethylsulfoxide (DMSO), soluble VEGFR-1 and soluble VEGFR-2 were purchased from R&D Systems. AAC789 was kindly provided by Dr. Jeanette Wood (Novartis).26
Flow Cytometry
Cells were labeled with FITC-conjugated mouse anti-human CD31 (Ancell, Bayport, MN) after permeabilization with 0.5% saponin (Sigma-Aldrich). Flow cytometry was performed on a BD FACScan. Data were analyzed using FlowJo software.
Quantitative Real-Time PCR
RNA was extracted using RNeasy Mini Kit (Qiagen, Valencia, CA). cDNA synthesis was performed with iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA). Primers used are listed in Table 1. All reactions were performed for 35 cycles with the following temperature profiles: 95°C for 2 minutes (initiation; 30 seconds per cycle thereafter), annealing step for 25 seconds, and an extension step at 72°C for 30 seconds.
Table 1.
Primers Used for Quantitative Real-Time PCR
| Primer | Forward | Reverse |
|---|---|---|
| NRP1 | 5′-ACACCTGAGCTGCGGACTTT-3′ | 5′-GGCCTGGTCGTCATCACA-3′ |
| NRP2 | 5′-GCGCAAGTTCAAAGTCTCCT-3′ | 5′-TCACAGCCCAGCACCTC-3′ |
| VE-cadherin | 5′-CCTTGGGTCCTGAAGTGACCT-3′ | 5′-CAGGGCCTTCCTTCTGCAA-3′ |
| VEGF-B167 | 5′-AGTCCGGATGCAGATCCTC-3′ | 5′-CTGCAGGTGTCTGGGTTG-3′ |
| VEGF-B186 | 5′-CCCTTGACTGTGGAGCTCAT-3′ | 5′-GGAGTGGCAGCCCTGTCT-3′ |
| VEGFR-1 | 5′-GCTCCTATTAACCCTCTTTATCC-3′ | 5′-AGTGATTTGCCCAGTTTAAGTC-3′ |
| VEGFR-2 | 5′-TCAAAGGAGAAGCAGAGCCATGTG-3′ | 5′-GCACTCTTCCTCCAACTGCCAATA-3′ |
| GAPDH | 5′-TGCACCACCAACTGCTTAG-3′ | 5′-GATGCAGGGATGATGTTC-3′ |
In Vivo Model of Infantile Hemangioma
Experiments were performed with 2 to 4 × 106 cells per animal as described.23, 24 Briefly, cells were suspended in 200 μL of Matrigel (BD Bioscience, Bedford, MA) and injected subcutaneously on the back of 6- to 7-week-old male athymic nu/nu mice (Massachusetts General Hospital, Boston, MA).
Microvessel Density
For the assessment of microvessel density, 10 fields from mid-Matrigel hematoxylin and eosin (H&E)–stained sections of each of the animals in the group were quantified by counting luminal structures containing red blood cells. MVD was expressed as vessels/mm2.
Immunohistochemistry
Paraffin sections of Matrigel explants or frozen sections of proliferating IH were used. The sections were stained with the following antibodies: human-specific CD31 monoclonal antibody (1:50; DakoCytomation, Carpinteria, CA, or Santa Cruz, Santa Cruz, CA) for detection of microvessels, and phosphoERK1/2 (1:100; Cell Signaling, Danvers, MA). Incubation with primary antibody was followed by peroxidase-labeled secondary antibody (1:200; Vector Laboratories, Burlingame, CA), and 3,3′-diaminobenzidine (DAB) staining (Vector Laboratories). Images were taken with Axiophot II microscope (Zeiss, Oberkochen, Germany) equipped with AxioCam MRc5 (Zeiss). Images were taken at room temperature (∼20 °C) and files always exported in 8-bit format. A list of all of the IH specimens used to test for phosphoERK expression is shown in Supplemental Table S2 (available at http://ajp.amjpathol.org).
Immunofluorescence
Frozen sections of proliferating hemangioma, and placenta were fixed in acetone and stained with anti–VEGF-B antibody (R&D Systems) followed by fluorescein isothiocyanate (FITC)–conjugated secondary antibody (Vector Labs). Images were taken with Leica TCS SP2 Acousto-Optical Beam Splitter confocal system equipped with DMIRE2 inverted microscope (diode, 405 nm; argon, 488 nm; HeNe, 594 nm; Leica Microsystems, Wetzlar, Germany), Leica Confocal Software Version 2.61, Build 1537. Images were taken at room temperature (∼20 °C), and files were always exported in 8-bit format.
Immunoblots
Cells were lysed with RIPA buffer (Boston Bioproducts, Ashland, MA), containing a protease inhibitor cocktail (Complete Mini Tablet; Roche, Indianapolis, IN). Lysates were subjected to sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to Immobilon-P membrane. Membranes were incubated with antibodies to detect NRP2, NRP1, VEGFR2 (1:1000; 1:500, and 1:500; Santa Cruz), phosphoVEGFR-2, and ERK, phospho-ERK, AKT, phospho-AKT, and p38, phospho-p38 (each of them at 1:1000; Cell Signaling). Loading control was performed with anti-human Tubulin (1:10000, Sigma-Aldrich). Membranes were incubated with HRP-conjugated secondary antibodies (1:5000; peroxidase-conjugated anti-specie isotype; Vector Laboratories). Antigen–antibody complexes were visualized using Lumiglo and chemiluminescent-sensitive film.
Statistical Analysis
Data were expressed as mean ± SD or as mean ± SEM, and were examined by analysis of variance followed by Student's t-test where appropriate. Differences were considered significant at P values <0.05.
Results
VEGF-B and Endogenous VEGF-A Induce HemSC-to-EC Differentiation
IH-derived stem cells, referred to as HemSCs, were isolated from IH tissue and selected based on the expression of the progenitor cell marker CD133. HemSCs do not express specific endothelial markers, and exhibit a mesenchymal morphology.24 To induce EC differentiation, HemSCs were cultured at high density (2 × 104 cells/cm2) for 8 days in an endothelial-differentiation medium (serum-free medium, plus VEGF-B 10 ng/mL). CD31 expression was detected in VEGF-B treated HemSCs, whereas no expression was detected in HemSCs cultured in serum-free medium with PDGF-BB plus EGF (both at 10 ng/mL) (Figure 1A). In a second experiment, we detected increasing mRNA levels of VE-cadherin and neuropilin 2 (NRP2) at days 5 and 12 (Figure 1A), 15 and three times greater, respectively, compared with HemSCs cultured in control conditions (serum-free medium with PDGF-BB plus EGF). We also detected NRP2 protein levels at day 5 and 10 when HemSCs were cultured in endothelial differentiation medium (Figure 1A). Some NRP2 expression was also detected in the differentiation medium without growth factors. We speculate that the high levels of VEGF-A expressed by HemSCs23 (see Supplemental Figure S1A at http://ajp.amjpathol.org) could act in an autocrine fashion to induce some HemSC-to-EC differentiation, even in absence of VEGF-B. Consistent with this idea, treating the cells with endothelial-differentiation medium, both with or without VEGF-B, resulted in a shift in morphology from spindle-shaped to cobblestone/endothelial-like (Figure 1B).
Figure 1.
