

Abstract
Parkinson's disease (PD) is characterized by α-synuclein–containing Lewy bodies (LBs) and loss of melanized neurons in the substantia nigra (SN). Recently, a link between apolipoprotein E (ApoE) expression, α-synuclein aggregation, and neurodegeneration was suggested. Here, we report on ApoE expression appearing in melanized neurons of the SN and in LBs in both PD and incidental LB disease cases. Interestingly, increased expression of the low-density lipoprotein receptor-related protein 1 (the receptor for ApoE) was also observed in incidental LB disease and PD. Our data suggest that alterations in lipoprotein homeostasis/signaling in melanized neurons of the SN are an early event during PD pathogenesis.
Parkinson's disease (PD) is characterized by progressive degeneration of melanized neurons in the substantia nigra (SN).1 In addition, accumulation of aggregated forms of α-synuclein protein in cytoplasmic neuronal inclusions, known as Lewy bodies (LBs),2 is associated with this neurodegeneration. However, the mechanisms underlying α-synuclein aggregation and degeneration of melanized neurons and their relevance to PD in pathogenesis remain largely unclear.
Apolipoprotein E (ApoE) is the main lipid-transport protein present in the brain,3 and inheritance of one or two copies of the APOE ε4 allele is associated with increased risk for, and an earlier age of onset of, late Alzheimer's disease.4 Intriguingly, recent discoveries in transgenic animal models suggest for the first time a molecular link between α-synuclein aggregation, neurodegeneration, and ApoE expression.5 ApoE deletion alleviated α-synuclein–induced neurodegeneration and boosted overall survival.5 These findings connect ApoE with PD pathogenesis, in particular neuronal cell loss and α-synuclein aggregation. However, information on ApoE's distribution and association with α-synuclein pathology in human PD brain is lacking.
Incidental LB disease (iLBD) subjects are cases with LB pathology in the brain without clinically documented signs of parkinsonism or dementia,6 and can be considered as a presymptomatic stage of PD.7 iLBD subjects have usually not been exposed to any forms of anti-PD therapy, and thus may provide a unique and unbiased opportunity for indentifying early events during PD pathogenesis.
As a first step in our quest to identify ApoE as a possible novel therapeutic target to ameliorate neurodegeneration in PD, we started with analyzing the expression of ApoE and its primary neuronal uptake receptor,8 the low-density lipoprotein receptor-related protein 1 (LRP1), in melanized neurons of SN in brain tissue of control, PD, and iLBD cases using immunohistochemistry. Our data showed the presence of ApoE in melanized neurons of the SN in both PD and iLBD cases, but not in controls, and clear ApoE immunoreactivity in LBs. Moreover, compared to controls, in melanized neurons in iLBD and PD, overall LRP1 immunoreactivity was increased and showed a change in distribution pattern. Our data suggest that alterations in lipoprotein homeostasis in melanized neurons of the SN are early events during PD pathogenesis.
Materials and Methods
Post-Mortem Brain Tissue
Human brain specimens were obtained from the VU University Medical Center, Department of Pathology. Informed consent for brain autopsy and the use of material and clinical information was obtained from the donor or the next of kin. Neuropathological evaluation was performed on disease-relevant brain areas. Distribution and density of LBs and neurofibrillary tangles was determined using Bodian staining, and immunohistochemistry for α-synuclein (BD Biosciences, San Jose, CA) or hyperphosphorylated tau (clone AT8; Pierce, Rockford, IL), respectively. Sex, age, postmortem delay, clinical diagnosis or cause of death, and Braak stage for neurofibrillary tangles are detailed in Table 1.
Table 1.
