

Abstract
Nonalcoholic fatty liver disease (NAFLD) is the hepatic manifestation of obesity and metabolic syndrome and contributes to increased risk of cardiovascular disease and liver-related morbidity and mortality. Indeed, obese patients with metabolic syndrome generate greater amounts of thrombin, an indication of coagulation cascade activation. However, the role of the coagulation cascade in Western diet–induced NAFLD has not been investigated. Using an established mouse model of Western diet–induced NAFLD, we tested whether the thrombin receptor protease-activated receptor 1 (PAR-1) and hematopoietic cell–derived tissue factor (TF) contribute to hepatic steatosis. In association with hepatic steatosis, plasma thrombin-antithrombin levels and hepatic fibrin deposition increased significantly in C57Bl/6J mice fed a Western diet for 3 months. PAR-1 deficiency reduced hepatic inflammation, particularly monocyte chemoattractant protein-1 expression and macrophage accumulation. In addition, PAR-1 deficiency was associated with reduced steatosis in mice fed a Western diet, including reduced liver triglyceride accumulation and CD36 expression. Similar to PAR-1 deficiency, hematopoietic cell TF deficiency was associated with reduced inflammation and reduced steatosis in livers of low-density lipoprotein receptor–deficient mice fed a Western diet. Moreover, hematopoietic cell TF deficiency reduced hepatic fibrin deposition. These studies indicate that PAR-1 and hematopoietic cell TF are required for liver inflammation and steatosis in mice fed a Western diet.
Obesity is a risk factor for the development of metabolic syndrome (MetS).1 Nonalcoholic fatty liver disease (NAFLD), the hepatic manifestation of MetS, is estimated to affect at least 25% of the Western population.2 NAFLD manifests as an excess accumulation of triglycerides in hepatocytes (ie, steatosis) but can transition to the more severe nonalcoholic steatohepatitis, which, if left unchecked, may lead to fibrosis and cirrhosis.3, 4 There is increasing evidence to suggest that hepatic steatosis is not just a benign precursor of nonalcoholic steatohepatitis but rather a central component of multiple diseases.5, 6 Indeed, disruption of key hepatic functions, including glucose and lipid homeostasis7, 8 and altered synthesis of proteins involved in hemostasis9, 10 and inflammation,11 may contribute not only to NAFLD but also to an increased risk of cardiovascular disease11, 12 and diabetes.5
Many studies examining the development of steatosis focus on changes in lipid metabolism. In patients with NAFLD, increased hepatic free fatty acid uptake, de novo synthesis of free fatty acids in the liver, and the conversion of free fatty acids to triglycerides that are stored in hepatocytes8, 13 yield a liver histologic profile typified by macrovesicular and microvesicular steatosis.14, 15 Although hepatocytes synthesize and store lipids in NAFLD, several studies indicate that proinflammatory cytokines, such as tumor necrosis factor α (TNFα) and monocyte chemoattractant protein-1 (MCP-1), are responsible for mediating changes in gene expression that contribute to the development of steatosis.16, 17, 18, 19, 20 For example, expression of the fatty acid transporter CD36 in livers of obese mice is MCP-1 dependent.21 These studies indicate that inflammatory mediators are among the necessary stimuli for lipid accumulation in hepatocytes.
One potential modifier of the hepatic inflammatory response in NAFLD is the coagulation cascade. The coagulation cascade is initiated by tissue factor (TF), resulting in generation of the serine protease thrombin. Thrombin cleaves circulating fibrinogen to fibrin monomers that are cross-linked into insoluble fibrin clots.22 In addition to its classic role in hemostasis, thrombin also triggers intracellular signaling by activating protease-activated receptor 1 (PAR-1), a tethered ligand G protein–coupled receptor.23 Obesity and MetS are associated with coagulation cascade activation in patients, as indicated by increased thrombin generation.24, 25, 26 Moreover, weight loss is associated with decreased TF expression and a reduction in thrombin generation.27, 28 An imbalance in hepatic synthesis of several coagulation factors causing a procoagulant state has been observed in patients with NAFLD.9, 10, 29, 30, 31 Increased coagulation cascade activation in patients with NAFLD is often viewed as potentially increasing the risk of cardiovascular events. In contrast, the possibility that the coagulation cascade acts as a critical determinant of diet-induced hepatic steatosis has not been investigated in detail.
One potential mechanism whereby thrombin could contribute to fatty liver disease is by increasing the production of inflammatory mediators responsible for steatosis. PAR-1 contributes to TNFα and MCP-1 expression in mice fed a methionine/choline–deficient (MCD) diet.32 The MCD diet elicits hepatic changes consistent with nonalcoholic steatohepatitis, but it is not an appropriate model of NAFLD associated with MetS.33 The mechanism of hepatic steatosis triggered by an MCD diet is vastly different from that triggered by a Western diet. For example, MCP-1 contributes to steatosis in mice fed a Western diet18, 19 but does not contribute to steatosis in mice fed an MCD diet.34 In agreement with these studies, we showed previously that PAR-1 deficiency did not affect steatosis in mice fed an MCD diet, despite marked inhibition of MCP-1 expression.32 The role of the coagulation cascade in regulating inflammation and steatosis in Western diet–induced NAFLD is not known.
We tested the hypothesis that coagulation cascade activation is increased in mice fed a Western diet and contributes to the development of steatosis. We evaluated the possibility that thrombin signaling through PAR-1 contributes to liver inflammation and steatosis in mice fed a Western diet using PAR-1–deficient mice. Moreover, using bone marrow transplantation, we tested the hypothesis that TF expressed by hematopoietic cells contributes to hepatic coagulation, inflammation, and steatosis in mice fed a Western diet.
Materials and Methods
Mice, Bone Marrow Transplantation, and Experimental Diets
Wild-type C57Bl/6J mice purchased from The Jackson Laboratory (Bar Harbor, ME) were fed a control diet [AIN-93M (10% kcal from fat); Dyets Inc., Bethlehem, PA] or a Western diet [Diet #100244 (40% kcal from milk fat); Dyets Inc.] ad libitum for 12 weeks. PAR-1+/+ and PAR-1−/− mice35 backcrossed eight generations onto a C57Bl/6J background were maintained by homozygous breeding. Age-matched PAR-1+/+ and PAR-1−/− mice were fed a control diet or a Western diet ad libitum for 12 weeks. Average food intake was measured weekly for each cage of mice, and mice were weighed weekly.
Bone marrow transplantation was performed as described previously.36 Male 8-week-old low-density lipoprotein receptor–deficient (LDLr−/−) mice were irradiated with 11 Gy (two doses of 550 rad each, 4 hours apart) using a Cs137 irradiator (JL Shepherd, San Fernando, CA). Irradiated mice were repopulated with bone marrow cells isolated from heterozygous control mice (mTF+/−hTF+ mice; n = 10 recipients) or “low TF” mice (mTF−/−hTF+ mice; n = 9 recipients) via retro-orbital injection of 2 × 106 cells. Low-TF mice express human TF at a level approximately 1% of wild-type mice, and this low level of expression rescues the embryonic lethality.37 Four weeks after injection of donor cells, mice were fed a Western diet [Teklad TD.88137 (40% kcal from milk fat); Harlan Laboratories, Madison, WI] ad libitum for 12 weeks. This diet has a formulation identical to Diet #100244 from Dyets Inc. The bone marrow of recipient mice was genotyped to verify successful repopulation of donor cells as described previously.37 All the studies were approved by the Animal Care and Use Committee of the different institutions and complied with National Institutes of Health guidelines.
Sample Collection
Under isoflurane anesthesia, blood was collected from the caudal vena cava into sodium citrate (final concentration, 0.38%) for the collection of plasma and into an empty syringe for the collection of serum. Citrated whole blood was subjected to centrifugation at 4000 × g for 10 minutes, plasma was collected, and the buffy coat was collected into TRI Reagent (Molecular Research Center Inc., Cincinnati, OH). Sections of liver from the left lateral lobe were fixed in 10% neutral-buffered formalin for 48 hours and embedded in paraffin. The right medial lobe was affixed to a cork using optimal cutting temperature compound (Fisher Scientific, Pittsburgh, PA) and immersed for approximately 3 minutes in liquid nitrogen–chilled isopentane. The remaining liver was snap frozen in liquid nitrogen.
Histopathologic Analysis, Clinical Chemistry, and Determination of Hepatic Lipid Levels
Paraffin-embedded livers were sectioned at 5 μm and were stained with H&E. The severity of liver steatosis was evaluated by a pathologist (O.T.) in a blinded manner. Oil Red O staining was performed as previously described.38 Plasma thrombin-antithrombin (TAT) levels were determined using a commercially available enzyme-linked immunosorbent assay kit (Siemens Healthcare Diagnostics, Deerfield, IL). Lipids were extracted from 100 mg of snap-frozen liver as described,32 and hepatic and serum triglyceride and cholesterol levels were determined using commercially available reagents (Pointe Scientific Inc., Canton, MI; and Wako, Richmond, VA).