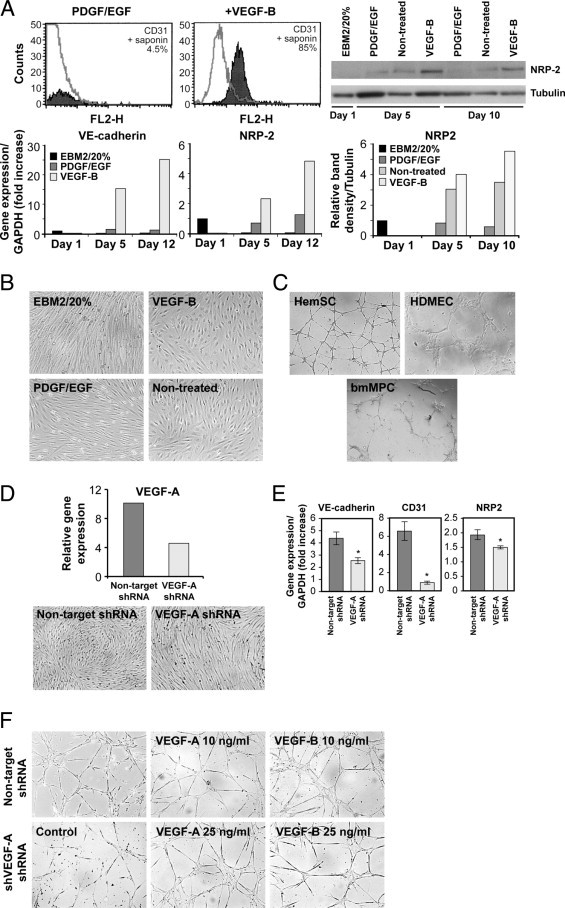
VEGF-B and endogenous VEGF-A induce HemSC-to-EC differentiation. A: Histogram (shaded gray) represents CD31 expression in HemSCs after 8 days of exposure to PDGF/EGF (both at 10 ng/mL) or VEGF-B (10 ng/mL) in serum-free medium; gray line, matching IgG control (top left). Immunoblot shows NRP-2 in HemSCs at day 1 in EBM2/20% FBS (EBM2/20%), and after 5 and 10 days in endothelial differentiation medium with PDGF-BB plus EGF (each at 10 ng/mL), or no treatment or VEGF-B (10 ng/mL). Tubulin is shown as a loading control (top right). Relative immunoblot band intensities (NRP2/Tubulin) shown in the graph below. On the bottom left, RT-PCR for VE-cadherin and NRP-2 HemSCs in EBM2/20%, in endothelial differentiation medium with PDGF plus EGF, or VEGF-B, at days 1, 5 and 12, normalized to day 1 in EBM2/20%. B: HemSCs cultured for 8 days in EBM2/20% or endothelial differentiation medium with addition of VEGF-B, PDGF/EGF, or without factors (nontreated). C: HemSCs, HDMECs, and bmMPCs plated on Matrigel for 18 hours. D: RT-PCR for VEGF-A in HemSCs infected with lentivirus containing nontarget shRNA and VEGF-A shRNA (top). Cells cultured for 5 days in endothelial differentiation medium, and (E) RT-PCR shows levels of VE-cadherin, CD31, and NRP-2. F: Nontarget and VEGF-A shRNA HemSCs cultured on Matrigel for 18 hours. In four right panels, tube formation assessed in response to exogenous VEGF-A and VEGF-B (10 and 25 ng/mL).
A well-known feature of ECs is their ability to form capillary-like structures when plated on a thin layer of Matrigel. When tested in this assay, HemSCs formed a well-organized network of capillary-like structures, similarly to mature HDMEC (Figure 1C). In contrast, in our conditions, bone marrow mesenchymal progenitor cells (bmMPCs) failed to form tubular structures in Matrigel. This ability to form capillary-like structures, even before the onset of endothelial markers, suggests that the HemSC have a propensity for endothelial function.
VEGF-A is highly expressed in HemSCs compared with HemECs and bone marrow mesenchymal stem cells (bmMSC) (see Supplemental Figure S1 at http://ajp.amjpathol.org). For this reason, we tested whether down-regulation of the endogenous VEGF-A would affect HemSC-to-EC differentiation. HemSCs were infected with lentiviral construct expressing short hairpin (sh)RNA for VEGF-A; this resulted in a 57% reduction in VEGF-A transcripts (Figure 1D). HemSCs in which VEGF-A was suppressed (VEGF-A shRNA) failed to assume a cobblestone morphology in endothelial-differentiation medium (Figure 1D, bottom). Furthermore, HemSC–VEGF-A shRNA failed to show increased expression of VE-cadherin, CD31 and NRP2 in response to endothelial-differentiation medium (Figure 1E). HemSC–VEGF-A shRNA also lost the ability to form tubular structures in Matrigel (Figure 1F). Notably, the addition of exogenous VEGF-A or VEGF-B rescued the phenotype (Figure 1F).
VEGF-B Is Highly Expressed in Proliferating IH and HemECs
As VEGF-B induced HemSC-to-EC differentiation, we wanted to assess whether VEGF-B is also expressed in IH tissue. VEGF-B was detected, by immunofluorescence, around the blood vessels in proliferating IH (see Supplemental Figure S1B at http://ajp.amjpathol.org). Real-time PCR confirmed that both VEGF-B isoforms, VEGF-B167, and VEGF-B186 were expressed in IH (see Supplemental Figure S1C at http://ajp.amjpathol.org). We further analyzed VEGF-B expression in HemSCs and HemECs, and, for comparison, in HDMECs, cord blood endothelial progenitor cells (cbEPCs) and bmMSCs (see Supplemental Figure S1D at http://ajp.amjpathol.org). VEGF-B167, the isoform containing the heparin-binding domain, was overexpressed in HemECs (seven of eight screened), compared with HDMECs and cbEPCs. This suggests that VEGF-B produced by HemECs might have a role in stimulating HemSCs-to-EC differentiation in proliferating IH.
VEGF-B and VEGF-A Strongly Activate ERK Signaling in HemSCs
To better understand the signaling pathway(s) involved in the HemSC-to-EC differentiation, we studied MAPK activation. HemSCs exhibited a strong increase in ERK phosphorylation in response to VEGF-A or VEGF-B167. In contrast, only VEGF-A increased phospho-ERK levels in HDMECs (Figure 2A). Phospho-ERK was also detected in proliferating IH tissue: many but not all ECs stained positively (Figure 2B, top). There were also subsets of phospho-ERK–positive cells not organized or associated with luminal structures. We suggest that these cells are ligand-activated HemSCs undergoing EC differentiation. In contrast, involuting IH showed much less immunoreactivity to phospho-ERK antibody (Figure 2B, bottom). PhosphoERK staining of additional proliferating and involuting IH specimens is shown in Supplemental Figure S2A (available at http://ajp.amjpathol.org). In HDMECs, VEGF-B induced phosphorylation of ERK at 10 to 15 minutes, whereas in HemSCs phospho-ERK levels were upregulated at 2 minutes (Figure 2C). Results were confirmed with HemSCs from four additional IHs from different patients (see Supplemental Figure S2B at http://ajp.amjpathol.org). Furthermore, PlGF, a cytokine with similar affinity for VEGFR-1, also induced strong ERK phosphorylation after 2 minutes of treatment (see Supplemental Figure S2C at http://ajp.amjpathol.org).
Figure 2.
