

Abstract
Activated protein C (APC) is a natural anticoagulant that exerts anti-inflammatory and cytoprotective properties mediated through the protease activated receptor (PAR)-1. APC can also proteolytically cleave PAR-2, although subsequent function is unknown. On the basis of recent evidence that APC promotes wound healing, the aim of this study was to determine whether APC acts through PARs to heal murine excisional wounds or to regulate human cultured keratinocyte function and to determine the signaling mechanisms. Topical administration of APC accelerated wound healing in wild-type mice and, unexpectedly, in PAR-1 knockout mice. PAR-2 knockout mice healed significantly slower than wild-type mice, and healing was not altered by adding APC, indicating that APC acts through PAR-2 to heal wounds. In cultured human primary keratinocytes, APC enhanced PAR-2, stimulated proliferation, activated phosphatidylinositol 3-kinase/Src/Akt, and inhibited phosphorylated (P)-p38. Inhibiting PAR-1 or PAR-2, by small-interfering RNA or blocking antibody, reversed APC-induced keratinocyte proliferation and Akt activation. Blocking PAR-2, but not PAR-1, reversed the inhibition of P-p38 by APC. Furthermore, inhibition of P-p38 accelerated wound healing in wild-type mice. In summary, although APC acts through both PAR-1 and PAR-2 to activate Akt and to increase keratinocyte proliferation, APC-induced murine wound healing depends on PAR-2 activity and inhibition of P-p38.
Activated protein C (APC) is a plasma protease with anticoagulant activity and direct cytoprotective properties, including anti-inflammatory and antiapoptotic effects and endothelial barrier protection.1, 2, 3, 4 In humans, recombinant APC reduces the 28-day mortality in severe sepsis compared with placebo5 and promotes healing of chronic wounds.6 In animal models, APC is neuroprotective 4 hours after stroke onset,7 protects diabetic nephropathy,8 inhibits the development of lung fibrosis in bleomycin-induced lung injury,9 and reduces intestinal injury in necrotizing enterocolitis.10 In vitro, APC modulates keratinocyte and endothelial function, promoting wound healing by enhancing re-epithelialization and angiogenesis.11, 12, 13 On the basis of in vivo and in vitro studies,2, 11, 12, 13, 14 one paradigm for the signaling of APC in keratinocytes and endothelial cells is explained by its binding to endothelial cell protein C receptor (EPCR)15 and subsequent proteolytic cleavage and activation of the G protein–coupled receptor, protease activated receptor (PAR)-1, commonly referred to as the thrombin receptor.16 However, the exact receptors and intracellular signaling mechanism(s) to explain the wound-healing actions of APC are unknown. Xue et al12 have shown that EPCR facilitates the function of APC on keratinocytes via activation of the PAR-1 pathway. APC can also cleave PAR-217; however, its subsequent function is yet to be determined. Kaneider et al18 have shown that the barrier-protective effects of APC are abolished by silencing with small-interfering RNA (siRNA) specific for either PAR-1 or PAR-2, suggesting that these effects of APC require both PAR-1 and PAR-2.
Less is known about the intracellular signaling mechanisms of PAR-2 than PAR-1. PAR-2 activation strongly activates the extracellular signal-regulated kinase 1/2 and weakly stimulates p38, although c-Jun N-terminal kinase is not affected.19, 20, 21 Recent studies showed that activation of PAR-2 leads to rapid phosphorylation of extracellular signal-regulated kinase 1/2 but not p38 in HaCat cells, an immortalized keratinocyte cell line.22 PAR-2 is expressed by many cell types present in the skin (eg, fibroblasts, nerves, dendritic cells, endothelial cells, mast cells, leukocytes, and epidermal keratinocytes23) and participates in cutaneous homeostasis and tissue repair. In this report, we detail the receptor and signaling mechanisms responsible for APC-mediated stimulation of wound healing, using human primary keratinocytes and PAR-1 and PAR-2 knockout (KO) mice. Our results clearly show that PAR-2 is important for the promotion of APC-induced cutaneous wound healing.
Materials and Methods
Animals
Studies were performed in male wild-type (WT), PAR-1 KO, or PAR-2 KO mice. Mice were all in the C57BL/6J background and at 6 weeks of age when starting wounding protocol. Mice were obtained from and housed at Kearns Facility, Kolling Medical Research Institute, University of Sydney, under a 12-hour light/12-hour dark cycle at 22°C.
Wound Creation, Macroscopic Examination, and Analysis
Full-thickness skin wounds extending through the panniculus carnosus were made with an iris scissors, and a sterile 6-mm punch biopsy tool was used to outline a pattern on the dorsum of the mice as described before.24 APC (20 μL) or phosphate-buffered saline (PBS; control) was topically applied onto wounds once a day for 3 consecutive days. The animals were then kept under anesthesia for 20 minutes to allow absorption of the solution. The wounds were left open, and the animals were housed in individual cages. Wound healing was monitored by taking digital photographs and blindly measuring the wound area by tracing the wound perimeter with a thin-tipped marker onto sterile Visitrak Grid (Smith & Nephew, NSW, Australia).6 Tracings were then placed onto the wound measurement device and copied with an electronic stylus to obtain a digital reading of the wound area. The skin tissues were removed from mice after being sacrificed at indicated days and used for further analysis. All procedures were performed according to the guidelines of the local animal care and ethics committee.
Histology and Immunohistochemistry
Skin tissues were fixed with 10% neutral-buffered formalin and paraffin sections (4 μm) and were stained with H&E. For immunohistochemistry, sections were incubated with anti-PAR-2 antibody (1:50 dilution; SAM11; Santa Cruz Biotechnology Inc., Santa Cruz, CA) and isotype-matched anti-IgG2a antibody (1:50 dilution; Sigma-Aldrich, St. Louis, MO) and detected with the use of MACH 3 mouse horseradish peroxidase-polymer detection kit (Biocare Medical, Concord, CA.) and Nova RED (vector) according to manufacturers' protocol and counterstained with Mayer's hematoxylin and Scott's bluing solution. After mounting, sections were observed under a light microscope (ECLIPSE 80i; Nikon, Tokyo, Japan).