Patient Characteristics
| Group | Sex | Age (years) | Postmortem delay (hours) | Clinical diagnosis/cause of death | Braak stage for NFT |
|---|---|---|---|---|---|
| Control | F | 32 | 24 | Hemangioendothelioma | 0 |
| Control | F | 61 | 7 | Euthanasia | 0 |
| Control | M | 66 | 24 | Cardiac infarct | 0 |
| Control | M | 68 | 24 | Cardiac infarct | 0 |
| Control | F | 73 | 48 | Hodgkin's lymphoma | 0 |
| iLBD | F | 62 | 24 | Cardiac infarct | 0 |
| iLBD | M | 66 | 8 | Aortic rupture | 0 |
| iLBD | M | 66 | 72 | Heart failure | 0 |
| iLBD | M | 67 | 24 | Cardiac infarct | 0 |
| iLBD | M | 73 | 24 | Heart failure | II |
| iLBD | F | 73 | 7 | Respiratory failure | II |
| iLBD | M | 77 | 24 | Bradycardia | 0 |
| iLBD | F | 80 | 5 | Euthanasia | II |
| iLBD | M | 88 | 24 | Cardiac infarct | 0 |
| PD | M | 74 | 24 | PD | II |
| PD | M | 77 | 48 | PD/Pneumonia | II |
| PD | M | 77 | 24 | PD/Pneumonia | III |
| PD | M | 82 | 19 | PD/Pneumonia | IV |
| PD | M | 84 | 24 | PD | I |
| DLB | M | 64 | 24 | DLB/dehydration | 0 |
| DLB | F | 80 | 24 | DLB/pneumonia | II |
| DLB | M | 73 | 24 | DLB/COPD | 0 |
F, female; M, male; COPD, chronic obstructive pulmonary disease; DLB, dementia with Lewy bodies; iLBD, incidental Lewy body disease; NFT, neurofibrillary tangles; PD, Parkinson's disease.
Immunohistochemistry
Sections (5 μm thick) were fixed and stained as described previously.9 Mouse anti-rat α-synuclein was obtained from BD Biosciences (dilution 1:1000). Both mouse anti-human ApoE (dilution 1:400) and mouse anti-human LRP1 (dilution 1:500) were purchased from Abcam (Cambridge, UK). Rabbit anti-human receptor-associated protein (RAP) (dilution 1:2000) has been described previously.10 Negative controls for immunostainings were generated by omission of primary antibodies. Secondary antibodies used were: biotin-labeled goat anti-mouse (dilution 1:200), biotin-labeled goat anti-rabbit (dilution 1:200; Jackson Immunoresearch Europe Ltd., Newmarket, UK), and biotin-conjugated rabbit anti-mouse F(ab′)2 (dilution 1:500; DAKO, Glostrup, Denmark). Immunohistochemistry was performed according to the avidin-biotin-peroxidase method. For LRP1, RAP, and ApoE stainings, nickel-enhanced 3,3′-diaminobenzidine was used as the chromogen (Sigma, St. Louis, MO). For α-synuclein staining, sections were incubated with EnVision solution (goat anti-mouse horseradish peroxidase, undiluted; DAKO), and colored using 3-amino-9-ethylcarbazole (AEC, Zymed, San Francisco, CA) as chromogen. Sections were counterstained with hematoxylin.
Semiquantitative Analysis of the Immunohistochemical Stainings
Two of the authors (M.M.M.W. and J.J.M.H.) independently performed a semiquantitative analysis of the immunostainings in melanized neurons of the SN of control, iLBD, and PD cases, as described previously.9 Analysis was performed on the substantia nigra pars compacta and pars reticulata at the level of the third cranial nerve. The immunohistochemical score (IHC score) ranged from 0 to 3, with 0 indicating no, 1 weak, 2 moderate, and 3 strong immunoreactivity of the relevant antibody in the studied cells. For each antibody, staining was performed on tissue of all patients and controls listed in Table 1. The IHC score for each case was an average of at least four microscopic fields (magnification ×10). The final IHC score was an average of the IHC scores found within a patient group or the control group (Table 1). Staining analysis was performed blind as to the case identification. Interobserver scores were highly consistent (average interobserver consensus was 90%; differentially judged images were excluded).