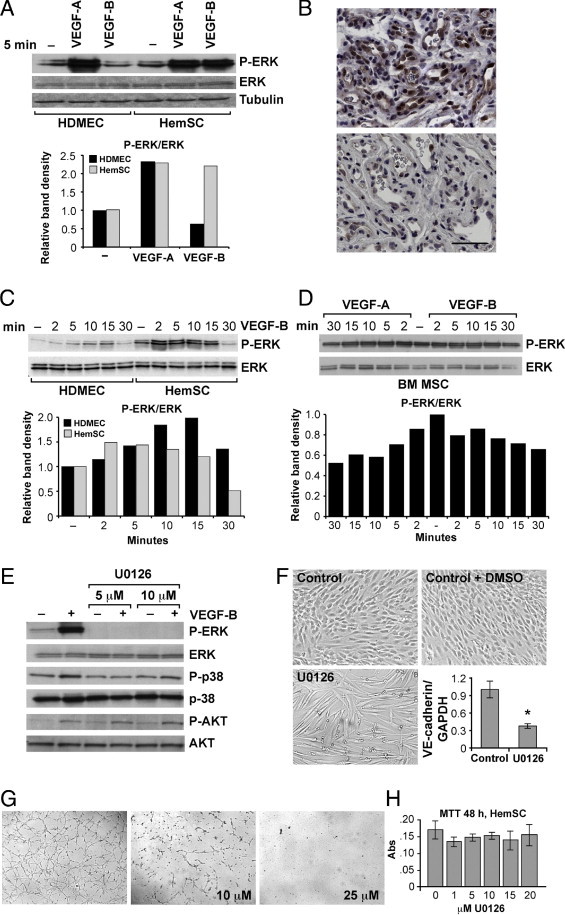
VEGF-B and VEGF-A activate ERK phosphorylation in HemSCs. Immunoblot of phospho-ERK (P-ERK) and ERK in HDMECs and HemSCs in response to (A) 5 minutes' exposure to VEGF-A (25 ng/mL) or VEGF-B (10 ng/mL). Relative immunoblot band intensities (phosphoERK/ERK) are shown in the graph. B: Proliferating (top panel) and involuting (bottom panel) IH specimen stained with anti-phosphoERK antibody. Scale bar = 20 μm. C: Immunoblot and graphs of relative band intensities of phospho-ERK and ERK in HDMECs and HemSCs in response to VEGF-B (25 ng/mL) at different time points, and (D) in bone marrow mesenchymal stem cells (bmMSC) after treatment with VEGF-A or VEGF-B (both at 25 ng/mL). E: HemSCs pretreated with U0126 (5 and 10 μmol/L) for 1 hour and incubated for 2 minutes with VEGF-B (25 ng/mL) subjected to immunoblot analysis for phospho-ERK, ERK, phosphor-p38, p38, and phosphoAKT, AKT. F: HemSCs cultured for 5 days in endothelial differentiation medium (Control), or with DMSO (Control+DMSO) or U0126, and RT-PCR analysis of VE-cadherin in control (DMSO) and U0126 (10 μmol/L) treated HemSCs. G: Tube formation in Matrigel assessed for HemSCs at 18 hours after control (DMSO) and U0126 (10 μmol/L and 25 μmol/L) treatment. H: Cell survival, measured with MTT assay, for HemSCs exposed to different concentrations of U0126. Data expressed as means ± SD.
Because bmMSCs express levels of VEGFR-1 and VEGFR-2 similar to HemSCs24 (see Supplemental Figure S2D at http://ajp.amjpathol.org), we tested bmMSCs for ERK phosphorylation in response to VEGF-A and VEGF-B. Neither elicited a response (Figure 2D) in bmMSCs indicating that strong VEGF-B-induced ERK activation at 2 minutes is a specific property of HemSCs. To test whether or not ERK phosphorylation is required for HemSC-to-EC differentiation, we used a specific phospho-ERK inhibitor, U0126. As expected, U0126-pretreatment inhibited ERK phosphorylation but did not alter the phosphorylation status of the MAP kinases p38 and AKT (Figure 2E). U0126 prevented HemSC-to-EC differentiation in vitro, as cells did not show the morphological transition to a cobblestone/endothelial-like phenotype and VE-cadherin expression was not upregulated in response to VEGF-B (Figure 2F). U0126 (10 and 25 mol/L) also affected the ability of HemSCs to form tubular structures in Matrigel (Figure 2G). Of note, U0126 did not affect cell viability at 48 hours at concentrations between 1 and 20 μmol/L (Figure 2H).
VEGFR-1Tyr1213 phoshorylation was reported to mediate the binding of phosphatidylinositol 3-kinase (PI3K).27 To study upstream regulation of ERK phosphorylation we used Wortmannin, an inhibitor of phosphoinositide 3-kinases (PI3K). We detected inhibition of VEGF-B– and VEGF-A–induced ERK phoshorylation when HemSCs were pretreated with Wortmannin (0.3 or 8 μmol/L) (see Supplemental Figure S2E at http://ajp.amjpathol.org). In contrast, Wortmannin pretreatment did not affect phospho-ERK levels in HDMECs. These results suggest that PI3K signaling acts as an upstream regulator of ERK1/2 activation in HemSCs.
NRP1 and VEGFR-2 Are Not involved in VEGF-Induced ERK Signaling in HemSCs
Because VEGF-A and VEGF-B both bind to neuropilin1 (NRP1) and to VEGFR-1, we wanted to identify which receptor was involved in HemSC-to-EC differentiation. As we had previously reported a relatively low level of VEGFR-1 expression in IH tissue,15, 22 we initially focused our attention on NRP1. We detected NRP1 expression in HemSCs and HemECs (see Supplemental Figure S3A at http://ajp.amjpathol.org). NRP1 expression was higher in HemSCs compared with HDMECs and HemECs. We performed siRNA to down-regulate NRP1 expression and measured VEGF-B induced phospho-ERK. siRNA transfection suppressed NRP1 in HDMECs and in HemSCs; however, ERK activation in response to VEGF-B 25 ng/mL did not change (Figure 3A), suggesting that NRP1 does not have a role in HemSC-to-EC differentiation.
Figure 3.
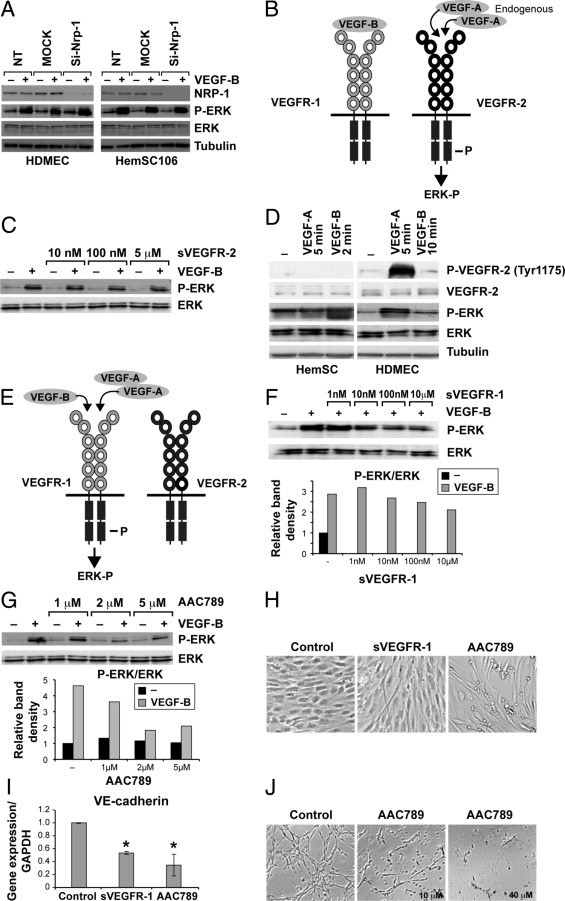
NRP1 and VEGFR-2 are not involved in HemSC-to EC differentiation. HDMECs and HemSCs subjected to siRNA to down-regulate NRP1 and immunoblot: A: NRP1, phosphoERK, and ERK levels in cells treated or not treated with VEGF-B (25 ng/mL); tubulin is loading control. B: Schematic of hypothesis of indirect role for VEGFR-1 in the HemSC-to-EC differentiation, as described in text. C: Immunoblot of phosphoERK and ERK in HemSCs pre-treated for 1 hour with soluble VEGFR-2 (sVEGFR-2) followed by stimulation with VEGF-B (25 ng/mL). D: HemSCs and HDMECs incubated with VEGF-A (25 ng/mL, 5 minutes) or VEGF-B (25 ng/mL, 2 minutes for HemSCs and 10 minutes for HDMECs). Cell lysates analyzed for phosphoVEGFR-2 (Tyr1175), total VEGFR-2, phosphoERK, ERK, and tubulin (loading control). E: Schematic of hypothesis of direct role for VEGFR-1 in the HemSC-to-EC differentiation, as described in in text. F: Immunoblot and graphs of relative band intensities for phosphoERK and ERK in HemSCs stimulated for 2 minutes with VEGF-B (25 ng/mL) after 1 hour of pretreatment with soluble VEGFR-1 (sVEGFR-1) (1 nmol to 10 μmol/L), or (G) with AAC789 (1–5 μmol/L). H: HemSCs cultured for 5 days in serum-free medium plus VEGF-B alone (Control), or VEGF-B plus sVEGFR-1 (5 μg/mL) or AAC789 (5 μmol/L), and (I) quantitative RT-PCR analysis of VE-cadherin in control, sVEGFR-1, and AAC789 treated HemSCs. J: Tube formation in Matrigel in HemSCs 18 hours after control (DMSO) and AAC789 (10 μmol/L and 40 μmol/L) treatment. Data are mean ± SD.