RNA Isolation, cDNA Synthesis, and Real-Time PCR
Total RNA was isolated from the buffy coat of blood samples (ie, white blood cells) and from approximately 100 mg of snap-frozen liver using TRI Reagent (Molecular Research Center Inc.) per the manufacturer's protocol. One microgram of RNA was used for the synthesis of cDNA using a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA) and a C1000 thermal cycler (Bio-Rad Laboratories, Hercules, CA). Levels of stearoyl-CoA desaturase 1 (SCD1), CD36, sterol regulatory element binding protein-1c (SREBP-1c), peroxisome proliferator-activated receptor-α (PPARα), fatty acid synthase (FAS), TNFα, MCP-1, TF mRNA, and 18S rRNA were determined using either TaqMan gene expression assays (Applied Biosystems) or TaqMan PrimeTime quantitative PCR assays (IDT, Coralville, IA), iTaq + ROX supermix (Bio-Rad Laboratories), and a StepOnePlus thermal cycler (Applied Biosystems). Mouse CD36 (NM_007643) primer sequences were 5′-GCGACATGATTAATGGCACAG-3′ (forward primer), 5′-GATCCGAACACAGCGTAGATAG-3′ (reverse primer), and 5′-/56-FAM/CAACAAAAG/ZEN/GTGGAAAGGAGGCTGC/3IABkFQ/-3′ (probe). Mouse SCD1 (NM_009127) primer sequences were 5′-CTGACCTGAAAGCCGAGAAG-3′ (forward primer), 5′-AGAAGGTGCTAACGAACAGG-3′ (reverse primer), and 5′-/56-FAM/TGTTTACAA/ZEN/AAGTCTCGCCCCAGCA/3IABkFQ/-3′ (probe). Mouse SREBP-1c (NM_011480) primer sequences were 5′-CCATCGACTACATCCGCTTC-3′ (forward primer), 5′-GCCCTCCATAGACACATCTG-3′ (reverse primer), and 5′-/56-FAM/TCTCCTGCT/ZEN/TGAGCTTCTGGTTGC/3IABkFQ/-3′ (probe). Mouse FAS (NM_007988) primer sequences were 5′-CCCCTCTGTTAATTGGCTCC-3′ (forward primer), 5′-TTGTGGAAGTGCAGGTTAGG-3′ (reverse primer), and 5′-/56-FAM/CAGGCTCAG/ZEN/GGTGTCCCATGTT/3IABkFQ/-3′ (probe). Mouse PPARα (NM_011144) primer sequences were 5′-CATTTCCCTGTTTGTGGCTG-3′ (forward primer), 5′-ATCTGGATGGTTGCTCTGC-3′ (reverse primer), and 5′-/56-FAM/ATAATTTGC/ZEN/TGTGGAGATCGGCCTGG/3IABkFQ/-3′ (probe). Mouse TF (F3) (NM_010171) primer sequences were 5′-CAGTTCATGGAGACGGAGAC-3′ (forward primer), 5′-CAACCACGTTCAGTTTTCTACC-3′ (reverse primer), and 5′-/56-FAM/AGACACAAA/ZEN/CCTCGGACAGCCAG/3IABkFQ/-3′ (probe). 18S (NM_003286) primer sequences were 5′-CTGTAGCCCTGTACTTCATCG-3′ (forward primer), 5′-CTACCACATATTCCTGACCATCC-3′ (reverse primer), and 5′-/56-FAM/CCTTCCTCC/ZEN/TTTTCATTGCCTGCTCT/3IABkFQ/-3′ (probe). CD36, SCD1, FAS, SREBP-1c, PPARα, TF, and 18S primers and probes were purchased from IDT. Mouse TNFα and MCP-1 gene expression assays were purchased from Applied Biosystems (TNFα, Assay ID Mm00443258_m1; MCP-1, Assay ID Mm00441242_m1). The expression of each gene was adjusted relative to 18S expression levels, and the relative expression level was determined using the comparative CT method.
Macrophage and Fibrin Staining
Frozen livers were sectioned at 8 μm for staining. Macrophages were identified in liver by CD68 and F4/80 staining, which was performed as previously described.32 Immunofluorescent staining for insoluble fibrin was performed as described previously39 using a rabbit anti-human fibrinogen antibody (A0080; Dako, Carpinteria, CA). The primary antibody was detected by the addition of goat anti-rabbit IgG conjugated to Alexa 594 (Invitrogen, Carlsbad, CA). Slides were then washed and counterstained with DAPI (Invitrogen).
Cytokine Enzyme-Linked Immunosorbent Assays
Total protein was isolated from approximately 100 mg of snap-frozen liver using PBS containing 0.1% Triton X-100 and Halt protease and phosphatase inhibitor cocktail (ThermoFisher Scientific, Waltham, MA). Samples were rotated for 30 minutes at 4°C and then were subjected to centrifugation at 12,000 × g for 15 minutes at 4°C. Protein concentrations were determined using a DC protein assay (ThermoFisher Scientific). The concentrations of TNFα and MCP-1 in 225 μg of protein were determined using commercially available DuoSet enzyme-linked immunosorbent assays (R&D Systems, Minneapolis, MN).
Multiplex Suspension Array
The concentration of TNFα and MCP-1 protein in serum was determined using a MilliPlex mouse metabolic magnetic bead kit (Millipore, Billerica, MA) and a Bio-Plex 200 System (Bio-Rad Laboratories). Phycoerythrin and xMAP bead fluorescence was detected by a dual-laser detector (532 and 635 nm) and Bio-Plex Manager 5.0 software (Bio-Rad Laboratories).
Western Blot Analysis
Total protein was isolated from approximately 100 mg of snap-frozen liver using radioimmunoprecipitation assay buffer containing Halt protease and phosphatase inhibitors (ThermoFisher Scientific). Protein concentrations were determined using a DC protein assay (ThermoFisher Scientific). Samples were reduced by heating to 95°C for 5 minutes in the presence of 2-mercaptoethanol, subjected to SDS-PAGE (Criterion 4% to 12% Bis-Tris gels; Bio-Rad Laboratories), and transferred to Immobilon polyvinylidene difluoride membrane (Millipore) by semidry transfer. The membranes were blocked for 1 hour at room temperature in 3% bovine serum albumin in Tris-buffered saline with Tween-20 and incubated overnight at 4°C with either anti-CD36 (1:1000) antibody or anti-PPARα (1:1000) antibody (BioVision, Mountain View, CA) diluted in 1% bovine serum albumin in Tris-buffered saline with Tween-20 or anti-SCD1 (1:1000) antibody (Cell Signaling Technology Inc., Danvers, MA) diluted in 5% bovine serum albumin in Tris-buffered saline with Tween-20. Membranes were then washed with Tris-buffered saline with Tween-20 and incubated for 1 hour with anti-rabbit horseradish peroxidase–conjugated secondary antibody (1:1000) (Jackson ImmunoResearch Laboratories Inc., West Grove, PA). Membranes were incubated with West Pico electrochemiluminescence reagent (ThermoFisher Scientific) before exposure to Blue Lite autoradiography film (ISC Bioexpress, Kaysville, UT). Before the addition of antibodies, membranes were stained with Ponceau S for 15 minutes and were destained with ddH2O to confirm equal loading of protein, as previously described.40, 41 Densitometry was performed using Quantity One 4.6.9 software (Bio-Rad Laboratories).
Statistics
Statistics were performed using SigmaPlot 11.0 software (Systat Software, Inc., San Jose, CA). Comparison of two groups was performed using a Student's t-test. Comparison of three or more groups was performed using two-way analysis of variance followed by the Student-Newman-Keuls post hoc test for multiple comparisons. Data are expressed as mean ± SEM. The criterion for statistical significance was P ≤ 0.05.
Results
Hepatic Steatosis and Coagulation Are Increased in C57Bl/6J Mice Fed a Western Diet
Mice fed a control diet gained 47.5% ± 2.5% body weight over 12 weeks, whereas mice fed a Western diet gained 92.1% ± 2.5% body weight (P ≤ 0.05). Microvesicular and macrovesicular steatosis was evident in livers of mice fed a Western diet but not in livers of mice fed a control diet, as indicated by histologic features (Figure 1, A and B). To determine whether the coagulation cascade was activated in mice fed a Western diet, we measured the plasma TAT level, which is a stable marker for thrombin generation (ie, coagulation cascade activation). Plasma TAT levels were significantly increased in mice fed a Western diet compared with mice fed a control diet (Figure 1E). To determine whether the coagulation cascade was activated in livers of mice fed a Western diet, we evaluated hepatic fibrin deposition, one consequence of thrombin generation. Fibrin deposition was increased in livers of mice fed a Western diet compared with mice fed a control diet (Figure 1, C and D), indicating a local increase in coagulation in the liver. Because previous studies suggested that hematopoietic cells, such as monocytes, could be a source of TF in obesity and MetS,42, 43 we measured TF mRNA levels in the buffy coat (white blood cells) isolated from blood of wild-type mice fed a control diet or a Western diet. White blood cell TF mRNA expression was increased significantly in mice fed a Western diet (Figure 1F).
Figure 1.