Results
ApoE Immunoreactivity in Melanized Neurons of the SN in iLBD and PD but Not in Control
In control brain, no α-synuclein staining was observed in the SN (Figure 1, A and E), whereas a weak cytoplasmic α-synuclein staining was found in a fraction of the melanized neurons of the SN in iLBD cases (Figure 1, B and E). In PD brain, however, the majority of neuromelanin-containing neurons of the SN demonstrated both strong diffuse cytoplasmic immunoreactivity (Figure 1, C and E) and LB staining for α-synuclein (Figure 1, D and E).
Figure 1.
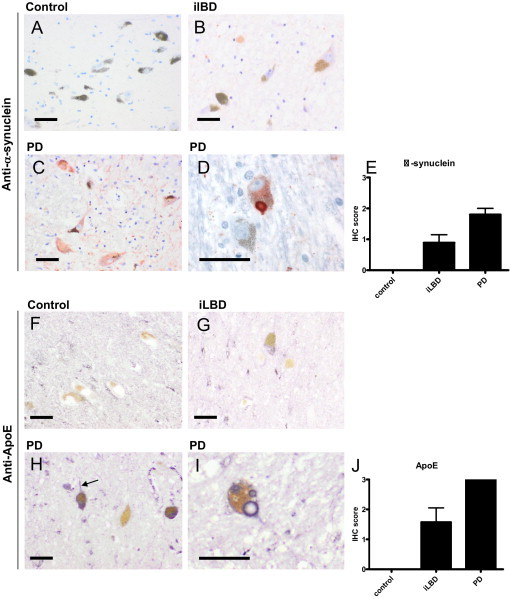
α-Synuclein and ApoE immunoreactivity in melanized neurons in SN of control, iLBD, and PD cases. Semiquantitative analysis of immunohistochemical (IHC) staining of α-synuclein or ApoE in melanized neurons of the SN of control, iLBD, and PD cases was performed as described in Materials and Methods. In neuromelanin-containing neurons of control cases, α-synuclein staining was absent (A and E; IHC score 0). In iLBD cases, a fraction of the neuromelanin-containing neurons demonstrated weak cytoplasmic α-synuclein staining (B and G; IHC score of 1 ± 0.8), whereas strong cytoplasmic α-synuclein staining (C and E; IHC score 2 ± 0.4) and α-synuclein–positive LBs (D) were observed in PD cases. In control cases, no ApoE immunoreactivity was observed in neuromelanin-containing neurons of the SN (F and J; IHC score 0). In iLBD cases, a fraction of the neuromelanin-containing neurons demonstrated weak cytoplasmic ApoE staining (G), and was scored as weak to moderate (J; IHC score of 1.5 ± 1.3). In PD cases, strong cytoplasmic ApoE staining was observed (H) in both cell body and neuronal extensions (H, arrow, and J; IHC score 3), and in LBs (I). Mean ± SD is shown (E and J), if large enough to be depicted. Scale bars: 20 μm (A–D and F–I).
In SN of control cases, ApoE staining was observed in cerebral vessels and astrocytes, as described previously.11 In addition, overall weak immunostaining of ApoE was observed in brain parenchyma, but not in melanized neurons (Figure 1, F and J). This general ApoE staining pattern found in SN of controls was also observed in iLBD and PD cases. Interestingly, however, in contrast to controls, weak ApoE staining was observed in a fraction of melanized neurons of the SN in all iLBD cases (Figure 1, G and J). Moreover, strong cytoplasmic ApoE staining in both cell body and neuronal extensions was found in almost all melanized neurons of the SN of PD cases (Figure 1, H and J), and ApoE staining was also observed in the outer rim of LBs (Figure 1I). In neurons and LBs in the neocortex of dementia with Lewy body cases, no ApoE was observed (not shown).