Cell Culture and Reagents
Normal keratinocytes were isolated from neonatal foreskins as described previously9 in accordance with the local ethics regulations. Extracted cells were cultured in keratinocyte-serum-free medium (Invitrogen, Carlsbad, CA). HaCat cells were cultured in Dulbecco's modified Eagle's medium (Sigma-Aldrich) containing 10% heat-inactivated fetal bovine serum, 100 U of penicillin, and 2 mmol/L l-glutamine at 37°C in a humidified atmosphere containing 5% CO2. When >70% were confluent, primary cultured cells and HaCat cells were trypsinized and used in experiments. Cells were seeded into either 24-well culture plates or 8-well Permanox slides (Nalge Nunc International, Rochester, NY) and cultured to confluence. The confluent cells were then treated with recombinant APC (Xigris; Eli Lilly & Co., Indianapolis, IN) at different time points in various concentrations as indicated in figures, and with RCR252 (EPCR blocking antibody), RCR92 (EPCR nonblocking antibody) (both anti-EPCR antibodies provided by Prof Kenji Fukudome, Department of Immunology, Saga Medical School, Nabeshima, Saga, Japan), PAR-2 blocking antibody (SAM11), matched isotype IgG2a (Sigma-Aldrich), Src family kinase inhibitor PP-1 (Calbiochem, San Diego, CA), or phosphatidylinositol 3 kinase (PI3K) inhibitors wortmannin and LY294002 (Cell Signaling Technology, Danvers, MA) at various concentrations. Cells and culture supernatant fluids were collected for detection of protein expression.
Knockdown of PAR-1 and PAR-2 through siRNA
PAR-1 and PAR-2 expression in primary keratinocytes was silenced with PAR-1 and PAR-2 siRNA. PAR-specific siRNAs were synthesized by Ambion (Austin, TX) and used to target the following mRNA sequences: PAR-1 siRNA, 5′-AGAUUAGUCUCCAUCAAUA-3′; PAR-2 siRNA, 5′-GGAAGAAGCCUUAUUGGUA-3′. The nonspecific siRNA, 5′-GGCUACGUCCAGGAGCGCACC-3′, was used as a negative control. The cells were transfected following the manufacturer's instructions with the PAR siRNAs with the use of siPORT NeoFX transfection reagent (Ambion, Austin, TX) in a 6-well plate (Nalge Nunc International) for 24 hours, then trypsinized and seeded into either 24-well plates (4 × 105 cells/mL) or 96-well plates (1 × 104 cells/mL), and incubated for 48 hours and 24 hours, respectively. PAR-1 and PAR-2 knockdown efficiency was confirmed by immunoblot analysis.
Cell Proliferation Assay
Cells (1 × 104 cells/well) were seeded into a 96-well microplate to a final volume of 200 μL and were incubated for 4 hours to allow cells to attach. Cells were then treated with APC at 0.01, 0.1, 1, or 10 μg/mL. After incubation for 24 hours, culture medium was removed, and cells were stained with 1 μg/mL crystal violet (Sigma-Aldrich) dissolved in PBS. The unbound dye was removed by washing with tap water, and cells were left to completely dry overnight. Bound crystal violet was solubilized with 0.1% SDS in PBS. The optical density of each well was determined at a wavelength of 550 nm. Results were expressed as percentages of controls.
Immunoblot Analysis
Immunoblot analysis was performed as described previously.13 Briefly, equal amounts of proteins were separated by 10% SDS-PAGE and transferred to a polyvinylidene difluoride membrane. The primary antibodies used were as follows: active caspase-3 (BioVision, Mountain View, CA); PAR-1 (Santa Cruz Biotechnology Inc.); PAR-2; anti-mouse EPCR (R&D Systems, Minneapolis, MN); rabbit polyclonal antibodies directed against phospho-Akt (Ser473), Akt, phospho-Src (tyr416), and Src (Cell Signaling Technology); and rabbit antiphosphorylated forms of p38 and p38 (Santa Cruz Biotechnology Inc.). Immunoreactivity was detected with the electrochemiluminescence detection system (Amersham Biosciences, Buckinghamshire, UK). Anti-human β-actin antibody was included to normalize against unequal loading.
Apoptosis Detection
PAR-1 and PAR-2 siRNA-induced apoptotic keratinocytes were detected with an in situ cell death detection kit according to manufacturer's instructions (Roche Diagnostics, Indianapolis, IN) as described before.13
Statistical Analysis
The data are summarized as the mean ± SD. Statistical analyses were performed with Student's t-test or analysis of variance. Statistical significance was accepted at P < 0.05.
Results
Topically Applied APC Accelerates Wound Healing in WT Mice
We first examined the effect of APC on cutaneous wound healing in mice. Full-thickness skin wounds treated with APC (10 μg/wound/day) for 3 consecutive days (beginning immediately after induction of wound) were markedly reduced in size and nearly completely healed on day 6 (Figure 1A). Compared with (PBS) control-treated wounds, APC significantly reduced wound size at each time point over the course of the healing process (Figure 1A). Time to complete wound closure was reduced by ∼3 days in APC-treated mice, compared with control mice (10.3 ± 1.7 versus 7.4 ± 1.2 days; P = 0.00016; Figure 1B). The three-treatment protocol was more effective than a single APC treatment (see Supplemental Figure S1 at http://ajp.amjpathol.org), as previously described in rats,24 and was used for all further experiments.
Figure 1.
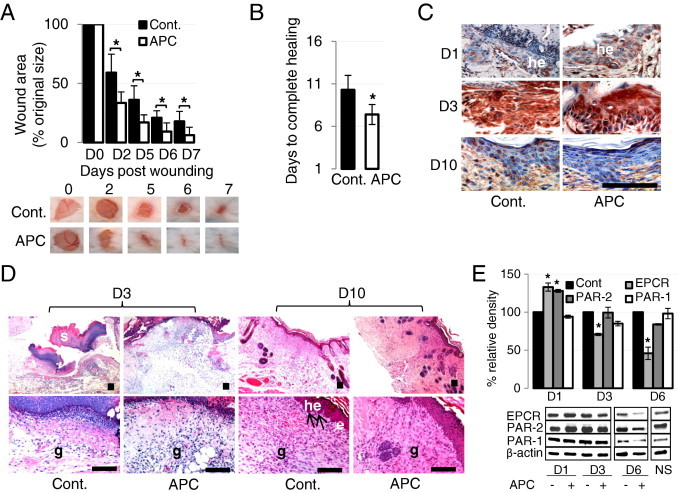
APC accelerates wound healing in wild-type (WT) mice. A and B: Full-thickness 6-mm diameter wounds were made and treated topically with 20 μL of phosphate-buffered saline or APC (10 μg) once a day for 3 consecutive days. A: Representative photographs of skin wounds on days after APC treatment. Percentage of wound area/initial area was calculated from tracing the wounds, measured with Visitrak. Values are mean ± SD; n = 10 wounds. *P < 0.05 by paired t-test. B: Time to complete wound closure Values are mean ± SD; n = 10 wounds. *P < 0.05 by t-test. C: Expression of PAR-2 by IHC on wounded skin in WT mice. he, hyperproliferative epithelium. Scale bar = 100 μm. D: H&E-stained paraffin sections from day 3 and day 10 wounds from WT mice. Arrows indicate the leading edge of the migrating epithelial tongue. e, epithelium; g, granulation tissue; he, hyperproliferative epithelium; s, scab. Scale bar = 100 μm. E: Expression of EPCR, PAR-1, and PAR-2 detected by immunoblotting from homogenate supernatants of wounded skin. NS, normal skin. The band intensity of the protein was normalized with β-actin, and each control was defined as 100%. Values are mean ± SD; n = 3. *P < 0.05 versus each control on each day by paired t-test.