We thereby hypothesized a role for VEGFR-1 in the HemSC-to-EC differentiation, and postulated two possible mechanisms of action to test. The first involved an indirect role for VEGFR-1, whereby VEGFR-1 would act as a decoy receptor and is occupied by exogenous VEGF-B. This would increase bioavailability of endogenous VEGF-A to bind and activate VEGFR-2 signaling (Figure 3B schematic). The second mechanism involved a direct role for VEGFR-1, whereby binding of VEGF-A or VEGF-B would elicit activation of the VEGFR-1 tyrosine kinase domain on the receptor and subsequent ERK phosphorylation (Figure 3E schematic).
To test the first hypothesis, we performed two different experiments. First, we used recombinant human soluble VEGFR-2 (sVEGFR-2), which binds only VEGF-A, to pretreat the HemSCs before VEGF-B stimulation. Increasing concentrations of sVEGFR-2 (10 nmol/L to 5 μmol/L) did not affect VEGF-B-induced ERK phosphorylation (Figure 3C). To further examine VEGFR-2 involvement, we tested whether VEGF-B or VEGF-A treatment induced phosphorylation of VEGFR-2. We used an antibody directed against the VEGFR-2 phosphorylation site (Tyr1175). VEGFR-2Tyr1175 phosphorylation was not detected (Figure 3D). These results suggest that VEGFR-2 is not involved in HemSC-to-EC differentiation.
We next tested the second hypothesis, that is, whether VEGFR-1 is directly involved in HemSC-to-EC differentiation through tyrosine kinase domain activation. We examined whether pretreatment of HemSCs with soluble VEGFR-1 (sVEGFR-1) inhibited VEGF-B–induced ERK phosphorylation. Indeed, sVEGFR-1 (10 nmol/L to 10 μmol/L) prevented ERK activation (Figure 3F).
Because VEGFR-1 in HemSCs is not highly expressed,24 we could not detect its phosphorylation with standard anti–VEGFR-1 immunoprecipitation followed by an anti-phosphoprotein immunoblot. The receptor tyrosine kinase inhibitor AAC789,26, 28 with selectivity for VEGFR-2 and VEGFR-1, was used to pretreat the HemSCs before stimulation with VEGF-B. Because activated VEGFR-2 was not detected in HemSCs, we assumed that AAC789 would specifically target VEGFR-1 in HemSCs. AAC789 pretreatment strongly reduced ERK phosphorylation (Figure 3G). Furthermore, HemSC-to-EC differentiation was blocked by sVEGFR-1 and AAC789, as evident from spindle-shaped cellular morphology (Figure 3H). VE-cadherin upregulation was significantly blocked in sVEGFR-1 and AAC789 treated cells in comparison to DMSO-treated controls (Figure 3I). Finally, AAC789 (10 and 40 μmol/L) inhibited tube formation in the Matrigel assay (Figure 3J). AAC789 affected HemSC viability only at concentrations of at least 40 μmol/L at 48 hours and at least 20 μmol/L at 72 hours (see Supplemental Figure S3, B and C, at http://ajp.amjpathol.org).
Silencing of VEGFR-1 Prevents HemSC-to-EC Differentiation in Vitro and Blood Vessel Formation in Vivo
Results obtained with AAC789 suggested involvement of VEGFR-1 in the HemSC-to-EC differentiation. To test this directly, we down-regulated VEGFR-1 using short-hairpin (sh)–RNA–mediated silencing. Five different shRNA sequences were tested, and the two that produced the lowest VEGFR-1 expression (F2 and F4) were used (Figure 4A). Cellular proliferation of HemSCs with VEGFR-1 suppression (VEGFR-1 shRNA) was significantly (P < 0.05) lower than control HemSCs at 72 hours in growth medium (EBM2/20%FBS), and at 48 and 72 hours in starvation medium (EBM2/1%BSA, no growth factors) (see Supplemental Figure S4 at http://ajp.amjpathol.org). HemSC-VEGFR-1 shRNA did not undergo endothelial differentiation in response to VEGF-B, as cell morphology remained spindle shaped (Figure 4B). HemSC-VEGFR-1 shRNA cultured in endothelial differentiation medium did not show significantly (P < 0.05) increased VE-cadherin mRNA expression (Figure 4C). In addition, in HemSC-VEGFR-1 shRNA, NRP2 was not upregulated in the endothelial differentiation medium with or without VEGF-B (Figure 4D). HemSC-VEGFR-1 shRNA also showed disrupted tubular structures in Matrigel (Figure 4E), whereas HemSC-nontarget shRNA retained this ability.
Figure 4.
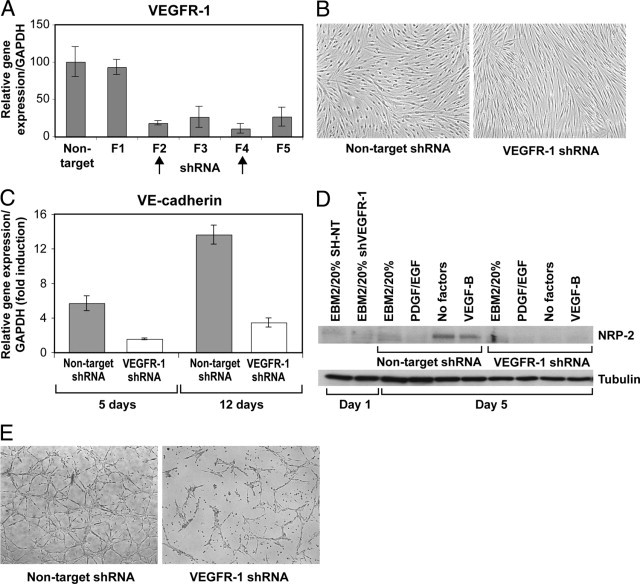
Silencing of VEGFR-1 prevents HemSC-to-EC differentiation in vitro. A: RT-PCR shows VEGFR-1 expression levels in HemSCs after infection with lentivirus encoding five different shRNA sequences (F1–F5) targeting VEGFR-1, and control nontarget sequence. B: Non-target and VEGFR-1 shRNA (F4) HemSCs cultured for 5 days in endothelial differentiation medium, and (C) quantitative RT-PCR illustrates relative VE-cadherin expression after 5 and 12 days in culture. D: Immunoblot for NRP-2 in HemSCs, nontarget and VEGFR-1 shRNA at days 1 and 5 after culture in regular growth medium (EBM2/20%) or in serum-free medium plus PDGF and EGF (10 ng/mL each), no factors, or VEGF-B (10 ng/mL); tubulin was used as loading control. E: Nontarget and VEGFR-1 shRNA HemSCs cultured in Matrigel thin layer for 18 hours. Data expressed as means ± SD.