Increased LRP1, but Not RAP, Immunoreactivity in Melanized Neurons of the SN in iLBD and PD
In SN of control, iLBD, and PD cases, comparable LRP1 staining was observed in astrocytes, neurons, and medium-sized parenchymal vessels, similar to the cellular distribution pattern described by us previously.12 In melanized neurons of controls, only weak diffuse cytoplasmic LRP1 staining was observed (Figure 2, A and C). Interestingly however, compared to controls, LRP1 immunoreactivity not only increased in melanized neurons of PD cases (Figure 2, B and C), but also changed into a granular pattern (Figure 2B). Analysis of LRP1 staining in melanized neurons in SN of iLBD cases demonstrated a similar granular pattern, although in a lower percentage of neurons and less pronounced than in PD cases (Figure 2C). In neurons and LB-containing neurons in the neocortex of dementia with Lewy body cases, no increased in LRP-1 immunoreactivity was observed (not shown).
Figure 2.
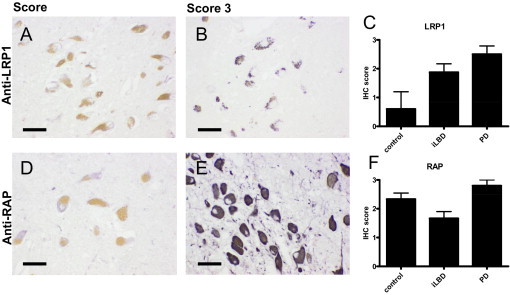
LRP1 and RAP immunoreactivity in melanized neurons in SN of control, iLBD, and PD cases. Immunohistochemical (IHC) staining of LRP1 (A and B) and RAP (D and E) in melanized neurons of the SN in control and PD cases. Semiquantitative analysis of the IHC staining for LRP1 (C) and RAP (F) in the SN of control, iLBD, and PD cases was performed as described in Materials and Methods. In melanized neurons of the SN in control cases, only weak, diffuse cytoplasmic LRP1 staining was observed (A and C; IHC score 0.5 ± 1.3), whereas a strong granular LRP1 staining was detected in PD cases (B and C; IHC score 2.5 ± 0.5). LRP1 immunoreactivity in melanized neurons of the SN in iLBD cases was scored as moderate (C; IHC score 1.9 ± 0.8). RAP staining in melanized neurons in SN of control, iLBD, and PD cases ranged between weak (D) to strong (E) cytoplasmic staining. RAP immunoreactivity demonstrated no apparent difference in immunoreactivity between control, iLBD, and PD cases (F). Mean ± SD is shown. Scale bars: 20 μm (A, B, D, and E).
To examine whether the altered LRP1 staining intensity and pattern in melanized neurons in SN of iLBD and PD may relate to aberrant processing of this large protein, immunoreactivity of the LRP1-specific chaperone RAP was studied. RAP serves two critical functions with respect to LRP1, first to ensure correct folding in the endoplasmic reticulum, and second to prevent premature tight binding of (other) protein ligands to the receptor before its proper insertion in the cell membrane.13, 14 In SN of control, iLBD, and PD cases, cytoplasmic RAP staining was observed in both neurons and glial cells. Moreover, in all cases, cytoplasmic RAP staining was found in melanized neurons of the SN (Figure 2, D and E). However, no clear differences in RAP immunoreactivity pattern or intensity were identified between control, iLBD, and PD patient groups (Figure 2F), which suggests no apparent differences in LRP1 processing between these groups.
Discussion
ApoE is a major lipid and cholesterol transporter in the brain. Evidence from both experimental and clinical investigations is mounting that alterations in lipid metabolism play an important role in PD pathogenesis, because direct cross talk between lipids and α-synuclein, influencing both lipid metabolism and α-synuclein aggregation, has been demonstrated.15 Recently, a direct link between increased ApoE levels in the nervous system and α-synuclein aggregation and neurotoxicity was demonstrated in transgenic mice.5 Our data provide the first evidence from human brain that altered neuronal ApoE levels and/or trafficking occurs in PD and may be involved in α-synuclein deposition.