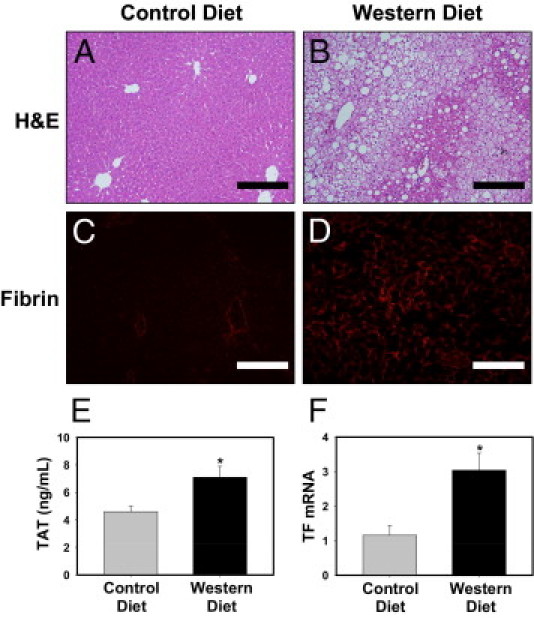
Procoagulant response associated with Western diet–induced steatosis in C57Bl/6J mice. Mice were fed either a control diet or a Western diet for 12 weeks. A–D: Representative photomicrographs showing H&E-stained (A and B) and fibrin-stained (red) (C and D) liver sections from mice fed a control diet (A and C) or a Western diet (B and D). Scale bars = 20 μm. E: Plasma TAT levels were determined. Data are expressed as mean ± SEM. F: The level of TF mRNA in the buffy coat (white blood cells) was determined by real-time PCR. Data are expressed as mean ± SEM and as fold change versus mice fed a control diet. n = 6 to 10 mice per group. *P ≤ 0.05 versus mice fed the control diet.
PAR-1 Deficiency Reduces Inflammation in Livers of Mice Fed a Western Diet
Previous studies have indicated a role for inflammation in the development of hepatic steatosis in mice fed a Western diet.18, 19, 44, 45, 46 One possible mechanism whereby thrombin could contribute to inflammation is by activating the receptor PAR-1. To evaluate the role of PAR-1 in liver inflammation induced by feeding a Western diet, we used PAR-1−/− mice. Plasma TAT levels tended to increase similarly in PAR-1+/+ and PAR-1−/− mice fed a Western diet, indicating that PAR-1 deficiency does not modulate activation of the coagulation cascade (data not shown). Expression of hepatic TNFα and MCP-1 mRNA was increased in PAR-1+/+ mice fed a Western diet, and this was significantly reduced by PAR-1 deficiency (Figure 2, A and B). Serum and liver MCP-1 protein concentrations were also significantly decreased in PAR-1−/− mice fed a Western diet compared with in PAR-1+/+ mice fed a Western diet (Figure 2, C and D). However, serum and liver TNFα protein levels were not significantly increased in mice fed a Western diet, and there was no difference in TNFα protein expression in PAR-1−/− mice compared with PAR-1+/+ mice fed a Western diet (data not shown). In addition, hepatic macrophage accumulation increased in PAR-1+/+ mice fed a Western diet, and this increase was largely prevented by PAR-1 deficiency (Figure 3, A, D, G, and J).
Figure 2.
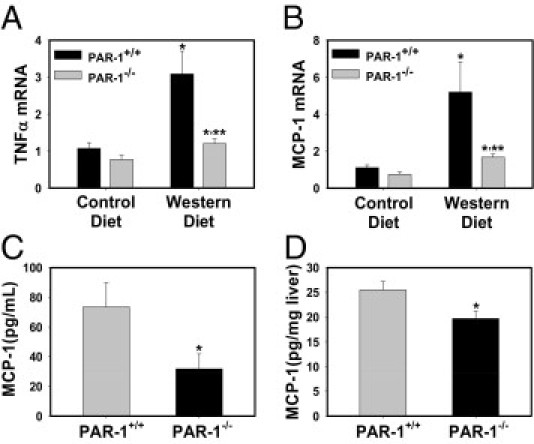
Effect of PAR-1 deficiency on inflammatory cytokine induction in mice fed a Western diet. PAR-1+/+ and PAR-1−/− mice were fed either a control diet or a Western diet for 12 weeks. Hepatic levels of (A) TNFα mRNA and (B) MCP-1 mRNA were determined by real-time PCR. Data are expressed as mean ± SEM and as a fold change versus PAR-1+/+ mice fed a control diet. *Significantly different from mice of the same genotype fed the control diet, P ≤ 0.05. **Significantly different from PAR-1+/+ mice fed the same diet, P ≤ 0.05. Serum (C) and hepatic (D) levels of MCP-1 protein in mice fed a Western diet were determined by Multiplex or enzyme-linked immunosorbent assay as stated in Materials and Methods. Data are expressed as mean ± SEM. n = 7 to 10 mice per group. Significantly different from PAR-1+/+ mice fed the same diet, P ≤ 0.05.
Figure 3.
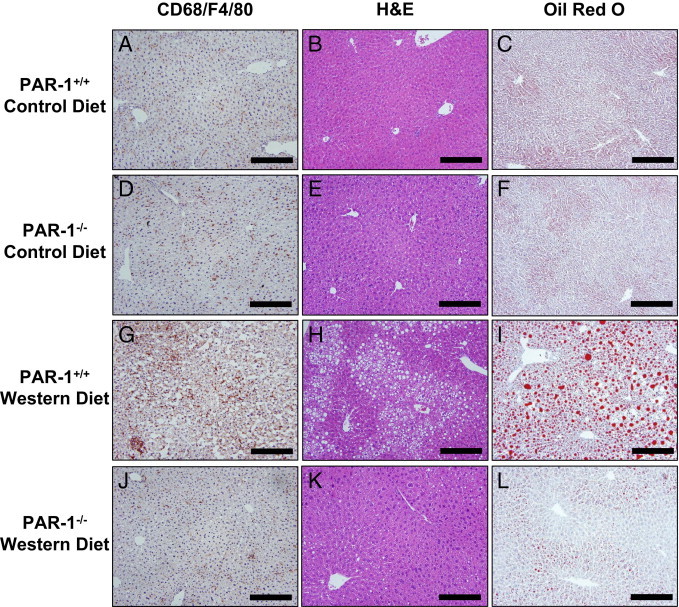
Effect of PAR-1 deficiency on macrophage accumulation and steatosis in livers of mice fed a Western diet. PAR-1+/+ and PAR-1−/− mice were fed either a control diet or a Western diet for 12 weeks. Representative photomicrographs of CD68 and F4/80–stained (brown) liver sections (A, D, G, and J), H&E-stained liver sections (B, E, H, and K), and Oil Red O–stained liver sections (C, F, I, and L) in PAR-1+/+ (A–C) and PAR-1−/− (D–F) mice fed a control diet and in PAR-1+/+ (G–I) and PAR-1−/− (J–L) mice fed a Western diet. Scale bars = 20 μm.
PAR-1 Deficiency Reduces Steatosis in Livers of Mice Fed a Western Diet
PAR-1 deficiency did not significantly affect food intake (data not shown) or weight gain. PAR-1+/+ mice fed a control diet gained 16.9% ± 1.4% body weight over 12 weeks, and PAR-1−/− mice fed a control diet gained 15.5% ± 1.2% body weight. In addition, PAR-1+/+ mice fed a Western diet gained 40.0% ± 3.4%* body weight over 12 weeks, and PAR-1−/− mice fed a Western diet gained 34.8% ± 2.2%* body weight (*P ≤ 0.05 compared with mice of the same genotype fed a control diet, not significant between PAR-1+/+ and PAR-1−/− mice fed a Western diet). To determine whether PAR-1 contributes to the development of steatosis in mice fed a Western diet, we evaluated several markers of steatosis in PAR-1+/+ and PAR-1−/− mice fed a Western diet. Microvesicular and macrovesicular steatosis occurred in livers of PAR-1+/+ mice fed a Western diet. In contrast, the severity of steatosis was greatly reduced in PAR-1−/− mice fed a Western diet (Figure 3, B and C, E and F, H and I, and K and L; also see Supplemental Table S1 at http://ajp.amjpathol.org). In agreement with liver histopathologic findings, hepatic triglyceride and cholesterol levels increased in livers of PAR-1+/+ mice fed a Western diet (Figure 4, A and B). The increase in liver triglyceride accumulation was prevented by PAR-1 deficiency, whereas cholesterol levels were not affected (Figure 4, A and B). Serum triglyceride levels were slightly increased in PAR-1−/− mice fed a Western diet compared with in PAR-1−/− mice fed a control diet (Figure 4C), and serum cholesterol levels were increased in PAR-1+/+ and PAR-1−/− mice fed a Western diet (Figure 4D). However, there was no significant effect of PAR-1 deficiency on serum triglyceride or cholesterol levels in mice fed a Western diet.
Figure 4.