Wound tissue from WT mice on days 1, 3, and 10 was collected for immunohistochemistry, histology, and immunoblot analysis. On day 1, but not day 3 or day 10, APC-treated wounds showed increased thickness of hyperproliferative epithelium and PAR-2 expression. Compared with control wounds (Figure 1C), H&E-stained sections on day 10 showed that all wounds were completely covered by neoepithelium; however, histologic differences were observed between control and APC-treated wounds (Figure 1D). APC-treated wounds were characterized by a reduced wound area, less inflammation, and enhanced granulation tissue compared with control wounds, whereas migrating epithelial tongues were consistently visible only in controls (Figure 1D). On day 10 more new blood vessels were present in control wounds, suggesting that regression of new blood vessels was delayed in control compared with APC-treated wounds (Figure 1D). These data indicate that, 10 days after injury, control wounds were still developing new tissue, whereas APC-treated wounds were in a more advanced tissue remodeling phase. Consistent with Figure 1C, immunoblot data suggest that, after wounding, APC increased PAR-2 expression on day 1 (24 hours after the first APC treatment) but decreased PAR-2 on day 6, whereas PAR-2 expression is the same in both groups on day 3 (Figure 1E). In contrast, APC had little effect on PAR-1 expression at any time point (Figure 1E). EPCR showed a similar response to PAR-2 (Figure 1E). WT, PAR-1 KO, and PAR-2 KO mice expressed similar levels of EPCR. No compensatory change was observed in the levels of PAR-2 in PAR-1 KO mice and vice versa (see Supplemental Figure S2 at http://ajp.amjpathol.org).
APC Accelerates Wound Healing via PAR-2, Not PAR-1
We used KO mice to elucidate the role of PAR-1 and PAR-2 in APC-induced wound healing. PAR-1 KO mice healed at the same rate as WT mice at all time points (Figure 2, A and B). In contrast, wound healing in PAR-2 KO mice was significantly delayed compared with WT mice (Figure 2C). Skin sections on day 3 showed that the wound area was wider in PAR-2 KO mice than in WT mice (Figure 2D). APC accelerated wound healing in PAR-1 KO mice (Figure 2B); however, it had negligible effect in PAR-2 KO mice (Figure 2, C and D). A significant reduction (P = 0.013 and P = 0.004 on days 3 and 6, respectively; n = 6) in wound area was observed in APC-treated WT mice compared with APC-treated PAR-2-KO mice (Figure 2C). These data indicate that APC uses PAR-2, and not PAR-1, to promote murine cutaneous wound healing. H&E-stained sections of wounded skin showed histologic differences between WT mice and PAR-2 KO mice (Figure 2D). On day 13, in both control and in response to APC, PAR-2 KO mice exhibited increased epithelial layers, a migrating epithelial tongue (Figure 2D), and disorganized dermis. In contrast, WT mice showed a normal thin epithelial layer, a more mature dermis, and lack of inflammatory cells when treated with APC.
Figure 2.
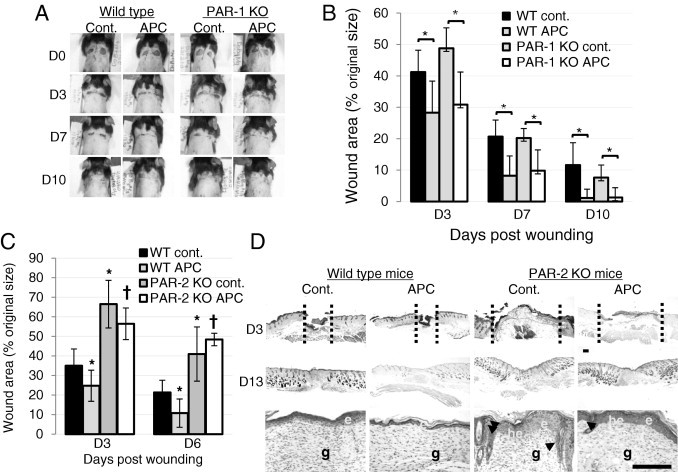
APC-treated PAR-1 knockout (KO) mice heal at the same rate as wild-type (WT) mice, whereas healing is significantly delayed in PAR-2 KO mice. A: Representative photographs of skin wound healing in WT and PAR-1 KO mice on days 0, 3, 7, and 10 after wounding. B: Percentage of wound areas/initial areas was calculated from the tracing wound areas, using Visitrak. Values are the mean ± SD; n = 6 wounds. *P < 0.05 by analysis of variance. C: Percentage of wound areas/initial areas was calculated from the tracing wound areas, using Visitrak. Values are the mean ± SD; n = 6 wounds. *P < 0.05 versus control WT mice; †P < 0.05 versus WT APC-treated mice by analysis of variance. D: H&E-stained paraffin sections from day 3 and day 10 wounds from control- and APC-treated WT mice and PAR-2 KO mice. Arrowheads indicate the migrating epithelial tongue. Wound gap is indicated by vertical dash line. e, epithelium; g, granulation tissue; he, hyperproliferative epithelium. Scale bar = 100 μm.
APC Stimulates Human Keratinocyte Proliferation via PAR-1 and PAR-2 and Reverses PAR-2 siRNA-Induced Apoptosis in Vitro
Consistent with in vivo results (Figure 1, C and E), APC enhanced the expression of PAR-2 in cultured human primary keratinocytes in a dose-dependent manner, after 24 hours of treatment (Figure 3A) and in HaCat keratinocytes (see Supplemental Figure S3A at http://ajp.amjpathol.org). When cells were pretreated with anti-PAR-2 blocking antibody (1 to 10 μg/mL), the increase in cell proliferation in response to APC was dose-dependently inhibited, compared with isotype control (Figure 3B). In addition, PAR-2 siRNA, which knocked down PAR-2 protein expression in human primary keratinocytes by >80% (see Supplemental Figure S3B at http://ajp.amjpathol.org), also suppressed cell growth, confirming that APC stimulates keratinocyte proliferation via PAR-2 (Figure 3C). Similarly, a significant inhibition of proliferation occurred in PAR-1 siRNA-treated cells (Figure 3D). Simultaneous knockdown of both PAR-1 and PAR-2 completely prevented APC from stimulating cell proliferation (Figure 3E). These data suggest that both PAR-1 and PAR-2 are required for APC-induced human keratinocyte proliferation in vitro.
Figure 3.