Another interesting aspect of our observations is that ApoE was detected intracellularly in melanized neurons of the SN in both iLBD and PD cases. With our approach, we cannot distinguish between uptake of ApoE from extraneuronal sources and intraneuronal synthesis. However, considering the general intensity of staining, we assume that neuronal synthesis is the most likely source of the ApoE in melanized neurons observed by us. This supposition is of interest because under normal conditions in the brain, ApoE is synthesized and released primarily by (astro)glial16 and perivascular cells.17 Neurons, however, are capable of producing ApoE when stressed.18 Although protective in nature, ApoE synthesis may adversely affect neuronal survival as a result of formation of a C-terminal truncated form that causes mitochondrial dysfunction and neurotoxicity.19 This holds true in particular for the ApoE4 isotype18 and is of major interest, considering the alleged role of mitochondrial impairment in PD pathogenesis and the still unclarified role of the ApoE genotype in PD disease development and/or progression.20
Acting as a shuttling receptor, LRP1 is responsible for ApoE trafficking in and out of cells, and within cells, and ApoE binds LRP1 only when it is associated with lipids and not when ApoE is devoid of lipids.21 Here, we observed, not only up-regulation of ApoE, but also up-regulation and redistribution of LRP1 in melanized neurons of the SN in both iLBD and symptomatic stages of PD, which might, therefore, reflect early alterations in lipid metabolism in these cells during PD pathogenesis. Our observation that RAP staining was not elevated in iLBD and/or PD, suggests that up-regulation and redistribution of LRP1 does not require apparent changes in its processing. However, since RAP expression is high in brain,22 sufficient levels of RAP might be present to mask alterations in LRP1 processing.
In conclusion, our data indicate that alterations in lipoprotein homeostasis in melanized neurons of the SN are an early event in the pathogenesis of PD. This phenomenon appears specific for melanized neurons in PD, since alterations in ApoE and LRP1 immunoreactivity were not found in neurons and LBs of dementia with Lewy body cases. Future studies need to address whether the initial up-regulation of ApoE in melanized neurons is a neuroprotective or neurotoxic response.
Footnotes
Supported by grants from the Stichting Internationaal Parkinson Fonds (M.M.M.W., B.D., and J.J.M.H.).
B.D. and J.J.M.H. contributed equally to this article.
References
- 1.Wolters E.C., Braak H. Parkinson's disease: premotor clinico-pathological correlations. J Neural Transm Suppl. 2006 doi: 10.1007/978-3-211-45295-0_47. [DOI] [PubMed] [Google Scholar]; (70):309–319
- 2.Braak H., Del Tredici K., Rub U., de Vos R.A., Jansen Steur E.N., Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 3.Mahley R.W. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988;240:622–630. doi: 10.1126/science.3283935. [DOI] [PubMed] [Google Scholar]
- 4.Corder E.H., Saunders A.M., Strittmatter W.J., Schmechel D.E., Gaskell P.C., Small G.W., Roses A.D., Haines J.L., Pericak-Vance M.A. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 5.Gallardo G., Schluter O.M., Sudhof T.C. A molecular pathway of neurodegeneration linking alpha-synuclein to ApoE and Abeta peptides. Nat Neurosci. 2008;11:301–308. doi: 10.1038/nn2058. [DOI] [PubMed] [Google Scholar]
- 6.Jellinger K.A. A critical evaluation of current staging of alpha-synuclein pathology in Lewy body disorders. Biochim Biophys Acta. 2009;1792:730–740. doi: 10.1016/j.bbadis.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 7.Dickson D.W., Fujishiro H., DelleDonne A., Menke J., Ahmed Z., Klos K.J., Josephs K.A., Frigerio R., Burnett M., Parisi J.E., Ahlskog J.E. Evidence that incidental Lewy body disease is pre-symptomatic Parkinson's disease. Acta Neuropathol. 2008;115:437–444. doi: 10.1007/s00401-008-0345-7. [DOI] [PubMed] [Google Scholar]