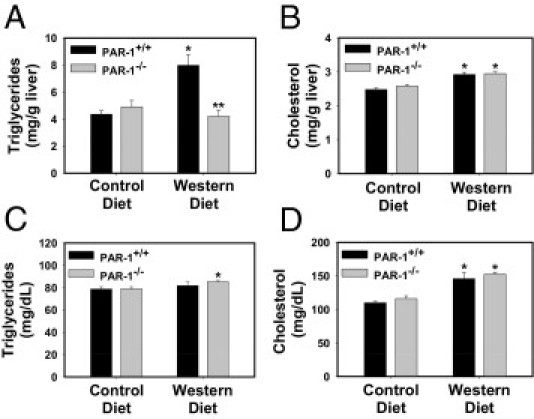
Effect of PAR-1 deficiency on lipid accumulation in mice fed a Western diet. PAR-1+/+ and PAR-1−/− mice were fed either a control diet or a Western diet for 12 weeks. Hepatic triglyceride (A), hepatic cholesterol (B), serum triglyceride (C), and serum cholesterol (D) levels were determined. Data are expressed as mean ± SEM. n = 7 to 10 mice per group. *P ≤ 0.05 versus mice of the same genotype fed a control diet; **P ≤ 0.05 versus PAR-1+/+ mice fed the same diet.
Expression of mRNAs encoding the lipogenic genes CD36, SCD1, SREBP-1c, and PPARα was increased in livers of PAR-1+/+ mice fed a Western diet (Figure 5, A–D). However, FAS mRNA expression was not changed in mice fed a Western diet (Figure 5E). PAR-1 deficiency reduced the hepatic expression of CD36, SCD1, and PPARα mRNAs but did not affect SREBP-1c mRNA expression in mice fed a Western diet (Figure 5, A–D). In addition, hepatic expression of CD36 and SCD1 protein was increased in PAR-1+/+ mice fed a Western diet (Figure 5F). In agreement with the reduction in CD36 mRNA in PAR-1−/− mice fed a Western diet, CD36 protein levels were reduced in PAR-1−/− mice fed a Western diet (Figure 5F). In contrast to expression of its mRNA, hepatic expression of SCD1 protein increased in PAR-1−/− mice fed a Western diet compared with in PAR-1+/+ mice fed a Western diet (Figure 5F). PPARα protein expression in liver was not affected by diet or genotype (data not shown). Taken together, the data indicate that PAR-1 is essential for steatosis in livers of mice fed a Western diet.
Figure 5.
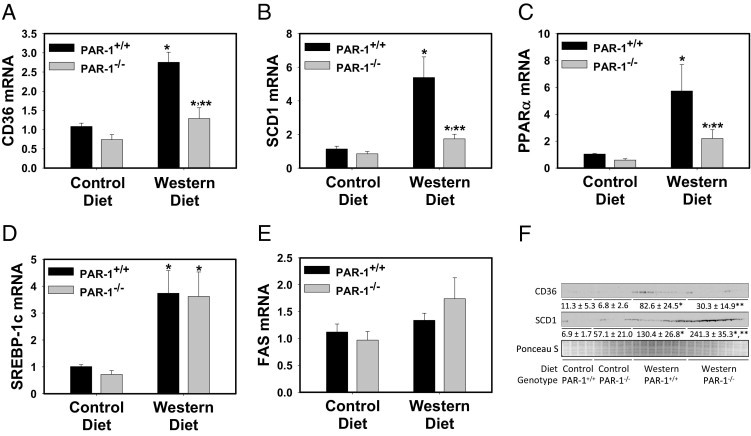
Effect of PAR-1 deficiency on lipogenic gene induction and protein expression in livers of mice fed a Western diet. PAR-1+/+ and PAR-1−/− mice were fed either a control diet or a Western diet for 12 weeks. Hepatic levels of CD36 (A), SCD1 (B), PPARα (C), SREBP-1c (D), and FAS (E) mRNA were determined by real-time PCR. Data are expressed as mean ± SEM of the fold change versus PAR-1+/+ mice fed a control diet. n = 7 to 10 mice per group. F: Hepatic CD36 and SCD1 protein levels were determined by Western blot analysis. Equal protein loading was determined by Ponceau S staining. Densitometry was performed using Quantity One software and is expressed as mean ± SEM and as intensity*mm2. *P ≤ 0.05 versus mice of the same genotype fed a control diet; **P ≤ 0.05 versus PAR-1+/+ mice fed the same diet.
Hematopoietic Cell TF Deficiency Reduces Inflammation in Livers of LDLr−/− Mice Fed a Western Diet
TF is the primary activator of the blood coagulation cascade, and hematopoietic cells, such as monocytes, could be a source of TF in obesity and MetS.42, 43 Indeed, TF expression by hematopoietic cells contributed to thrombin generation in LDLr−/− mice fed a Western diet,43 a mouse model of hyperlipidemia and atherosclerosis due to the inability to remove LDL cholesterol from the circulation. These mice also develop fatty liver disease resembling NAFLD associated with MetS.19, 47 To specifically assess the role of TF on hematopoietic cells in the development of steatosis and inflammation, LDLr−/− mice were irradiated, and the bone marrow was repopulated with bone marrow from either heterozygous (TF+/−) control mice or low-TF mice. Mice were then fed a Western diet for 12 weeks. To analyze local coagulation in the liver, we evaluated hepatic fibrin deposition. Fibrin deposition was reduced in livers of LDLr−/− mice with low-TF bone marrow compared with in LDLr−/− mice with heterozygous (TF+/−) control bone marrow after 3 months on a Western diet (Figure 6, A and B). To determine the effect of hematopoietic cell TF deficiency on hepatic inflammation, we evaluated hepatic cytokine expression and macrophage accumulation. Expression of TNFα and MCP-1 mRNAs was significantly reduced in LDLr−/− mice with low-TF bone marrow after being fed a Western diet (Figure 7, A and B). Hepatic TNFα and MCP-1 protein levels also tended to decrease in LDLr−/− mice with low-TF bone marrow after being fed a Western diet, although this difference did not achieve statistical significance (data not shown). Hepatic macrophage accumulation was also decreased in livers of LDLr−/− mice with low-TF bone marrow compared with LDLr−/− mice with TF+/− control bone marrow (Figure 6, C and D). The data indicate that hematopoietic cell–derived TF contributes to increased inflammation in livers of LDLr−/− mice fed a Western diet.
Figure 6.
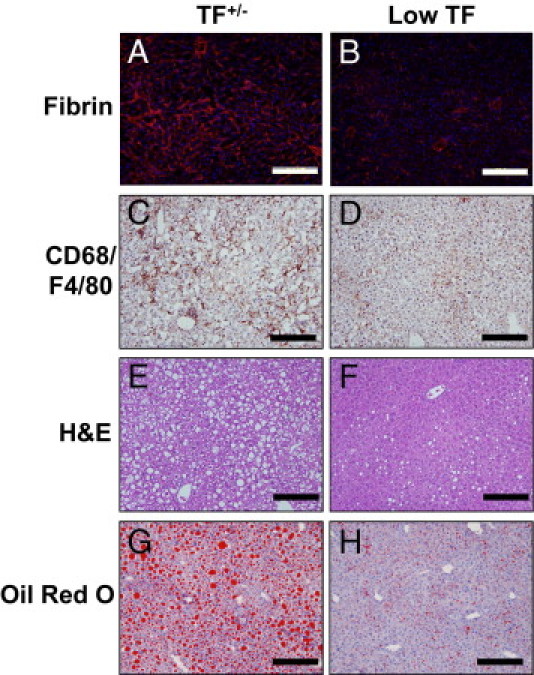
Effect of hematopoietic cell TF deficiency on coagulation, inflammation, and steatosis in livers of LDLr−/− mice fed a Western diet. LDLr−/− mice with TF+/− or low-TF bone marrow were fed a Western diet for 12 weeks. Representative photomicrographs showing fibrin staining (red) in liver sections counterstained with DAPI (blue) (A and B), CD68 and F4/80–stained (brown) liver sections (C and D), H&E-stained liver sections (E and F), and Oil Red O–stained liver sections (G and H) in mice with TF+/− (A, C, E, and G) or low-TF (B, D, F, and H) bone marrow. Scale bars = 20 μm.
Figure 7.
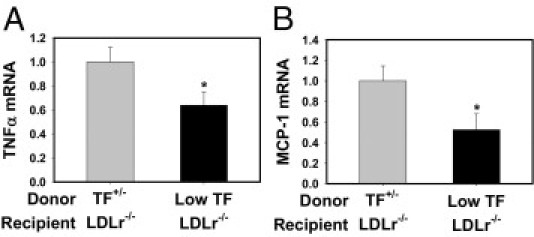
Effect of hematopoietic cell TF deficiency on hepatic inflammatory cytokine induction in LDLr−/− mice fed a Western diet. LDLr−/− mice with TF+/− or low-TF bone marrow were fed a Western diet for 12 weeks. Hepatic levels of TNFα (A) and MCP-1 (B) mRNA were determined by real-time PCR. Data are expressed as mean ± SEM and as fold change versus LDLr−/− mice with TF+/− bone marrow. n = 9 to 10 mice per group. *P ≤ 0.05 versus mice with TF+/− bone marrow.