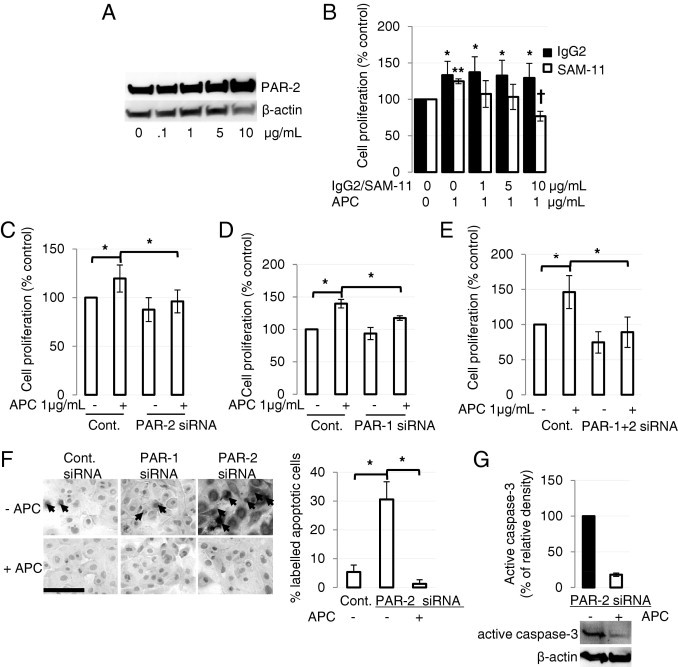
APC stimulates human keratinocyte proliferation via PAR-1 and PAR-2 and reverses PAR-2 siRNA-induced apoptosis in vitro. A: Human primary cultured keratinocytes were treated with or without APC for 24 hours, and PAR-2 was detected by immunoblot analysis. B: Cells were pretreated with the PAR-2 blocking antibody (10 μg/mL) or isotype control for 2 hours and then treated with1 μg/mL APC for 24 hours. Proliferation was measured by crystal violet assay. Values are mean ± SD; n = 4. *P < 0.05 versus control, **P < 0.05 versus control; †P < 0.05 versus APC-treated keratinocytes, with the use of analysis of variance. C–E: Keratinocyte proliferation, measured by crystal violet assay, after blocking PAR-2 (C), PAR-1 (D), or PAR-1+ PAR-2 (E) with the use of PAR-2 and PAR-1 specific siRNA (10 nmol/L each). Values are mean ± SD; n = 4. *P < 0.05 by analysis of variance. F: Keratinocytes were treated with PAR-2 siRNA (10 nmol/L). After 48 hours, cells were treated with or without of 1 μg/mL APC for 30 minutes and assessed with TUNEL assay to detect apoptotic cells (black arrows). Images represent one of three independent experiments. Scale bar = 100 μm. Graph shows the mean number of apoptotic cells per field of 5 fields. Values are mean ± SD; n = 3. *P < 0.05 by analysis of variance. (G) PAR-2 siRNA-transduced keratinocytes were treated without or with APC for 24 hours, and active caspase-3 was detected by immunoblot analysis. Data are expressed as mean ± SD; n = 2.
After siRNA knockdown of PAR-1 in human keratinocytes for 48 hours, a small percentage of apoptotic keratinocytes, similar to control, were detectable with an in situ death kit (Figure 3F). However, when cells were treated with PAR-2 siRNA, a marked induction of apoptotic cells was observed (Figure 3F). Consistent with a previous study,17 treatment with APC (1 μg/mL) for 30 minutes was sufficient to totally reverse apoptosis after 24 hours in all conditions (Figure 3F). These results were confirmed by a significant reduction (>80%) in active caspase-3 in APC-treated PAR-2 siRNA-transfected cells (Figure 3G). Our data show two interesting findings: first, PAR-2 itself is antiapoptotic, and, second, APC does not require PAR-1 or PAR-2 to exert its antiapoptotic effects in human keratinocytes.
APC Activates Akt and the Protein Tyrosine Kinase Src via PAR-1 and PAR-2 in Human Keratinocytes and Promotes Wound-Healing Activity Through the PI3K/Src/Akt Axis
The Src/Akt signaling pathway was assessed with immunoblot analysis. APC stimulated activation of Akt and Src but had minimal effect on total Akt or Src (Figure 4A). When keratinocytes were pretreated with PAR-2 siRNA, activation of Akt and Src by APC was significantly inhibited (Figure 4B). PAR-1 siRNA or combined PAR-1 and PAR-2 siRNAs showed a similar result (Figure 4C). These results show that APC activates Akt and Src via both PAR-1 and PAR-2. Activation of Src at tyrosine can signal through Akt to regulate numerous cellular processes, including cell proliferation.25 To test whether APC stimulates keratinocyte proliferation or Akt activation via Src, cells were stimulated with APC after pretreatment with PP-1, a Src inhibitor, or the phosphatidylinositol 3-kinase (PI3K) inhibitor, wortmannin, at concentrations that were not toxic to the cells (data not shown). When added separately, PP-1 and wortmannin completely blocked cell proliferation and Akt activation induced by APC (Figure 4, D and E). Taken together these results indicate that APC acts through Src, PI3K, and Akt to stimulate proliferation of human skin keratinocytes.
Figure 4.
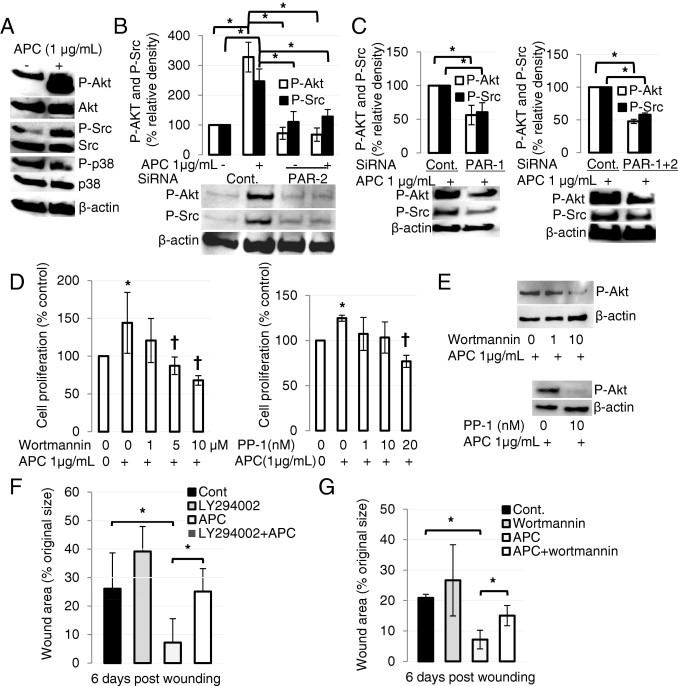
APC promotes wound healing via PI3K/Src/Akt axis. A: Confluent keratinocytes in 24-well plates were treated without or with 1 μg/mL APC for 30 minutes. Total and phosphorylated (P) (activated) forms of Akt, Src, and p38 were measured by immunoblot analysis in whole-cell lysates. β-Actin was used as an internal control. Images represent one of three independent experiments. B and C: Keratinocytes were treated with PAR-2 (B), PAR-1 or PAR-1 + PAR-2 (C) siRNA for 48 hours and then treated with 1 μg/mL APC for 30 minutes. Control siRNA was used for each experiment. Akt/Src kinases were detected by immunoblot analysis. β-Actin was used as an internal control. Images represent one of three independent experiments. Values show mean ± SD; n = 3. *P < 0.05 by analysis of variance. D: After pretreatment with wortmannin (left) or PP-1 (right) at indicated concentrations for 2 hours, cells were treated with 1 μg/mL APC for 24 hours, and proliferation was measured by crystal violet assay. Values are mean ± SD; n = 4. *P < 0.05 by analysis of variance; †P < 0.05 versus control. E: After pretreatment with wortmannin (top) or PP-1 (bottom) for 2 hours, cells were treated with 1 μg/mL APC for 30 minutes, and Akt was detected from whole-cell lysate by immunoblot analysis. β-Actin was used as an internal control. F and G: WT mice were treated with APC and PI3K inhibitors LY294002 (F) or wortmannin (G). Values are mean ± SD; n = 6 wounds. *P < 0.05.