- 8.Bu G. Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nat Rev Neurosci. 2009;10:333–344. doi: 10.1038/nrn2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilhelmus M.M., Verhaar R., Andringa G., Bol J.G., Cras P., Shan L., Hoozemans J.J., Drukarch B. Presence of tissue transglutaminase in granular endoplasmic reticulum is characteristic of melanized neurons in Parkinson's disease brain. Brain Pathol. 2011 doi: 10.1111/j.1750-3639.2010.00429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kanekiyo T., Bu G. Receptor-associated protein interacts with amyloid-beta peptide and promotes its cellular uptake. J Biol Chem. 2009;284:33352–33359. doi: 10.1074/jbc.M109.015032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Namba Y., Tomonaga M., Kawasaki H., Otomo E., Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer's disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991;541:163–166. doi: 10.1016/0006-8993(91)91092-f. [DOI] [PubMed] [Google Scholar]
- 12.Wilhelmus M.M., Otte-Holler I., van Triel J.J., Veerhuis R., Maat-Schieman M.L., Bu G., de Waal R.M., Verbeek M.M. Lipoprotein receptor-related protein-1 mediates amyloid-beta-mediated cell death of cerebrovascular cells. Am J Pathol. 2007;171:1989–1999. doi: 10.2353/ajpath.2007.070050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bu G., Geuze H.J., Strous G.J., Schwartz A.L. 39 kDa receptor-associated protein is an ER resident protein and molecular chaperone for LDL receptor-related protein. EMBO J. 1995;14:2269–2280. doi: 10.1002/j.1460-2075.1995.tb07221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Obermoeller L.M., Warshawsky I., Wardell M.R., Bu G. Differential functions of triplicated repeats suggest two independent roles for the receptor-associated protein as a molecular chaperone. J Biol Chem. 1997;272:10761–10768. doi: 10.1074/jbc.272.16.10761. [DOI] [PubMed] [Google Scholar]
- 15.Ruiperez V., Darios F., Davletov B. Alpha-synuclein, lipids and Parkinson's disease. Prog Lipid Res. 2010;49:420–428. doi: 10.1016/j.plipres.2010.05.004. [DOI] [PubMed] [Google Scholar]
- 16.Poirier J., Hess M., May P.C., Finch C.E. Astrocytic apolipoprotein E mRNA and GFAP mRNA in hippocampus after entorhinal cortex lesioning. Brain Res Mol Brain Res. 1991;11:97–106. doi: 10.1016/0169-328x(91)90111-a. [DOI] [PubMed] [Google Scholar]
- 17.Wilhelmus M.M., Otte-Holler I., Davis J., Van Nostrand W.E., de Waal R.M., Verbeek M.M. Apolipoprotein E genotype regulates amyloid-beta cytotoxicity. J Neurosci. 2005;25:3621–3627. doi: 10.1523/JNEUROSCI.4213-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mahley R.W., Weisgraber K.H., Huang Y. Apolipoprotein E4: a causative factor and therapeutic target in neuropathology, including Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:5644–5651. doi: 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chang S., ran Ma T., Miranda R.D., Balestra M.E., Mahley R.W., Huang Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc Natl Acad Sci U S A. 2005;102:18694–18699. doi: 10.1073/pnas.0508254102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Williams-Gray C.H., Goris A., Saiki M., Foltynie T., Compston D.A., Sawcer S.J., Barker R.A. Apolipoprotein E genotype as a risk factor for susceptibility to and dementia in Parkinson's disease. J Neurol. 2009;256:493–498. doi: 10.1007/s00415-009-0119-8. [DOI] [PubMed] [Google Scholar]
- 21.Zerbinatti C.V., Bu G. LRP and Alzheimer's disease. Rev Neurosci. 2005;16:123–135. doi: 10.1515/revneuro.2005.16.2.123. [DOI] [PubMed] [Google Scholar]
- 22.Bu G. The roles of receptor-associated protein (RAP) as a molecular chaperone for members of the LDL receptor family. Int Rev Cytol. 2001;209:79–116. doi: 10.1016/s0074-7696(01)09011-8. [DOI] [PubMed] [Google Scholar]