Hematopoietic Cell TF Deficiency Reduces Steatosis in Livers of LDLr−/− Mice Fed a Western Diet
In agreement with previous studies examining NAFLD development in LDLr−/− mice,19, 47, 48 marked panacinar microvesicular and macrovesicular steatosis occurred in LDLr−/− mice with TF+/− bone marrow fed a Western diet (Figure 6, E and G; also see Supplemental Table S1 at http://ajp.amjpathol.org). The severity of steatosis was markedly reduced in LDLr−/− mice with low-TF bone marrow fed a Western diet (Figure 6, F and H; also see Supplemental Table S1 at http://ajp.amjpathol.org). Mirroring the effect of PAR-1 deficiency on hepatic steatosis, liver triglyceride levels (Figure 8A), but not liver cholesterol levels (Figure 8B), were reduced in LDLr−/− mice with low-TF bone marrow fed a Western diet compared with in LDLr−/− mice with TF+/− bone marrow fed a Western diet. Expression of mRNAs encoding the lipogenic genes CD36, SCD1, PPARα, and FAS, but not SREBP-1c, was significantly reduced by hematopoietic cell TF deficiency (Figure 9, A–E). Similar to PAR-1 deficiency, TF deficiency significantly reduced CD36 protein expression in livers of LDLr−/− mice (Figure 9F). However, hepatic SCD1 protein expression was increased in livers of LDLr−/− mice with low-TF bone marrow (Figure 9F), and hepatic PPARα protein expression was unchanged between the two groups (data not shown). The data indicate that hematopoietic cell–derived TF contributes to the development of steatosis in livers of LDLr−/− mice fed a Western diet.
Figure 8.
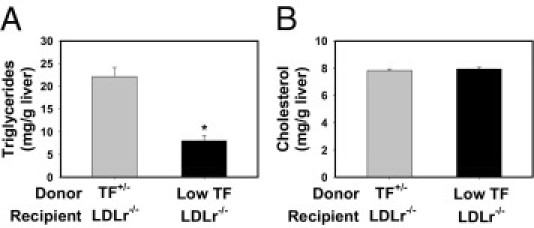
Effect of hematopoietic cell TF deficiency on hepatic lipid accumulation in livers of LDLr−/− mice fed a Western diet. LDLr−/− mice with TF+/− or low-TF bone marrow were fed a Western diet for 12 weeks. Hepatic triglyceride (A) and cholesterol (B) levels were determined. Data are expressed as mean ± SEM. n = 9 to 10 mice per group. *P ≤ 0.05 versus mice with TF+/− bone marrow.
Figure 9.
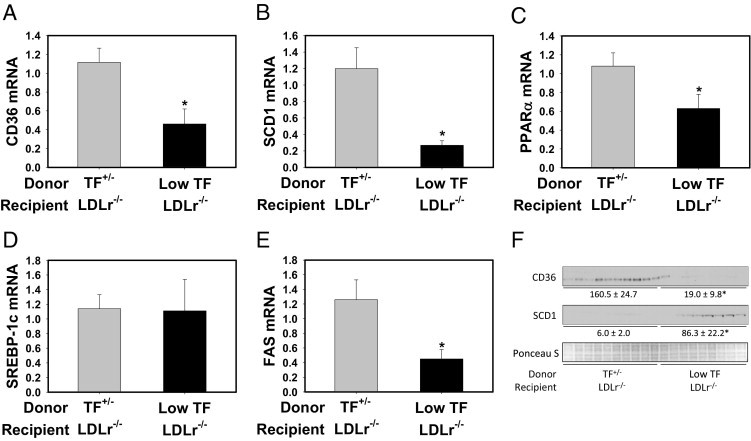
Effect of hematopoietic cell TF deficiency on hepatic lipogenic gene induction and protein expression in livers of LDLr−/− mice fed a Western diet. LDLr−/− mice with TF+/− or low-TF bone marrow were fed a Western diet for 12 weeks. Hepatic levels of CD36 (A), SCD1 (B), PPARα (C), SREBP-1c (D), and FAS (E) mRNA were determined by real-time PCR. Data are expressed as mean ± SEM and as fold change versus LDLr−/− mice with TF+/− bone marrow. n = 9 to 10 mice per group. F: Hepatic CD36 and SCD1 protein levels were determined by Western blot analysis. Equal protein loading was determined by Ponceau S staining. Densitometry was performed using Quantity One software and is expressed as mean ± SEM and as intensity*mm2. *P ≤ 0.05 versus mice with TF+/− bone marrow.
Discussion
A substantial increase in lipid accumulation occurs in livers of patients with obesity and/or MetS49, 50 and in livers of mice fed a Western diet, contributing to the development of NAFLD.18, 47, 51 Thrombin generation is also increased in patients with obesity and/or MetS,24, 27, 52, 53 and systemic coagulation and hepatic coagulation were increased in mice fed a Western diet (Figure 1). The finding that PAR-1 deficiency and hematopoietic cell TF deficiency each reduced steatosis in mice fed a Western diet strongly suggests a role for thrombin signaling in the hepatic manifestation of MetS. The studies presented herein demonstrate that increased coagulation cascade activation may have a functional consequence to promote an inflammatory environment that leads to the development of steatosis, as both the thrombin receptor PAR-1 and hematopoietic cell–derived TF contributed to macrophage infiltration and hepatic steatosis in mice fed a Western diet.
Several studies have shown that inflammatory cells and the mediators they produce are required for hepatic steatosis in mice fed a Western diet. For example, depletion of liver macrophages significantly reduced hepatic steatosis in mice fed a Western diet.16, 17 Moreover, inhibition of the monocyte/macrophage chemokine MCP-118, 19 or TNFα, a cytokine produced by Kupffer cells,46 significantly reduced hepatic steatosis. These studies strongly suggest that the recruitment and activation of liver macrophages is essential for the development of fatty liver disease in mice fed a Western diet. The present results indicate that PAR-1 contributes to hepatic expression of MCP-1 and is required for liver macrophage accumulation in mice fed a Western diet. Initiation or amplification of the inflammatory response represents one potential mechanism whereby thrombin activation of PAR-1 could contribute to hepatic steatosis in mice fed a Western diet (Figure 10).
Figure 10.
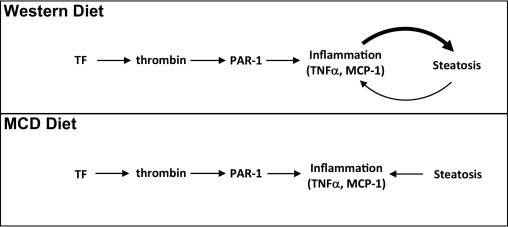
Model-dependent difference in the role of coagulation in hepatic steatosis. TF increases thrombin generation, and thrombin activation of PAR-1 increases inflammation, particularly MCP-1 expression. In mice fed a Western diet, inflammatory mediators contribute to increased steatosis in neighboring hepatocytes. However, in mice fed an MCD diet, coagulation-mediated increases in inflammatory mediators do not contribute to increased steatosis. In addition, increased hepatic steatosis contributes to further increases in inflammation in both models of NAFLD/nonalcoholic steatohepatitis.
Participation of the coagulation cascade in the inflammatory response accompanying hepatic steatosis seems to be conserved in at least two models. In previous studies, we found that TF and PAR-1 contributed to MCP-1 mRNA and protein expression in mice fed an MCD diet.32 Hepatic macrophage accumulation was significantly reduced in PAR-1−/− and low-TF mice fed an MCD diet. However, in contrast to the Western diet model, neither TF deficiency nor PAR-1 deficiency reduced hepatic steatosis in mice fed an MCD diet.32 The rapid development of severe hepatic steatosis in mice fed an MCD diet represents a complex biochemical defect not resembling the mechanism of steatosis in MetS.54 Unlike in mice fed a Western diet, inflammatory cytokines, such as MCP-1 and TNFα, do not contribute to hepatic steatosis in mice fed an MCD diet.34, 55 The fundamental contribution of inflammatory cytokines to steatosis induced by either an MCD diet or a Western diet could explain the striking difference in the role of the coagulation cascade in hepatic steatosis (Figure 10). Collectively, the present studies indicate that the coagulation cascade amplifies inflammatory cytokine expression in the MCD diet and Western diet models but also contributes to hepatic steatosis in the Western diet model, a more representative model of NAFLD in humans.
A proper balance of fatty acid uptake, triglyceride synthesis, and β-oxidation is required to maintain lipid homeostasis in the liver. Excess uptake or increased synthesis of fatty acids provokes triglyceride synthesis and storage by the liver, resulting in hepatic steatosis. Proteins involved in lipid metabolism are regulated at multiple levels, including gene expression, protein expression, and activation state, which all contribute to the overall handling and metabolism of lipids during a dyslipidemic state. The present studies suggest that the coagulation cascade, through PAR-1 activation, contributes to the regulation of genes involved in several facets of lipid metabolism, including fatty acid uptake (CD36), triglyceride synthesis (SCD1), and β-oxidation (PPARα). The decrease in PPARα mRNA levels is most likely a consequence of reduced steatosis and was not accompanied by a change in hepatic protein expression. In contrast to the expression of SCD1 mRNA, SCD1 protein expression increased in PAR-1−/− mice and in mice with low-TF bone marrow fed a Western diet. Indeed, previous studies have indicated that SCD1 is also regulated at the posttranscriptional level.56 Mice lacking SCD1 in all cells are protected from hepatic steatosis induced by a Western diet, whereas liver-specific SCD1 deficiency does not reduce hepatic steatosis in mice fed a Western diet.57 Taken together, these studies suggest that the effect of either TF deficiency or PAR-1 deficiency on hepatic steatosis is not a consequence of altered SCD1 expression. Further studies evaluating the role of the coagulation cascade in the dichotomous regulation of SCD1 mRNA and protein expression are needed.