We next assessed formation of blood vessels in vivo by implanting HemSC-VEGFR-1 shRNA and nontarget shRNA (control) into nude mice. The MVD, calculated as number of red blood cell–filled lumens, of implants containing HemSC-VEGFR-1 shRNA F2 or F4 was significantly (P = 0.04 and P = 0.01) lower than in controls (Figure 5A). We next stained the Matrigel explants for human specific CD31 and quantified the CD31+ MVD. VEGFR-1 down-regulation impaired the formation of human (HemSC)–derived blood vessels (Figure 5B). Furthermore, implants with HemSC-VEGFR-1 shRNA showed a reduction of phosphoERK immunoreactivity (Figure 5C). These results suggest that VEGFR-1 in HemSCs is responsible for the blood vessel formation and for ERK1/2 activation in the IH murine model.
Figure 5.
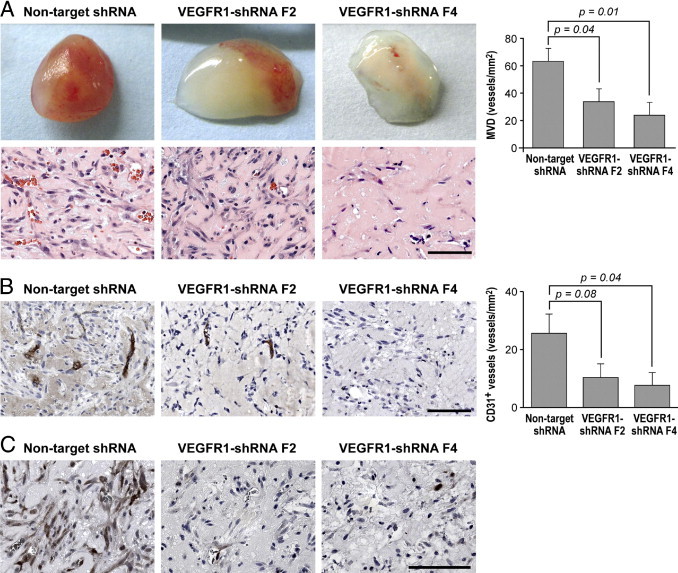
Silencing of VEGFR-1 prevents formation of blood vessels in vivo. A: Representative photographs of Matrigel explants at day 7 after injection of HemSCs nontarget and VEGFR-1 shRNA (F2 and F4), corresponding sections stained for H&E, and quantification of total microvessel density (MVD) as microvessels/mm2. B: Anti-human-CD31 staining in Matrigel explants from experiment in A, graph shows quantification of human CD31+ vessels. C: PhosphoERK1/2 staining in Matrigel explants from experiment in A. Scale bar = 100 μm. Data are mean ± SEM.
Discussion
In this study, we show that VEGFR-1 is required for HemSC-to-EC differentiation and for formation of blood vessels in an in vivo model of IH. Furthermore, we present evidence that HemSC-to-EC differentiation is mediated by VEGF-A or VEGF-B induced ERK1/2 phosphorylation. In the murine IH model, down-regulation of VEGFR-1 in HemSCs resulted in decreased vasculogenesis and ERK1/2 phosphorylation. Therefore, we suggest that, in our model, ERK1/2 is phosphorylated in response to VEGFR-1 activation.
We used IH, the most common tumor of infancy, as a biological system to study pathological disruption of human vascular development. IH is a lesion of disorganized vasculature20, 21, 29 that originates from tumor-derived stem cells (HemSCs).24 HemSCs are selected based on the expression of the progenitor cell marker CD133,30 and they exhibit stem cell properties.24 Similarly to HemSCs, CD133+ cancer stem-like cells in the highly angiogenic tumor glioblastoma can differentiate into endothelial cells.31, 32 In the glioblastoma model, the VEGF-A blocking antibody bevacizumab prevented maturation of tumor endothelial progenitors into endothelium. Furthermore VEGFR-1 expression was increased in the CD133+/CD144+ cell population compared with the CD133+/CD144-, suggesting a role for VEGFR-1 in the endothelial differentiation of the glioblastoma stem-like cells. In summary, HemSC-to EC differentiation in IH is a prototype for this recently identified endothelial differentiation ability of glioblastoma cancer stem cell.
Interestingly, VEGFR-1 expression in HemECs is low.15 The mechanism for this low expression was shown to be caused by sequestration of β1-integrin in a multiprotein complex composed by TEM8 and VEGFR-2 that inhibits NFAT-mediated VEGFR-1 transcription. VEGFR-1 has been shown to act as a decoy receptor in HemECs, such that low VEGFR-1 is responsible for the constitutive phosphorylation of VEGFR-2 and increased endothelial proliferation. In contrast, it is interesting to note that, even if expressed at low levels, VEGFR-1 in HemSCs can be activated and can induce ERK1/2 signaling. Thus, the functions of VEGFR-1 are very likely dependent on and modulated by the cellular context.
The signaling pathways leading to endothelial differentiation are still poorly understood. Embryonic stem cells and vascular progenitors differentiate into endothelial cells when exposed to VEGF-A in serum-free conditions.33, 34 We report that addition of exogenous VEGF-A can increase HemSC-to-EC differentiation, but endogenous VEGF-A is also sufficient when cells are in a serum-free medium. Furthermore, we showed that both VEGF-A and VEGF-B activate ERK1/2 and rescue the tube-forming ability of HemSCs with suppressed VEGF-A expression.
In mouse embryonic stem cell–derived-VEGFR-2+ cells, VEGFR-2Tyr1175 phosphorylation was crucial for endothelial specification.35, 36 In addition, Nrp1 has been shown to have an essential role in the induction of EC differentiation and vascular formation in response to VEGF-A.37, 38 We investigated the role of VEGFR-2 and NRP1 in the HemSC-to-EC differentiation. We did not detect VEGFR-2Tyr1175 phosphorylation, and NRP1 suppression did not inhibit ERK1/2 activation, indicating that VEGFR-2 and NRP1 are not involved in HemSC-to-EC differentiation.
VEGF-B and VEGF-A induced strong ERK1/2 phosphorylation in HemSCs after 2 to 5 minutes. In contrast, HDMEC showed weak VEGF-B-induced ERK1/2 activation only after 10 to 15 minutes (Figure 2C). VEGF-B does not bind to VEGFR-2, thereby we hypothesize that VEGF-B added to HDMECs acts by occupying VEGFR-1 binding sites and rendering endogenous VEGF-A available for binding to VEGFR-2 because the high affinity binding sites on VEGFR-1 are occupied.39 This indirect mechanism may explain why ERK 1/2 activation in response to VEGF-B occurs more slowly in HDMECs compared with HemSCs.
We showed that VEGF-induced ERK1/2 phosphorylation is needed for HemSC-to-EC differentiation using the ERK inhibitor U0126. U0126 prevented HemSC-to-EC differentiation and impaired tube formation in Matrigel, without affecting survival of HemSCs. We also observed phosphoERK1/2 expression in patient-derived IH tissue, around the immature blood vessels and in some cells outside the endothelium that could be nascent ECs. ERK1/2 activation has been implicated in the differentiation of various cell types in vivo and in vitro, including neuronal cells, ECs, and cells of the visual cortex.33, 40, 41, 42, 43 EC differentiation from murine multipotent adult progenitor cells is induced by VEGF-A and resulted in a sustained phosphorylation of the p42 subunit of ERK33; however, in this study, they did not identify the VEGF-A receptor involved in the signaling pathway.
Based on the IH model of vasculogenesis, we propose that VEGF-A produced by the HemSCs promotes ERK1/2 mediated endothelial differentiation in an autocrine fashion. In turn HemSC-derived ECs secrete VEGF-B, which can then potentiate further HemSC-to-EC differentiation (Figure 6).
Figure 6.