When APC-treated WT mice were pretreated with PI3K inhibitors, LY294002 (Figure 4F) or wortmannin (Figure 4G), wound closure was significantly delayed, compared with APC treatment alone, on day 6 (P = 0.007, n = 6, or P = 0.006, n = 6, respectively). Neither inhibitor had any significant effect on wound closure when used alone (Figure 4, F and G). Consistent with in vitro studies (Figure 4E), these data suggest that PI3K is required for APC's promotion of wound healing in mice.
Inhibition of p38 by APC Accelerates Wound Healing
To further determine the downstream mechanism(s) underlying PARs mediated wound healing–promoting activity of APC, we tested the consequences of proinflammatory signaling molecule, p38 mitogen-activated protein kinase activation by APC. In vitro, APC reduced activation of p38 by 80% in human primary keratinocytes, and this effect was completely reversed by RCR252 (EPCR blocking antibody), SAM11 (PAR-2 blocking antibody), or PAR-2 siRNA, but not by RCR-92 (EPCR nonblocking antibody) or PAR-1 siRNA (Figure 5, A and B). This suggests that APC inhibits activation of p38 via EPCR and PAR-2 activation in vitro. In vivo, APC-treated wounds had substantially less activated p38 than control on day 6 in WT mice and PAR-1 KO mice but not in PAR-2 KO mice (Figure 5, C and D). Consistently, when mice were pretreated with the p38 inhibitor, SB203580, a significant reduction in wound area was observed in WT mice but not in PAR-2 KO mice on day 7, compared with control (Figure 5E).
Figure 5.
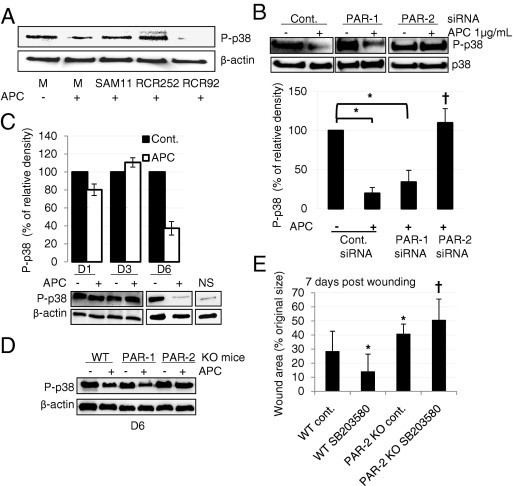
p38 is inhibited by APC via EPCR-PAR-2 which can accelerate wound healing. A: Cells were pretreated with SAM-11 (10 μg/mL), RCR 252 (10 μg/mL), or RCR 92 (10 μg/mL) for 2 hours and then treated with 1 μg/mL APC for 30 minutes. Activation of p38 was detected by immunoblot analysis. B: Keratinocytes were treated with PAR-1 or PAR-2 siRNA for 48 hours, and then treated with 1 μg/mL APC for 30 minutes. Control siRNA was used for each experiment. Activated and total p38 were detected by immunoblot analysis. Values are mean ± SD; n = 3. *P < 0.05, †P < 0.05 versus control (cont.) siRNA+APC with analysis of variance. C and D: Tissue homogenates were obtained from control and APC-treated wound samples from wild-type (WT) mice on days 1, 3, and 6 (C), as well as from PAR-1 and PAR-2 KO mice on day 6 (D), and phosphorylated (P)-p38 was detected by immunoblot analysis. β-Actin was used as an internal control. Images represent one of two independent experiments. Values are mean ± SD; n = 2. E: WT and PAR-2 KO mice were wounded and treated with the p38 inhibitor, SB203580 (10 μmol/L) on days 0, 1, and 3 before wound area was measured on day 8. Percentage of wound areas/initial areas (initial areas was set as 100 on day 0) was calculated from the tracing wound areas, using Visitrak. Values are mean ± SD; n = 6 wounds. *P < 0.05 versus control WT mice; †P < 0.05 versus WT SB203580-treated mice by analysis of variance.
Discussion
Previously, we have shown that APC promotes wound healing both in vitro in cultured keratinocytes and in vivo in rats and humans 6, 11, 12, 13, 24. Our new findings identify the wound-healing capacity of APC in mice and show four important new findings. First, APC stimulates wound healing in PAR-1, but not PAR-2, KO mice. Second, APC acts through both PAR-1 and PAR-2 to stimulate the PI3K/Akt pathway and keratinocyte proliferation. Third, neither PAR-1 nor PAR-2 is required for APC to inhibit apoptosis. Finally, APC requires PAR-2, but not PAR-1, to inhibit activation of p38 in human keratinocytes.
Cutaneous wound healing requires a complex and dynamic array of partially overlapping phases: inflammation, angiogenesis, new granulation tissue formation, re-epithelialization, and tissue remodeling. The initial events need to cease before the deposition of new granulation tissue can be completed, and excessive inflammation is thought to be a causal factor in the pathogenesis of chronic wounds.26 Consistent with our previous study,24 histologic sections of wounds on days 10 and 13 clearly show that the infiltration of inflammatory cells is decreased in response to APC. Furthermore, regression of new blood vessels was delayed in control mice, compared with APC-treated mice on day 10. These results suggest that wounds treated with APC are in a more mature stage of healing.