Expression of the fatty acid transporter CD36 increased significantly in mice fed a Western diet, and the increase in CD36 mRNA and protein was attenuated by deficiency in PAR-1 or hematopoietic cell–derived TF. Previous studies identified an association between hepatic steatosis and CD36 expression in livers of patients with NAFLD,58 and the expression of CD36 increased in livers of mice fed a Western diet (Figure 5, Figure 9). Moreover, hepatic overexpression of CD36 caused hepatic steatosis.59 Therefore, the coagulation cascade could potentially contribute to hepatic steatosis by increasing CD36 expression. One potential mechanism whereby PAR-1 activation could contribute to CD36 expression is indirectly via MCP-1 up-regulation because administration of an antagonist of the MCP-1 receptor CCR2 reduced CD36 expression in livers of obese mice.21 However, the mechanism whereby the coagulation cascade regulates CD36 expression requires further investigation.
The present studies suggest that thrombin activation of PAR-1 is vital to the development of hepatic steatosis in mice fed a Western diet. PAR-1 expression was not detected on rodent or human hepatocytes in previous studies,60, 61 suggesting a requirement of PAR-1 expressed by nonparenchymal cells in liver steatosis. Nonparenchymal cells of the hepatic microcirculation, including Kupffer cells, endothelial cells, and stellate cells, express PAR-1.61, 62, 63 Although the contribution of each nonparenchymal cell population in the development of steatosis is not known, several studies indicate that Kupffer cells contribute to steatosis in rodent models by producing inflammatory mediators.16, 17 MCP-1 is expressed by thrombin-stimulated nonparenchymal cells,63 and MCP-1 stimulates lipid accumulation in cultured hepatocytes.44 Taken together, the results indicate a complex mechanism of steatosis development coordinated by the coagulation cascade by stimulating the production of paracrine inflammatory mediators by hepatic nonparenchymal cells that then cause lipid accumulation in hepatocytes (Figure 10).
Identifying cell types that express TF in patients with NAFLD could lead to a better understanding of the mechanisms whereby the coagulation cascade is activated in MetS. One potential cellular source of TF in mice fed a Western diet is the monocyte/macrophage. Indeed, TF expression is increased on monocytes and/or Kupffer cells in several rodent models of liver injury/disease and in patients with liver disease, obesity, and MetS.42, 64, 65, 66, 67, 68, 69 We found that TF mRNA expression was increased in the buffy coat (ie, white blood cells) from mice fed a Western diet. In related studies, we found that hematopoietic cell TF deficiency reduced plasma microparticle TF activity and TAT levels in LDLr−/− mice fed a Western diet.43 Moreover, hematopoietic cell TF deficiency reduced hepatic fibrin deposition in mice fed a Western diet, suggesting reduced thrombin generation in the liver. Although the contribution of TF from other cellular sources cannot be excluded, the finding that TF deficiency in bone marrow–derived cells nearly prevented steatosis in LDLr−/− mice fed a Western diet suggests a critical role for TF expressed by this cell population in hepatic steatosis. The levels of several mediators that induce TF expression in monocytes/macrophages, including endotoxin,64, 69 proinflammatory cytokines,64 and oxidized LDL,70 are increased in patients with obesity or MetS. Moreover, several of these mediators have been implicated in the development of liver steatosis.18, 19, 45, 46 Additional studies evaluating the mechanism whereby TF expression increases on monocytes/macrophages in MetS are needed.
In summary, the results of the studies presented herein indicate that PAR-1 and hematopoietic cell TF are required for hepatic inflammation and steatosis in mice fed a Western diet. These studies reveal an as yet unidentified role of the coagulation cascade in promoting Western diet–induced hepatic steatosis and highlight the importance of circulating mediators in stimulating lipid accumulation in parenchymal cells in NAFLD. The contribution of the coagulation cascade to the development of NAFLD as well as atherosclerosis and other cardiac disorders may provide a connection between multiple facets of MetS.
Acknowledgments
We thank Lai Peng and Alyson Baker for their technical assistance and Dr. Daniel Kirchhofer (Genentech Inc.) for supplying the anti-mouse TF antibody.
Footnotes
Supported by American Heart Association Scientist Development grant 0835121G (J.P.L.) and by NIH grants R01 ES017537 (J.P.L.), P20 RR021940 (G.L.G.), R01 HL095096 (N.M.), and F32 HL099175 (A.P.O.).
K.M.K. and A.P.O. contributed equally to this work.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.07.015.
Supplementary data
References
- 1.Larter C.Z., Chitturi S., Heydet D., Farrell G.C. A fresh look at NASH pathogenesis, part 1: the metabolic movers. J Gastroenterol Hepatol. 2010;25:672–690. doi: 10.1111/j.1440-1746.2010.06253.x. [DOI] [PubMed] [Google Scholar]
- 2.Clark J.M., Brancati F.L., Diehl A.M. Nonalcoholic fatty liver disease. Gastroenterology. 2002;122:1649–1657. doi: 10.1053/gast.2002.33573. [DOI] [PubMed] [Google Scholar]
- 3.Farrell G.C., Larter C.Z. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43:S99–S112. doi: 10.1002/hep.20973. [DOI] [PubMed] [Google Scholar]
- 4.Friedman S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kotronen A., Yki-Jarvinen H. Fatty liver: a novel component of the metabolic syndrome. Arterioscler Thromb Vasc Biol. 2008;28:27–38. doi: 10.1161/ATVBAHA.107.147538. [DOI] [PubMed] [Google Scholar]
- 6.Day C.P., James O.F. Hepatic steatosis: innocent bystander or guilty party? Hepatology. 1998;27:1463–1466. doi: 10.1002/hep.510270601. [DOI] [PubMed] [Google Scholar]
- 7.Schwimmer J.B., Pardee P.E., Lavine J.E., Blumkin A.K., Cook S. Cardiovascular risk factors and the metabolic syndrome in pediatric nonalcoholic fatty liver disease. Circulation. 2008;118:277–283. doi: 10.1161/CIRCULATIONAHA.107.739920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fabbrini E., Sullivan S., Klein S. Obesity and nonalcoholic fatty liver disease: biochemical, metabolic, and clinical implications. Hepatology. 2010;51:679–689. doi: 10.1002/hep.23280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Targher G., Chonchol M., Miele L., Zoppini G., Pichiri I., Muggeo M. Nonalcoholic fatty liver disease as a contributor to hypercoagulation and thrombophilia in the metabolic syndrome. Semin Thromb Hemost. 2009;35:277–287. doi: 10.1055/s-0029-1222606. [DOI] [PubMed] [Google Scholar]
- 10.Cigolini M., Targher G., Agostino G., Tonoli M., Muggeo M., De Sandre G. Liver steatosis and its relation to plasma haemostatic factors in apparently healthy men: role of the metabolic syndrome. Thromb Haemost. 1996;76:69–73. [PubMed] [Google Scholar]
- 11.Ndumele C.E., Nasir K., Conceicao R.D., Carvalho J.A., Blumenthal R.S., Santos R.D. Hepatic steatosis, obesity, and the metabolic syndrome are independently and additively associated with increased systemic inflammation. Arterioscler Thromb Vasc Biol. 2011;31:1927–1932. doi: 10.1161/ATVBAHA.111.228262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Targher G., Bertolini L., Poli F., Rodella S., Scala L., Tessari R., Zenari L., Falezza G. Nonalcoholic fatty liver disease and risk of future cardiovascular events among type 2 diabetic patients. Diabetes. 2005;54:3541–3546. doi: 10.2337/diabetes.54.12.3541. [DOI] [PubMed] [Google Scholar]
- 13.Choi S.S., Diehl A.M. Hepatic triglyceride synthesis and nonalcoholic fatty liver disease. Curr Opin Lipidol. 2008;19:295–300. doi: 10.1097/MOL.0b013e3282ff5e55. [DOI] [PubMed] [Google Scholar]
- 14.Tiniakos D.G., Vos M.B., Brunt E.M. Nonalcoholic fatty liver disease: pathology and pathogenesis. Annu Rev Pathol. 2010;5:145–171. doi: 10.1146/annurev-pathol-121808-102132. [DOI] [PubMed] [Google Scholar]