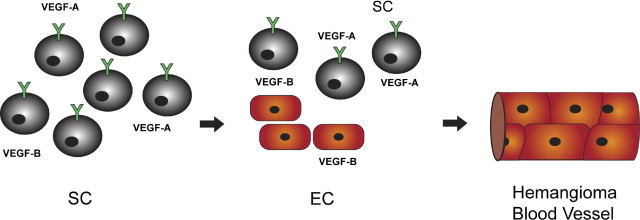
Hypothesis of HemSC-to-EC differentiation in IH tissue. HemSCs produce VEGF-A, which in turn mediates endothelial differentiation in an autocrine fashion by binding to VEGFR1. HemSC-derived ECs secrete VEGF-B, which can then potentiate further HemSC-to-EC differentiation leading to IH blood vessel formation.
In summary, we showed that HemSCs isolated from IH recapitulate endothelial differentiation in vitro and in vivo in a VEGFR-1–dependent manner. We demonstrated that the ligands VEGF-A and VEGF-B act by stimulating ERK1/2 phosphorylation though VEGFR-1. Preventing VEGFR-1 and subsequent ERK1/2 activation could be a therapeutic strategy to slow IH progression.
Acknowledgments
We thank the Dana-Farber/Harvard Cancer Center for Specialized Histopathology Core; Jill Wylie-Sears, Shoshana Greenberger, Lan Huang, and Camille L. Stewart for technical assistance, David Smadja for critical suggestions, and Kristin Johnson and Danielle Stanton for preparation of the figures.
Footnotes
Supported by NIH grant HL096384-01 and the by the Garrett Smith Foundation (E.B.).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.07.040
Supplementary data
A: Real-time PCR (RT-PCR) (left) and protein levels measured by enzyme-linked immunosorbent assay (right) for VEGF-A expression in different cell types, from normal tissues (HDMEC, cbEPC, bmMPC) and IH-derived (HemSC, HemEC). B: VEGF-B immunostaining (green) in proliferating IH and placenta, IgG was used as negative control in proliferating IH tissue. Nuclei are counterstained with DAPI (blue). Scale bar, 100 μm. (C) RT-PCR for VEGF-B167 and VEGF-B186 in different tissues: placenta, foreskin, proliferating IH (75, 76, 82) involuting IH (I-39, I-47, I-52), and involuted IH (ID3, ID16). HEOMA, hemangioendothelioma; NICH, noninvoluting congenital hemangioma; RICH, rapidly involuting congenital hemangioma. (D) RT-PCR for VEGF-B167 and VEGF-B186 in endothelial and mesenchymal/stem cells from normal tissues and IH. bmMSC, bone marrow–derived mesenchymal stem cells; cbEPC, cord blood–derived endothelial progenitor cells; HDMEC, Human Dermal Microvascular Endothelial Cells; HemEC, hemangioma-derived endothelial cells; HemSC, hemangioma-derived stem cells. Data are mean ± SEM.
A: Proliferating and involuting IH specimens stained with anti-phosphoERK antibody (dark brown). B: Immunoblot of phospho-ERK and ERK expression in HDMECs and HemSCs obtained from four different patient-derived HIs (numbers 97, 106, 124, 125) in response to 0, 2, and 5 minutes' treatment with VEGF-B (25 ng/mL). C: Immunoblot of phosphoERK and ERK expression in HemSC in response to PlGF at different concentrations (10–100 ng/mL) and to PLGF at 10 ng/mL for different treatment times. D: RT-PCR analyses for VEGFR-1 and VEGFR-2 expression levels in HDMEC, HemECs from three different IH specimens, numbers 26, 131, 133, bmMPC, and HemSC from four different IH specimens (numbers 97, 125, 131, 133). E: HDMECs and HemSCs pretreated for 1 hour with PI3K inhibitor Wortmannin and then with VEGF-B (25 ng/mL) (left) or with VEGF-A (25 ng/mL) (right). Cell lysates analyzed by immunoblot for phospho-ERK and ERK expression.
A: Immunoblot (left) and RT-PCR (right) for NRP-1 in HDMECs, and IH-derived endothelial cells (HemEC) and IH-derived stem cells (HemSC); PAE, porcine aortic endothelial cells; PAE-NRP-1, PAEs transfected with vector containing NRP-1 gene sequence; PAE-NT, PAEs transfected with empy vector. B: Cell survival at 48 and (C) 72 hours, measured with MTT assay, for HemSCs treated with different concentrations (0, 10, 20, 40 μmol/L) of AAC789. Data are as mean ± SD.
A: Cell proliferation of HemSC nontarget shRNA (Control) and VEGFR-1 shRNA (shVEGFR-1) (F2 and F4) at 0, 24, 48, and 72 hours, in regular growth medium (EBM2/20%FBS) and (B) in reduced medium EBM2/0.1%BSA without growth factors. Data are mean ± SD.
References
- 1.Fong G.H., Rossant J., Gertsenstein M., Breitman M.L. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:66–70. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- 2.Hiratsuka S., Maru Y., Okada A., Seiki M., Noda T., Shibuya M. Involvement of Flt-1 tyrosine kinase (vascular endothelial growth factor receptor-1) in pathological angiogenesis. Cancer Res. 2001;61:1207–1213. [PubMed] [Google Scholar]
- 3.Kaplan R.N., Riba R.D., Zacharoulis S., Bramley A.H., Vincent L., Costa C., MacDonald D.D., Jin D.K., Shido K., Kerns S.A., Zhu Z., Hicklin D., Wu Y., Port J.L., Altorki N., Port E.R., Ruggero D., Shmelkov S.V., Jensen K.K., Rafii S., Lyden D. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature. 2005;438:820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fischer C., Mazzone M., Jonckx B., Carmeliet P. FLT1 and its ligands VEGFB and PlGF: drug targets for anti-angiogenic therapy? Nat Rev Cancer. 2008;8:942–956. doi: 10.1038/nrc2524. [DOI] [PubMed] [Google Scholar]
- 5.Carmeliet P., Ferreira V., Breier G., Pollefeyt S., Kieckens L., Gertsenstein M., Fahrig M., Vandenhoeck A., Harpal K., Eberhardt C., Declercq C., Pawling J., Moons L., Collen D., Risau W., Nagy A. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 6.Kanda M., Nomoto S., Nishikawa Y., Sugimoto H., Kanazumi N., Takeda S., Nakao A. Correlations of the expression of vascular endothelial growth factor B and its isoforms in hepatocellular carcinoma with clinico-pathological parameters. J Surg Oncol. 2008;98:190–196. doi: 10.1002/jso.21095. [DOI] [PubMed] [Google Scholar]
- 7.Shintani S., Ishikawa T., Nonaka T., Li C., Nakashiro K., Wong D.T., Hamakawa H. Growth-regulated oncogene-1 expression is associated with angiogenesis and lymph node metastasis in human oral cancer. Oncology. 2004;66:316–322. doi: 10.1159/000078333. [DOI] [PubMed] [Google Scholar]
- 8.Bry M., Kivela R., Holopainen T., Anisimov A., Tammela T., Soronen J., Silvola J., Saraste A., Jeltsch M., Korpisalo P., Carmeliet P., Lemstrom K.B., Shibuya M., Yla-Herttuala S., Alhonen L., Mervaala E., Andersson L.C., Knuuti J., Alitalo K. Vascular endothelial growth factor-B acts as a coronary growth factor in transgenic rats without inducing angiogenesis, vascular leak, or inflammation. Circulation. 2010;122:1725–1733. doi: 10.1161/CIRCULATIONAHA.110.957332. [DOI] [PubMed] [Google Scholar]
- 9.Wei S.C., Tsao P.N., Yu S.C., Shun C.T., Tsai-Wu J.J., Wu C.H., Su Y.N., Hsieh F.J., Wong J.M. Placenta growth factor expression is correlated with survival of patients with colorectal cancer. Gut. 2005;54:666–672. doi: 10.1136/gut.2004.050831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parr C., Watkins G., Boulton M., Cai J., Jiang W.G. Placenta growth factor is over-expressed and has prognostic value in human breast cancer. Eur J Cancer. 2005;41:2819–2827. doi: 10.1016/j.ejca.2005.07.022. [DOI] [PubMed] [Google Scholar]