Knockdown of PAR-2 in keratinocytes induced significant caspase-3 activation and apoptosis, whereas PAR-1 knockdown had minimal effect on apoptosis (Figure 3, F and G). PAR-2 has been shown to prevent apoptosis in several cell types27, 28, 29; for example, agonist activation of PAR-2 rescues astrocytes and neutrophils from apoptotic cell death.28, 29 The antiapoptotic effects of APC are mediated through PAR-1 in endothelial cells stressed with high glucose 8 and both PAR-1 and PAR-3 in neuronal apoptosis.30 Surprisingly, we found that neither PAR-1 nor PAR-2 knockdown, alone or together, prevented the antiapoptotic effects of APC in human primary keratinocytes, suggesting that APC may act through another receptor(s). APC can exert protective effects via epidermal growth factor receptor and Tie2 in human keratinocytes and endothelial cells31, 32 and CD11b in macrophages,33 although at least some of these interactions require PAR-1. APC can also cleave and signal through ApoER2 in a monocyte cell line in the absence of PARs.34
A main finding from this work is the vital role of PAR-2 in murine wound healing. PAR-2 can be proteolytically cleaved by serine proteinases, such as tryptase or trypsin,35, 36 and act in a protective manner to induce bronchodilatation,37 inhibit lipopolysaccharide-induced pulmonary neutrophilia,38 diminish formation of gastric ulcers39 and experimental colitis,40 protect against ischemia-perfusion injury in the heart,41 and potentiate reparative angiogenesis.42 However, PAR-2 can mediate inflammation43 and contribute to the pathogenesis of chronic arthritis, inflammatory pain, and colitis.35, 44
In 2002, Riewald et al17 found that APC could equally activate PAR-1 and PAR-2 in endothelial cells; however, subsequent mitogen-activated protein kinase phosphorylation and function depended on PAR-1 activation, and the function of PAR-2 has remained an enigma. In the current report, not only did PAR-2 mediate APC's stimulation of murine wound healing, but also wound healing was significantly delayed in PAR-2 KO mice in the absence of exogenous APC, suggesting that this receptor is required for normal wound healing. PAR-2 KO mice appeared to have an inherent skin defect because their skin was thinner and they healed more slowly than WT mice, which may be because of impaired cell growth or increased apoptosis (Figure 3), although the exact mechanisms need to be determined.
PAR-1 is required for APC to stimulate keratinocyte proliferation.12 In the current study, inhibition of either PAR-1 or PAR-2 reversed the stimulatory effect of APC on proliferation and Akt in human primary keratinocytes. When PAR-1 and PAR-2 were simultaneously inhibited, there was no significant additional effect. These data indicate that both PAR-1 and PAR-2 are required for APC-induced keratinocyte proliferation. One possible scenario to explain these findings is that APC triggers transactivation of PARs. Evidence that the thrombin-cleaved tethered ligand of PAR-1 can robustly transactivate PAR-2 was obtained with COS-7 cells coexpressing PAR-2 with a PAR-1 variant that can be cleaved, but not activated by thrombin.45 Kaneider et al18 found that PAR-1 can be protective in sepsis only when it transactivated PAR-2 signaling. They concluded that therapeutics that selectively activate PAR-1/PAR-2 complexes may be beneficial in the treatment of sepsis. Transactivation of PARs implies that PAR-1 and PAR-2 are located sufficiently close to each other in the endothelial cell plasma membrane that the cleaved NH2 terminus of PAR-1 can access PAR-2.18
Akt protein kinase modulates cell survival and proliferation46 and is activated by various growth and survival factors to function in a wortmannin-sensitive pathway involving PI3K.47 Mice lacking both Akt1 (usually referred to as Akt) and Akt2 display translucent skin with reduced hair follicles and reduced number of cells in each epidermal layer.48 In reconstituted human epidermis in vitro, knockdown of Akt, but not Akt2, results in premature keratinocyte apoptosis and an abnormal epidermis in skin cultures.49 In the current study, inhibition experiments confirmed that Src and PI3K are upstream of Akt activation and human keratinocyte proliferation in response to APC. Furthermore, the delay in wound healing imposed by wortmannin and LY294002 in WT mice (Figure 4) indicates that similar mechanisms may be involved in wound-healing activity of APC in vivo. Thus, Akt appears to be a key signaling component in APC-induced keratinocyte proliferation and wound healing.
In agreement with our previous report showing that endogenous protein C/APC inhibits phosphorylated-p38,13 the current work shows that exogenous APC prevents activation of p38 in keratinocytes. Inhibition of p38 has broad anti-inflammatory effects in an experimental model of human endotoxemia50 and improves survival after cecal ligation and puncture and in lipopolysaccharide models of sepsis.51, 52 Inhibition of p38 activation also reduces apoptosis in various cell types.27, 53 Our results with PAR KO mice indicate that inhibition of p38 by APC may have a profound effect on wound healing. The inhibition of p38 activation of APC in keratinocytes requires PAR-2, but not PAR-1 (Figure 5B), and PAR-1 KO mice healed similarly to WT mice; PAR-2 KO mice did not respond to APC. This may explain why PAR-1 was not required for cutaneous wound healing in mice in response to APC, whereas PAR-2 was essential to relay the wound-healing effects of APC.
In conclusion, this study shows a novel function for the PAR-2 receptor, as providing the key to APC's ability to heal wounds in mice (Figure 6). APC can cleave PAR-1 and PAR-2 to activate the PI3K/Src/Akt pathway that stimulates keratinocyte proliferation and contributes to, but is not sufficient for, accelerated wound healing. Instead, the action of APC on PAR-2 to suppress p38 activation appears to be a fundamental requirement for promotion of wound healing.
Figure 6.
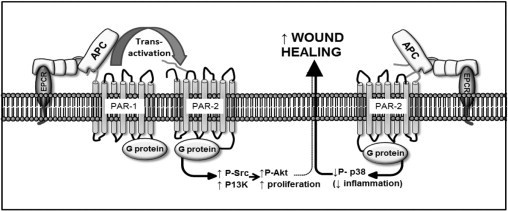
Proposed mechanism of murine wound healing by APC. First, APC can transactivate PAR-1 or PAR-2 as described in Discussion, which activates PI3K and Src to phosphorylate Akt and to stimulate keratinocyte proliferation, which contributes to wound healing but is not sufficient to accelerate healing. Second, APC can activate PAR-2 alone which reduces p38 activity, which can accelerate wound healing possibly by reducing inflammation.
Acknowledgments
We thank Shu-oi Chow and Yee-ka Agnes Chan for technical assistance and Dr. Gordon Campbell (Sydney Adventist Hospital) for providing foreskins.
Footnotes
Supported by a NHMRC Career Development Award, NHMRC project grant, the Rebecca Cooper Foundation and Isabelle Millner and family.
C.J.J. and P.N.S. hold a patent application for the use of APC to treat chronic wounds.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.07.024.
Supplementary data
Topically applied treatment 3 consecutive days was more effective than a single APC treatment in WT mice. Full-thickness 6-mm diameter wounds were treated topically with 10 μg of APC once a day for 1 day (black column) or 3 consecutive days (white column). Percentage of wound area/initial area was calculated from tracing the wounds, measured with Visitrak. Values are mean ± SD; n = 4 wounds.