- 15.Kleiner D.E., Brunt E.M., Van Natta M., Behling C., Contos M.J., Cummings O.W., Ferrell L.D., Liu Y.C., Torbenson M.S., Unalp-Arida A., Yeh M., McCullough A.J., Sanyal A.J. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 16.Stienstra R., Saudale F., Duval C., Keshtkar S., Groener J.E., van Rooijen N., Staels B., Kersten S., Muller M. Kupffer cells promote hepatic steatosis via interleukin-1β-dependent suppression of peroxisome proliferator-activated receptor α activity. Hepatology. 2010;51:511–522. doi: 10.1002/hep.23337. [DOI] [PubMed] [Google Scholar]
- 17.Huang W., Metlakunta A., Dedousis N., Zhang P., Sipula I., Dube J.J., Scott D.K., O'Doherty R.M. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes. 2010;59:347–357. doi: 10.2337/db09-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tamura Y., Sugimoto M., Murayama T., Minami M., Nishikaze Y., Ariyasu H., Akamizu T., Kita T., Yokode M., Arai H. C-C chemokine receptor 2 inhibitor improves diet-induced development of insulin resistance and hepatic steatosis in mice. J Atheroscler Thromb. 2010;17:219–228. doi: 10.5551/jat.3368. [DOI] [PubMed] [Google Scholar]
- 19.Rull A., Rodriguez F., Aragones G., Marsillach J., Beltran R., Alonso-Villaverde C., Camps J., Joven J. Hepatic monocyte chemoattractant protein-1 is upregulated by dietary cholesterol and contributes to liver steatosis. Cytokine. 2009;48:273–279. doi: 10.1016/j.cyto.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 20.Wobser H., Dorn C., Weiss T.S., Amann T., Bollheimer C., Buttner R., Scholmerich J., Hellerbrand C. Lipid accumulation in hepatocytes induces fibrogenic activation of hepatic stellate cells. Cell Res. 2009;19:996–1005. doi: 10.1038/cr.2009.73. [DOI] [PubMed] [Google Scholar]
- 21.Tamura Y., Sugimoto M., Murayama T., Ueda Y., Kanamori H., Ono K., Ariyasu H., Akamizu T., Kita T., Yokode M., Arai H. Inhibition of CCR2 ameliorates insulin resistance and hepatic steatosis in db/db mice. Arterioscler Thromb Vasc Biol. 2008;28:2195–2201. doi: 10.1161/ATVBAHA.108.168633. [DOI] [PubMed] [Google Scholar]
- 22.Mackman N. Tissue-specific hemostasis in mice. Arterioscler Thromb Vasc Biol. 2005;25:2273–2281. doi: 10.1161/01.ATV.0000183884.06371.52. [DOI] [PubMed] [Google Scholar]
- 23.Coughlin S.R. How the protease thrombin talks to cells. Proc Natl Acad Sci U S A. 1999;96:11023–11027. doi: 10.1073/pnas.96.20.11023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fritsch P., Kleber M., Rosenkranz A., Fritsch M., Muntean W., Mangge H., Reinehr T. Haemostatic alterations in overweight children: associations between metabolic syndrome, thrombin generation, and fibrinogen levels. Atherosclerosis. 2010;212:650–655. doi: 10.1016/j.atherosclerosis.2010.06.028. [DOI] [PubMed] [Google Scholar]
- 25.Romano M., Guagnano M.T., Pacini G., Vigneri S., Falco A., Marinopiccoli M., Manigrasso M.R., Basili S., Davi G. Association of inflammation markers with impaired insulin sensitivity and coagulative activation in obese healthy women. J Clin Endocrinol Metab. 2003;88:5321–5326. doi: 10.1210/jc.2003-030508. [DOI] [PubMed] [Google Scholar]
- 26.Sola E., Navarro S., Medina P., Vaya A., Estelles A., Hernandez-Mijares A., Espana F. Activated protein C levels in obesity and weight loss influence. Thromb Res. 2009;123:697–700. doi: 10.1016/j.thromres.2008.07.017. [DOI] [PubMed] [Google Scholar]
- 27.Ay L., Kopp H.P., Brix J.M., Ay C., Quehenberger P., Schernthaner G.H., Pabinger I., Schernthaner G. Thrombin generation in morbid obesity: significant reduction after weight loss. J Thromb Haemost. 2010;8:759–765. doi: 10.1111/j.1538-7836.2010.03766.x. [DOI] [PubMed] [Google Scholar]
- 28.Kopp C.W., Kopp H.P., Steiner S., Kriwanek S., Krzyzanowska K., Bartok A., Roka R., Minar E., Schernthaner G. Weight loss reduces tissue factor in morbidly obese patients. Obes Res. 2003;11:950–956. doi: 10.1038/oby.2003.131. [DOI] [PubMed] [Google Scholar]
- 29.Targher G., Bertolini L., Scala L., Zoppini G., Zenari L., Falezza G. Non-alcoholic hepatic steatosis and its relation to increased plasma biomarkers of inflammation and endothelial dysfunction in non-diabetic men: role of visceral adipose tissue. Diabet Med. 2005;22:1354–1358. doi: 10.1111/j.1464-5491.2005.01646.x. [DOI] [PubMed] [Google Scholar]
- 30.de Larranaga G., Wingeyer S.P., Graffigna M., Belli S., Bendezu K., Alvarez S., Levalle O., Fainboim H. Plasma plasminogen activator inhibitor-1 levels and nonalcoholic fatty liver in individuals with features of metabolic syndrome. Clin Appl Thromb Hemost. 2008;14:319–324. doi: 10.1177/1076029607304094. [DOI] [PubMed] [Google Scholar]
- 31.Kotronen A., Joutsi-Korhonen L., Sevastianova K., Bergholm R., Hakkarainen A., Pietilainen K.H., Lundbom N., Rissanen A., Lassila R., Yki-Jarvinen H. Increased coagulation factor VIII, IX, XI and XII activities in non-alcoholic fatty liver disease. Liver Int. 2011;31:176–183. doi: 10.1111/j.1478-3231.2010.02375.x. [DOI] [PubMed] [Google Scholar]
- 32.Luyendyk J.P., Sullivan B.P., Guo G.L., Wang R. Tissue factor-deficiency and protease activated receptor-1-deficiency reduce inflammation elicited by diet-induced steatohepatitis in mice. Am J Pathol. 2010;176:177–186. doi: 10.2353/ajpath.2010.090672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rinella M.E., Green R.M. The methionine-choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J Hepatol. 2004;40:47–51. doi: 10.1016/j.jhep.2003.09.020. [DOI] [PubMed] [Google Scholar]
- 34.Kassel K.M., Guo G.L., Tawfik O., Luyendyk J.P. Monocyte chemoattractant protein-1 deficiency does not affect steatosis or inflammation in livers of mice fed a methionine-choline-deficient diet. Lab Invest. 2010;90:1794–1804. doi: 10.1038/labinvest.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Connolly A.J., Suh D.Y., Hunt T.K., Coughlin S.R. Mice lacking the thrombin receptor, PAR1, have normal skin wound healing. Am J Pathol. 1997;151:1199–1204. [PMC free article] [PubMed] [Google Scholar]
- 36.Pawlinski R., Wang J.G., Owens A.P., III, Williams J., Antoniak S., Tencati M., Luther T., Rowley J.W., Low E.N., Weyrich A.S., Mackman N. Hematopoietic and nonhematopoietic cell tissue factor activates the coagulation cascade in endotoxemic mice. Blood. 2010;116:806–814. doi: 10.1182/blood-2009-12-259267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parry G.C., Erlich J.H., Carmeliet P., Luther T., Mackman N. Low levels of tissue factor are compatible with development and hemostasis in mice. J Clin Invest. 1998;101:560–569. doi: 10.1172/JCI814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tanaka Y., Aleksunes L.M., Yeager R.L., Gyamfi M.A., Esterly N., Guo G.L., Klaassen C.D. NF-E2-related factor 2 inhibits lipid accumulation and oxidative stress in mice fed a high-fat diet. J Pharmacol Exp Ther. 2008;325:655–664. doi: 10.1124/jpet.107.135822. [DOI] [PubMed] [Google Scholar]
- 39.Copple B.L., Banes A., Ganey P.E., Roth R.A. Endothelial cell injury and fibrin deposition in rat liver after monocrotaline exposure. Toxicol Sci. 2002;65:309–318. doi: 10.1093/toxsci/65.2.309. [DOI] [PubMed] [Google Scholar]
- 40.Behari J., Yeh T.H., Krauland L., Otruba W., Cieply B., Hauth B., Apte U., Wu T., Evans R., Monga S.P. Liver-specific β-catenin knockout mice exhibit defective bile acid and cholesterol homeostasis and increased susceptibility to diet-induced steatohepatitis. Am J Pathol. 2010;176:744–753. doi: 10.2353/ajpath.2010.090667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cieply B., Zeng G., Proverbs-Singh T., Geller D.A., Monga S.P. Unique phenotype of hepatocellular cancers with exon-3 mutations in β-catenin gene. Hepatology. 2009;49:821–831. doi: 10.1002/hep.22695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakagomi A., Sasaki M., Ishikawa Y., Morikawa M., Shibui T., Kusama Y., Atarashi H., Mizuno K. Upregulation of monocyte tissue factor activity is significantly associated with low-grade chronic inflammation and insulin resistance in patients with metabolic syndrome. Circ J. 2010;74:572–577. doi: 10.1253/circj.cj-09-0835. [DOI] [PubMed] [Google Scholar]
- 43.Owens A.P., Temel R.E., Barcel D.A., Marshall S.M., McDaniel A.L., Mackman N. Hyperlipidemia increases microparticle tissue factor and the activation of coagulation: a tale of mice and monkeys. Circulation. 2010;122:A20151. (abstract) [Google Scholar]