- 11.Fischer C., Jonckx B., Mazzone M., Zacchigna S., Loges S., Pattarini L., Chorianopoulos E., Liesenborghs L., Koch M., De Mol M., Autiero M., Wyns S., Plaisance S., Moons L., van Rooijen N., Giacca M., Stassen J.M., Dewerchin M., Collen D., Carmeliet P. Anti-PlGF inhibits growth of VEGF(R)-inhibitor-resistant tumors without affecting healthy vessels. Cell. 2007;131:463–475. doi: 10.1016/j.cell.2007.08.038. [DOI] [PubMed] [Google Scholar]
- 12.Hiratsuka S., Minowa O., Kuno J., Noda T., Shibuya M. Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc Natl Acad Sci USA. 1998;95:9349–9354. doi: 10.1073/pnas.95.16.9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kearney J.B., Kappas N.C., Ellerstrom C., DiPaola F.W., Bautch V.L. The VEGF receptor flt-1 (VEGFR-1) is a positive modulator of vascular sprout formation and branching morphogenesis. Blood. 2004;103:4527–4535. doi: 10.1182/blood-2003-07-2315. [DOI] [PubMed] [Google Scholar]
- 14.Roberts D.M., Kearney J.B., Johnson J.H., Rosenberg M.P., Kumar R., Bautch V.L. The vascular endothelial growth factor (VEGF) receptor Flt-1 (VEGFR-1) modulates Flk-1 (VEGFR-2) signaling during blood vessel formation. Am J Pathol. 2004;164:1531–1535. doi: 10.1016/S0002-9440(10)63711-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jinnin M., Medici D., Park L., Limaye N., Liu Y., Boscolo E., Bischoff J., Vikkula M., Boye E., Olsen B.R. Suppressed NFAT-dependent VEGFR1 expression and constitutive VEGFR2 signaling in infantile hemangioma. Nat Med. 2008;14:1236–1246. doi: 10.1038/nm.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bae D.G., Kim T.D., Li G., Yoon W.H., Chae C.B. Anti-flt1 peptide, a vascular endothelial growth factor receptor 1-specific hexapeptide, inhibits tumor growth and metastasis. Clin Cancer Res. 2005;11:2651–2661. doi: 10.1158/1078-0432.CCR-04-1564. [DOI] [PubMed] [Google Scholar]
- 17.Luttun A., Tjwa M., Moons L., Wu Y., Angelillo-Scherrer A., Liao F., Nagy J.A., Hooper A., Priller J., De Klerck B., Compernolle V., Daci E., Bohlen P., Dewerchin M., Herbert J.M., Fava R., Matthys P., Carmeliet G., Collen D., Dvorak H.F., Hicklin D.J., Carmeliet P. Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nat Med. 2002;8:831–840. doi: 10.1038/nm731. [DOI] [PubMed] [Google Scholar]
- 18.Autiero M., Waltenberger J., Communi D., Kranz A., Moons L., Lambrechts D., Kroll J., Plaisance S., De Mol M., Bono F., Kliche S., Fellbrich G., Ballmer-Hofer K., Maglione D., Mayr-Beyrle U., Dewerchin M., Dombrowski S., Stanimirovic D., Van Hummelen P., Dehio C., Hicklin D.J., Persico G., Herbert J.M., Shibuya M., Collen D., Conway E.M., Carmeliet P. Role of PlGF in the intra- and intermolecular cross talk between the VEGF receptors Flt1 and Flk1. Nat Med. 2003;9:936–943. doi: 10.1038/nm884. [DOI] [PubMed] [Google Scholar]
- 19.Kami J., Muranaka K., Yanagi Y., Obata R., Tamaki Y., Shibuya M. Inhibition of choroidal neovascularization by blocking vascular endothelial growth factor receptor tyrosine kinase. Jpn J Ophthalmol. 2008;52:91–98. doi: 10.1007/s10384-007-0506-6. [DOI] [PubMed] [Google Scholar]
- 20.Mulliken J.B. Cutaneous vascular anomalies. Semin Vasc Surg. 1993;6:204–218. [PubMed] [Google Scholar]
- 21.Chang L.C., Haggstrom A.N., Drolet B.A., Baselga E., Chamlin S.L., Garzon M.C., Horii K.A., Lucky A.W., Mancini A.J., Metry D.W., Nopper A.J., Frieden I.J. Growth characteristics of infantile hemangiomas: implications for management. Pediatrics. 2008;122:360–367. doi: 10.1542/peds.2007-2767. [DOI] [PubMed] [Google Scholar]
- 22.Picard A., Boscolo E., Khan Z.A., Bartch T.C., Mulliken J.B., Vazquez M.P., Bischoff J. IGF-2 and FLT-1/VEGF-R1 mRNA levels reveal distinctions and similarities between congenital and common infantile hemangioma. Pediatr Res. 2008;63:263–267. doi: 10.1203/PDR.0b013e318163a243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greenberger S., Boscolo E., Adini I., Mulliken J.B., Bischoff J. Corticosteroid suppression of VEGF-A in infantile hemangioma-derived stem cells. N Engl J Med. 2010;362:1005–1013. doi: 10.1056/NEJMoa0903036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khan Z.A., Boscolo E., Picard A., Psutka S., Melero-Martin J.M., Bartch T.C., Mulliken J.B., Bischoff J. Multipotential stem cells recapitulate human infantile hemangioma in immunodeficient mice. J Clin Invest. 2008;118:2592–2599. doi: 10.1172/JCI33493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Khan Z.A., Melero-Martin J.M., Wu X., Paruchuri S., Boscolo E., Mulliken J.B., Bischoff J. Endothelial progenitor cells from infantile hemangioma and umbilical cord blood display unique cellular responses to endostatin. Blood. 2006;108:915–921. doi: 10.1182/blood-2006-03-006478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wood J.M., Bold G., Buchdunger E., Cozens R., Ferrari S., Frei J., Hofmann F., Mestan J., Mett H., O'Reilly T., Persohn E., Rosel J., Schnell C., Stover D., Theuer A., Towbin H., Wenger F., Woods-Cook K., Menrad A., Siemeister G., Schirner M., Thierauch K.H., Schneider M.R., Drevs J., Martiny-Baron G., Totzke F. PTK787/ZK 222584, a novel and potent inhibitor of vascular endothelial growth factor receptor tyrosine kinases, impairs vascular endothelial growth factor-induced responses and tumor growth after oral administration. Cancer Res. 2000;60:2178–2189. [PubMed] [Google Scholar]
- 27.Yu Y., Hulmes J.D., Herley M.T., Whitney R.G., Crabb J.W., Sato J.D. Direct identification of a major autophosphorylation site on vascular endothelial growth factor receptor Flt-1 that mediates phosphatidylinositol 3′-kinase binding. Biochem J. 2001;358:465–472. doi: 10.1042/0264-6021:3580465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bold G., Frei J., Furet P., Manley P., Bruggen J., Cozens R., Ferrari S., Hofmann F., Martiny-Baron G., Mestan J., Meyer T., Wood J. CGP 79787D (PTK787/ZK22584): CGP 84738, NVP-AAC789, NVP-AAD777 and related 1-anilino-(4-pyridylmethyl)phthalazines as inhibitors of VEGF- and bFGF-induced angiogenesis. Drugs Future. 2002;27:43–55. [Google Scholar]
- 29.Boscolo E., Bischoff J. Vasculogenesis in infantile hemangioma. Angiogenesis. 2009;12:197–207. doi: 10.1007/s10456-009-9148-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yin A.H., Miraglia S., Zanjani E.D., Almeida-Porada G., Ogawa M., Leary A.G., Olweus J., Kearney J., Buck D.W. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. 1997;90:5002–5012. [PubMed] [Google Scholar]