Relative expression of EPCR (A) on WT, PAR-1 KO, and PAR-2 KO mice; PAR-2 (B) expression on WT and PAR-1 KO mice; and PAR-1 (C) expression on WT and PAR-2 KO mice. Expression of EPCR, PAR-1, and PAR-2 were detected by immunoblot analysis from homogenates of normal skin of WT, PAR-1 KO, and PAR-2 KO mice. β-Actin was used as an internal control.
A: Dose-dependent stimulation of PAR-2 in HaCat keratinocytes. Confluent cells were treated with or without APC for 24 hours, and PAR-2 was detected by immunoblot analysis. B: PAR-2 expression in human primary keratinocytes was assayed by immunoblot analysis after 48 hours of treatment with PAR-2 siRNA.
References
- 1.Mosnier L.O., Zlokovic B.V., Griffin J.H. The cytoprotective protein C pathway. Blood. 2007;109:3161–3172. doi: 10.1182/blood-2006-09-003004. [DOI] [PubMed] [Google Scholar]
- 2.Jackson C.J., Xue M. Activated protein C–an anticoagulant that does more than stop clots. Int J Biochem Cell Biol. 2008;40:2692–2697. doi: 10.1016/j.biocel.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 3.Esmon C.T. New mechanisms for vascular control of inflammation mediated by natural anticoagulant proteins. J Exp Med. 2002;196:561–564. doi: 10.1084/jem.20021088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Esmon C.T. Crosstalk between inflammation and thrombosis. Maturitas. 2004;47:305–314. doi: 10.1016/j.maturitas.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 5.Bernard G.R., Vincent J.L., Laterre P.F., LaRosa S.P., Dhainaut J.F., Lopez-Rodriguez A., Steingrub J.S., Garber G.E., Helterbrand J.D., Ely E.W., Fisher C.J. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 6.Whitmont K., Reid I., Tritton S., March L., Xue M., Lee M., Fulcher G., Sambrook P., Slobedman E., Cooper A., Jackson C. Treatment of chronic leg ulcers with topical activated protein C. Arch Dermatol. 2008;144:1479–1483. doi: 10.1001/archderm.144.11.1479. [DOI] [PubMed] [Google Scholar]
- 7.Zlokovic B.V., Zhang C., Liu D., Fernandez J., Griffin J.H., Chopp M. Functional recovery after embolic stroke in rodents by activated protein C. Ann Neurol. 2005;58:474–477. doi: 10.1002/ana.20602. [DOI] [PubMed] [Google Scholar]
- 8.Isermann B., Vinnikov I.A., Madhusudhan T., Herzog S., Kashif M., Blautzik J., Corat M.A.F., Zeier M., Blessing E., Oh J., Gerlitz B., Berg D.T., Grinnell B.W., Chavakis T., Esmon C.T., Weiler H., Bierhaus A., Nawroth P.P. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat Med. 2007;13:1349–1358. doi: 10.1038/nm1667. [DOI] [PubMed] [Google Scholar]
- 9.Yasui H., Gabazza E.C., Tamaki S., Kobayashi T., Hataji O., Yuda H., Shimizu S., Suzuki K., Adachi Y., Taguchi O. Intratracheal administration of activated protein C inhibits bleomycin-induced lung fibrosis in the mouse. Am J Respir Crit Care Med. 2001;163:1660–1668. doi: 10.1164/ajrccm.163.7.9911068. [DOI] [PubMed] [Google Scholar]
- 10.Kumral A., Yesilirmak D.C., Tugyan K., Baskin H., Tekman I., Duman N., Ozkan H. Activated protein C reduces intestinal injury in an experimental model of necrotizing enterocolitis. J Pediatr Surg. 2010;45:483–489. doi: 10.1016/j.jpedsurg.2009.07.077. [DOI] [PubMed] [Google Scholar]
- 11.Xue M., Thompson P., Kelso I., Jackson C. Activated protein C stimulates proliferation, migration and wound closure, inhibits apoptosis and upregulates MMP-2 activity in cultured human keratinocytes. Exp Cell Res. 2004;299:119–127. doi: 10.1016/j.yexcr.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 12.Xue M., Campbell D., Sambrook P.N., Fukudome K., Jackson C.J. Endothelial protein C receptor and protease-activated receptor-1 mediate induction of a wound-healing phenotype in human keratinocytes by activated protein C. J Invest Dermatol. 2005;125:1279–1285. doi: 10.1111/j.0022-202X.2005.23952.x. [DOI] [PubMed] [Google Scholar]
- 13.Xue M., Campbell D., Jackson C.J. Protein C is an autocrine growth factor for human skin keratinocytes. J Biol Chem. 2007;282:13610–13616. doi: 10.1074/jbc.M610740200. [DOI] [PubMed] [Google Scholar]
- 14.Ruf W. Protease-activated receptor signaling in the regulation of inflammation. Crit Care Med. 2004;32:S287–S292. doi: 10.1097/01.ccm.0000126364.46191.12. [DOI] [PubMed] [Google Scholar]
- 15.Esmon C.T. The endothelial protein C receptor. Curr Opin Hematol. 2006;13:382–385. doi: 10.1097/01.moh.0000239712.93662.35. [DOI] [PubMed] [Google Scholar]
- 16.Vu T.K., Hung D.T., Wheaton V.I., Coughlin S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- 17.Riewald M., Petrovan R.J., Donner A., Mueller B.M., Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002;296:1880–1882. doi: 10.1126/science.1071699. [DOI] [PubMed] [Google Scholar]
- 18.Kaneider N.C., Leger A.J., Agarwal A., Nguyen N., Perides G., Derian C., Covic L., Kuliopulos A. ‘Role reversal’ for the receptor PAR1 in sepsis-induced vascular damage. Nat Immunol. 2007;8:1303–1312. doi: 10.1038/ni1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Belham C.M., Tate R.J., Scott P.H., Pemberton A.D., Miller H.R., Wadsworth R.M., Gould G.W., Plevin R. Trypsin stimulates proteinase-activated receptor-2-dependent and -independent activation of mitogen-activated protein kinases. Biochem J. 1996;320(Pt 3):939–946. doi: 10.1042/bj3200939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeFea K.A., Zalevsky J., Thoma M.S., Dery O., Mullins R.D., Bunnett N.W. beta-Arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J Cell Biol. 2000;148:1267–1281. doi: 10.1083/jcb.148.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu Z., Ahmad S., Schwartz J.L., Banville D., Shen S.H. Protein-tyrosine phosphatase SHP2 is positively linked to proteinase-activated receptor 2-mediated mitogenic pathway. J Biol Chem. 1997;272:7519–7524. doi: 10.1074/jbc.272.11.7519. [DOI] [PubMed] [Google Scholar]