- 44.Clement S., Juge-Aubry C., Sgroi A., Conzelmann S., Pazienza V., Pittet-Cuenod B., Meier C.A., Negro F. Monocyte chemoattractant protein-1 secreted by adipose tissue induces direct lipid accumulation in hepatocytes. Hepatology. 2008;48:799–807. doi: 10.1002/hep.22404. [DOI] [PubMed] [Google Scholar]
- 45.Endo M., Masaki T., Seike M., Yoshimatsu H. TNF-α induces hepatic steatosis in mice by enhancing gene expression of sterol regulatory element binding protein-1c (SREBP-1c) Exp Biol Med (Maywood) 2007;232:614–621. [PubMed] [Google Scholar]
- 46.De Taeye B.M., Novitskaya T., McGuinness O.P., Gleaves L., Medda M., Covington J.W., Vaughan D.E. Macrophage TNF-α contributes to insulin resistance and hepatic steatosis in diet-induced obesity. Am J Physiol Endocrinol Metab. 2007;293:E713–E725. doi: 10.1152/ajpendo.00194.2007. [DOI] [PubMed] [Google Scholar]
- 47.MacDonald M.L., Singaraja R.R., Bissada N., Ruddle P., Watts R., Karasinska J.M., Gibson W.T., Fievet C., Vance J.E., Staels B., Hayden M.R. Absence of stearoyl-CoA desaturase-1 ameliorates features of the metabolic syndrome in LDLR-deficient mice. J Lipid Res. 2008;49:217–229. doi: 10.1194/jlr.M700478-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coenen K.R., Hasty A.H. Obesity potentiates development of fatty liver and insulin resistance, but not atherosclerosis, in high-fat diet-fed agouti LDLR-deficient mice. Am J Physiol Endocrinol Metab. 2007;293:E492–E499. doi: 10.1152/ajpendo.00171.2007. [DOI] [PubMed] [Google Scholar]
- 49.Kang H., Greenson J.K., Omo J.T., Chao C., Peterman D., Anderson L., Foess-Wood L., Sherbondy M.A., Conjeevaram H.S. Metabolic syndrome is associated with greater histologic severity, higher carbohydrate, and lower fat diet in patients with NAFLD. Am J Gastroenterol. 2006;101:2247–2253. doi: 10.1111/j.1572-0241.2006.00719.x. [DOI] [PubMed] [Google Scholar]
- 50.Vanni E., Bugianesi E., Kotronen A., De Minicis S., Yki-Jarvinen H., Svegliati-Baroni G. From the metabolic syndrome to NAFLD or vice versa? Dig Liver Dis. 2010;42:320–330. doi: 10.1016/j.dld.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 51.Newberry E.P., Xie Y., Kennedy S.M., Luo J., Davidson N.O. Protection against Western diet-induced obesity and hepatic steatosis in liver fatty acid-binding protein knockout mice. Hepatology. 2006;44:1191–1205. doi: 10.1002/hep.21369. [DOI] [PubMed] [Google Scholar]
- 52.Hickman I.J., Sullivan C.M., Flight S., Campbell C., Crawford D.H., Masci P.P., O'Moore-Sullivan T.M., Prins J.B., Macdonald G.A. Altered clot kinetics in patients with non-alcoholic fatty liver disease. Ann Hepatol. 2009;8:331–338. [PubMed] [Google Scholar]
- 53.Beijers H.J., Ferreira I., Spronk H.M., Bravenboer B., Dekker J.M., Nijpels G., Ten C.H., Stehouwer C.D. Body composition as determinant of thrombin generation in plasma: the Hoorn study. Arterioscler Thromb Vasc Biol. 2010;30:2639–2647. doi: 10.1161/ATVBAHA.110.211946. [DOI] [PubMed] [Google Scholar]
- 54.Anstee Q.M., Goldin R.D. Mouse models in non-alcoholic fatty liver disease and steatohepatitis research. Int J Exp Pathol. 2006;87:1–16. doi: 10.1111/j.0959-9673.2006.00465.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dela Pena A., Leclercq I., Field J., George J., Jones B., Farrell G. NF-κB activation, rather than TNF, mediates hepatic inflammation in a murine dietary model of steatohepatitis. Gastroenterology. 2005;129:1663–1674. doi: 10.1053/j.gastro.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 56.Flowers M.T., Ntambi J.M. Role of stearoyl-coenzyme A desaturase in regulating lipid metabolism. Curr Opin Lipidol. 2008;19:248–256. doi: 10.1097/MOL.0b013e3282f9b54d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Miyazaki M., Flowers M.T., Sampath H., Chu K., Otzelberger C., Liu X., Ntambi J.M. Hepatic stearoyl-CoA desaturase-1 deficiency protects mice from carbohydrate-induced adiposity and hepatic steatosis. Cell Metab. 2007;6:484–496. doi: 10.1016/j.cmet.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 58.Miquilena-Colina M.E., Lima-Cabello E., Sanchez-Campos S., Garcia-Mediavilla M.V., Fernandez-Bermejo M., Lozano-Rodriguez T., Vargas-Castrillon J., Buque X., Ochoa B., Aspichueta P., Gonzalez-Gallego J., Garcia-Monzon C. Hepatic fatty acid translocase CD36 upregulation is associated with insulin resistance, hyperinsulinaemia and increased steatosis in non-alcoholic steatohepatitis and chronic hepatitis C. Gut. 2011;60:1394–1402. doi: 10.1136/gut.2010.222844. [DOI] [PubMed] [Google Scholar]
- 59.Koonen D.P., Jacobs R.L., Febbraio M., Young M.E., Soltys C.L., Ong H., Vance D.E., Dyck J.R. Increased hepatic CD36 expression contributes to dyslipidemia associated with diet-induced obesity. Diabetes. 2007;56:2863–2871. doi: 10.2337/db07-0907. [DOI] [PubMed] [Google Scholar]
- 60.Rullier A., Senant N., Kisiel W., Bioulac-Sage P., Balabaud C., Le Bail B., Rosenbaum J. Expression of protease-activated receptors and tissue factor in human liver. Virchows Arch. 2006;448:46–51. doi: 10.1007/s00428-005-0078-0. [DOI] [PubMed] [Google Scholar]
- 61.Copple B.L., Moulin F., Hanumegowda U.M., Ganey P.E., Roth R.A. Thrombin and protease-activated receptor-1 agonists promote lipopolysaccharide-induced hepatocellular injury in perfused livers. J Pharmacol Exp Ther. 2003;305:417–425. doi: 10.1124/jpet.102.046391. [DOI] [PubMed] [Google Scholar]
- 62.Gaca M.D., Zhou X., Benyon R.C. Regulation of hepatic stellate cell proliferation and collagen synthesis by proteinase-activated receptors. J Hepatol. 2002;36:362–369. doi: 10.1016/s0168-8278(01)00285-9. [DOI] [PubMed] [Google Scholar]
- 63.Fiorucci S., Antonelli E., Distrutti E., Severino B., Fiorentina R., Baldoni M., Caliendo G., Santagada V., Morelli A., Cirino G. PAR1 antagonism protects against experimental liver fibrosis: role of proteinase receptors in stellate cell activation. Hepatology. 2004;39:365–375. doi: 10.1002/hep.20054. [DOI] [PubMed] [Google Scholar]
- 64.Mackman N. Regulation of tissue factor gene expression in human monocytic and endothelial cells. Haemostasis. 1996;26(Suppl 1):17–19. doi: 10.1159/000217234. [DOI] [PubMed] [Google Scholar]
- 65.Saliola M., Lorenzet R., Ferro D., Basili S., Caroselli C., Santo A.D., Sallese M., Violi F. Enhanced expression of monocyte tissue factor in patients with liver cirrhosis. Gut. 1998;43:428–432. doi: 10.1136/gut.43.3.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Arai M., Mochida S., Ohno A., Ogata I., Obama H., Maruyama I., Fujiwara K. Blood coagulation equilibrium in rat liver microcirculation as evaluated by endothelial cell thrombomodulin and macrophage tissue factor. Thromb Res. 1995;80:113–123. doi: 10.1016/0049-3848(95)00157-m. [DOI] [PubMed] [Google Scholar]
- 67.Mochida S., Arai M., Ohno A., Yamanobe F., Ishikawa K., Matsui A., Maruyama I., Kato H., Fujiwara K. Deranged blood coagulation equilibrium as a factor of massive liver necrosis following endotoxin administration in partially hepatectomized rats. Hepatology. 1999;29:1532–1540. doi: 10.1002/hep.510290533. [DOI] [PubMed] [Google Scholar]
- 68.Kobayashi Y., Yoshimura N., Nakamura K., Yamagishi H., Oka T. Expression of tissue factor in hepatic ischemic-reperfusion injury of the rat. Transplantation. 1998;66:708–716. doi: 10.1097/00007890-199809270-00004. [DOI] [PubMed] [Google Scholar]
- 69.Guha M., O'Connell M.A., Pawlinski R., Hollis A., McGovern P., Yan S.F., Stern D., Mackman N. Lipopolysaccharide activation of the MEK-ERK1/2 pathway in human monocytic cells mediates tissue factor and tumor necrosis factor α expression by inducing Elk-1 phosphorylation and Egr-1 expression. Blood. 2001;98:1429–1439. doi: 10.1182/blood.v98.5.1429. [DOI] [PubMed] [Google Scholar]
- 70.Emekli-Alturfan E., Basar I., Alturfan A.A., Ayan F., Koldas L., Balci H., Emekli N. The relation between plasma tissue factor and oxidized LDL levels in acute coronary syndromes. Pathophysiol Haemost Thromb. 2007;36:290–297. doi: 10.1159/000296279. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.