- 31.Wang R., Chadalavada K., Wilshire J., Kowalik U., Hovinga K.E., Geber A., Fligelman B., Leversha M., Brennan C., Tabar V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. 2010;468:824–838. doi: 10.1038/nature09624. [DOI] [PubMed] [Google Scholar]
- 32.Ricci-Vitiani L., Pallini R., Biffoni M., Todaro M., Invernici G., Cenci T., Maira G., Parati E.A., Stassi G., Larocca L.M., De Maria R. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;468:829–833. doi: 10.1038/nature09557. [DOI] [PubMed] [Google Scholar]
- 33.Xu J., Liu X., Jiang Y., Chu L., Hao H., Liua Z., Verfaillie C., Zweier J., Gupta K., Liu Z. MAPK/ERK signalling mediates VEGF-induced bone marrow stem cell differentiation into endothelial cell. J Cell Mol Med. 2008;12:2395–2406. doi: 10.1111/j.1582-4934.2008.00266.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.James D., Nam H.S., Seandel M., Nolan D., Janovitz T., Tomishima M., Studer L., Lee G., Lyden D., Benezra R., Zaninovic N., Rosenwaks Z., Rabbany S.Y., Rafii S. Expansion and maintenance of human embryonic stem cell-derived endothelial cells by TGFbeta inhibition is Id1 dependent. Nature Biotechnol. 2010;28:161–166. doi: 10.1038/nbt.1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sase H., Watabe T., Kawasaki K., Miyazono K., Miyazawa K. VEGFR2-PLCgamma1 axis is essential for endothelial specification of VEGFR2+ vascular progenitor cells. J Cell Sci. 2009;122:3303–3311. doi: 10.1242/jcs.049908. [DOI] [PubMed] [Google Scholar]
- 36.Kawasaki K., Watabe T., Sase H., Hirashima M., Koide H., Morishita Y., Yuki K., Sasaoka T., Suda T., Katsuki M., Miyazono K., Miyazawa K. Ras signaling directs endothelial specification of VEGFR2+ vascular progenitor cells. J Cell Biol. 2008;181:131–141. doi: 10.1083/jcb.200709127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gualandris A., Noghero A., Geuna M., Arese M., Valdembri D., Serini G., Bussolino F. Microenvironment drives the endothelial or neural fate of differentiating embryonic stem cells coexpressing neuropilin-1 and Flk-1. FASEB J. 2009;23:68–78. doi: 10.1096/fj.08-112847. [DOI] [PubMed] [Google Scholar]
- 38.Yamamizu K., Kawasaki K., Katayama S., Watabe T., Yamashita J.K. Enhancement of vascular progenitor potential by protein kinase A through dual induction of Flk-1 and Neuropilin-1. Blood. 2009;114:3707–3716. doi: 10.1182/blood-2008-12-195750. [DOI] [PubMed] [Google Scholar]
- 39.Shibuya M., Claesson-Welsh L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp Cell Res. 2006;312:549–560. doi: 10.1016/j.yexcr.2005.11.012. [DOI] [PubMed] [Google Scholar]
- 40.Di Cristo G., Berardi N., Cancedda L., Pizzorusso T., Putignano E., Ratto G.M., Maffei L. Requirement of ERK activation for visual cortical plasticity. Science. 2001;292:2337–2340. doi: 10.1126/science.1059075. [DOI] [PubMed] [Google Scholar]
- 41.Stavridis M.P., Lunn J.S., Collins B.J., Storey K.G. A discrete period of FGF-induced Erk1/2 signalling is required for vertebrate neural specification. Development. 2007;134:2889–2894. doi: 10.1242/dev.02858. [DOI] [PubMed] [Google Scholar]
- 42.Lu J., Zhao J., Liu K., Yang H., Huang Y., Qin Z., Bai R., Li P., Ma J., Yan W., Zhao M., Dong Z. MAPK/ERK1/2 signaling mediates endothelial-like differentiation of immature DCs in the microenvironment of esophageal squamous cell carcinoma. Cell Mol Life Sci. 2010;67:2091–2106. doi: 10.1007/s00018-010-0316-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cowley S., Paterson H., Kemp P., Marshall C.J. Activation of MAP kinase kinase is necessary and sufficient for PC12 differentiation and for transformation of NIH 3T3 cells. Cell. 1994;77:841–852. doi: 10.1016/0092-8674(94)90133-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A: Real-time PCR (RT-PCR) (left) and protein levels measured by enzyme-linked immunosorbent assay (right) for VEGF-A expression in different cell types, from normal tissues (HDMEC, cbEPC, bmMPC) and IH-derived (HemSC, HemEC). B: VEGF-B immunostaining (green) in proliferating IH and placenta, IgG was used as negative control in proliferating IH tissue. Nuclei are counterstained with DAPI (blue). Scale bar, 100 μm. (C) RT-PCR for VEGF-B167 and VEGF-B186 in different tissues: placenta, foreskin, proliferating IH (75, 76, 82) involuting IH (I-39, I-47, I-52), and involuted IH (ID3, ID16). HEOMA, hemangioendothelioma; NICH, noninvoluting congenital hemangioma; RICH, rapidly involuting congenital hemangioma. (D) RT-PCR for VEGF-B167 and VEGF-B186 in endothelial and mesenchymal/stem cells from normal tissues and IH. bmMSC, bone marrow–derived mesenchymal stem cells; cbEPC, cord blood–derived endothelial progenitor cells; HDMEC, Human Dermal Microvascular Endothelial Cells; HemEC, hemangioma-derived endothelial cells; HemSC, hemangioma-derived stem cells. Data are mean ± SEM.
A: Proliferating and involuting IH specimens stained with anti-phosphoERK antibody (dark brown). B: Immunoblot of phospho-ERK and ERK expression in HDMECs and HemSCs obtained from four different patient-derived HIs (numbers 97, 106, 124, 125) in response to 0, 2, and 5 minutes' treatment with VEGF-B (25 ng/mL). C: Immunoblot of phosphoERK and ERK expression in HemSC in response to PlGF at different concentrations (10–100 ng/mL) and to PLGF at 10 ng/mL for different treatment times. D: RT-PCR analyses for VEGFR-1 and VEGFR-2 expression levels in HDMEC, HemECs from three different IH specimens, numbers 26, 131, 133, bmMPC, and HemSC from four different IH specimens (numbers 97, 125, 131, 133). E: HDMECs and HemSCs pretreated for 1 hour with PI3K inhibitor Wortmannin and then with VEGF-B (25 ng/mL) (left) or with VEGF-A (25 ng/mL) (right). Cell lysates analyzed by immunoblot for phospho-ERK and ERK expression.
A: Immunoblot (left) and RT-PCR (right) for NRP-1 in HDMECs, and IH-derived endothelial cells (HemEC) and IH-derived stem cells (HemSC); PAE, porcine aortic endothelial cells; PAE-NRP-1, PAEs transfected with vector containing NRP-1 gene sequence; PAE-NT, PAEs transfected with empy vector. B: Cell survival at 48 and (C) 72 hours, measured with MTT assay, for HemSCs treated with different concentrations (0, 10, 20, 40 μmol/L) of AAC789. Data are as mean ± SD.
A: Cell proliferation of HemSC nontarget shRNA (Control) and VEGFR-1 shRNA (shVEGFR-1) (F2 and F4) at 0, 24, 48, and 72 hours, in regular growth medium (EBM2/20%FBS) and (B) in reduced medium EBM2/0.1%BSA without growth factors. Data are mean ± SD.