- 22.Rattenholl A., Seeliger S., Buddenkotte J., Schon M., Schon M.P., Stander S., Vergnolle N., Steinhoff M. Proteinase-activated receptor-2 (PAR2): a tumor suppressor in skin carcinogenesis. J Invest Dermatol. 2007;127:2245–2252. doi: 10.1038/sj.jid.5700847. [DOI] [PubMed] [Google Scholar]
- 23.Steinhoff M., Corvera C.U., Thoma M.S., Kong W., McAlpine B.E., Caughey G.H., Ansel J.C., Bunnett N.W. Proteinase-activated receptor-2 in human skin: tissue distribution and activation of keratinocytes by mast cell tryptase. Exp Dermatol. 1999;8:282–294. doi: 10.1111/j.1600-0625.1999.tb00383.x. [DOI] [PubMed] [Google Scholar]
- 24.Jackson C.J., Xue M., Thompson P., Davey R.A., Whitmont K., Smith S., Buisson-Legendre N., Sztynda T., Furphy L.J., Cooper A., Sambrook P., March L. Activated protein C prevents inflammation yet stimulates angiogenesis to promote cutaneous wound healing. Wound Repair Regen. 2005;13:284–294. doi: 10.1111/j.1067-1927.2005.00130311.x. [DOI] [PubMed] [Google Scholar]
- 25.Fresno Vara J.A., Casado E., de C.J., Cejas P., Belda-Iniesta C., Gonzalez-Baron M. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev. 2004;30:193–204. doi: 10.1016/j.ctrv.2003.07.007. [DOI] [PubMed] [Google Scholar]
- 26.Schafer M., Werner S. Cancer as an overhealing wound: an old hypothesis revisited. Nat Rev Mol Cell Biol. 2008;9:628–638. doi: 10.1038/nrm2455. [DOI] [PubMed] [Google Scholar]
- 27.Greenwood SM and Bushell T.J. Astrocytic activation and an inhibition of MAP kinases are required for proteinase-activated receptor-2-mediated protection from neurotoxicity. J Neurochem. 2010;113:1471–1480. doi: 10.1111/j.1471-4159.2010.06737.x. [DOI] [PubMed] [Google Scholar]
- 28.Shpacovitch V.M., Seeliger S., Huber-Lang M., Balkow S., Feld M., Hollenberg M.D., Sarma V.J., Ward P.A., Strey A., Gerke V., Sommerhoff C.P., Vergnolle N., Steinhoff M. Agonists of proteinase-activated receptor-2 affect transendothelial migration and apoptosis of human neutrophils. Exp Dermatol. 2007;16:799–806. doi: 10.1111/j.1600-0625.2007.00605.x. [DOI] [PubMed] [Google Scholar]
- 29.Wang Y., Luo W., Reiser G. Activation of protease-activated receptors in astrocytes evokes a novel neuroprotective pathway through release of chemokines of the growth-regulated oncogene/cytokine-induced neutrophil chemoattractant family. Eur J Neurosci. 2007;26:3159–3168. doi: 10.1111/j.1460-9568.2007.05938.x. [DOI] [PubMed] [Google Scholar]
- 30.Guo H., Liu D., Gelbard H., Cheng T., Insalaco R., Fernandez J.A., Griffin J.H., Zlokovic B.V. Activated protein C prevents neuronal apoptosis via protease activated receptors 1 and 3. Neuron. 2004;41:563–572. doi: 10.1016/s0896-6273(04)00019-4. [DOI] [PubMed] [Google Scholar]
- 31.Xue M., Chow S.O., Dervish S., Chan Y.K., Julovi S.M., Jackson C.J. Activated protein C enhances human keratinocyte barrier integrity via sequential activation of epidermal growth factor receptor and tie2. J Biol Chem. 2011;286:6742–6750. doi: 10.1074/jbc.M110.181388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Minhas N., Xue M., Fukudome K., Jackson C.J. Activated protein C utilizes the angiopoietin/Tie2 axis to promote endothelial barrier function. FASEB J. 2010;24:873–881. doi: 10.1096/fj.09-134445. [DOI] [PubMed] [Google Scholar]
- 33.Cao C., Gao Y., Li Y., Antalis T.M., Castellino F.J., Zhang L. The efficacy of activated protein C in murine endotoxemia is dependent on integrin CD11b. J Clin Invest. 2010;120:1971–1980. doi: 10.1172/JCI40380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang X.V., Banerjee Y., Fernandez J.A., Deguchi H., Xu X., Mosnier L.O., Urbanus R.T., de Groot P.G., White-Adams T.C., McCarty O.J., Griffin J.H. Activated protein C ligation of ApoER2 (LRP8) causes Dab1-dependent signaling in U937 cells. Proc Natl Acad Sci U S A. 2009;106:274–279. doi: 10.1073/pnas.0807594106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Steinhoff M., Vergnolle N., Young S.H., Tognetto M., Amadesi S., Ennes H.S., Trevisani M., Hollenberg M.D., Wallace J.L., Caughey G.H., Mitchell S.E., Williams L.M., Geppetti P., Mayer E.A., Bunnett N.W. Agonists of proteinase-activated receptor 2 induce inflammation by a neurogenic mechanism. Nat Med. 2000;6:151–158. doi: 10.1038/72247. [DOI] [PubMed] [Google Scholar]
- 36.Molino M., Barnathan E.S., Numerof R., Clark J., Dreyer M., Cumashi A., Hoxie J.A., Schechter N., Woolkalis M., Brass L.F. Interactions of mast cell tryptase with thrombin receptors and PAR-2. J Biol Chem. 1997;272:4043–4049. doi: 10.1074/jbc.272.7.4043. [DOI] [PubMed] [Google Scholar]
- 37.Cocks T.M., Fong B., Chow J.M., Anderson G.P., Frauman A.G., Goldie R.G., Henry P.J., Carr M.J., Hamilton J.R., Moffatt J.D. A protective role for protease-activated receptors in the airways. Nature. 1999;398:156–160. doi: 10.1038/18223. [DOI] [PubMed] [Google Scholar]
- 38.Moffatt J.D., Jeffrey K.L., Cocks T.M. Protease-activated receptor-2 activating peptide SLIGRL inhibits bacterial lipopolysaccharide-induced recruitment of polymorphonuclear leukocytes into the airways of mice. Am J Respir Cell Mol Biol. 2002;26:680–684. doi: 10.1165/ajrcmb.26.6.4693. [DOI] [PubMed] [Google Scholar]
- 39.Kawabata A., Kinoshita M., Nishikawa H., Kuroda R., Nishida M., Araki H., Arizono N., Oda Y., Kakehi K. The protease-activated receptor-2 agonist induces gastric mucus secretion and mucosal cytoprotection. J Clin Invest. 2001;107:1443–1450. doi: 10.1172/JCI10806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fiorucci S., Mencarelli A., Palazzetti B., Distrutti E., Vergnolle N., Hollenberg M.D., Wallace J.L., Morelli A., Cirino G. Proteinase-activated receptor 2 is an anti-inflammatory signal for colonic lamina propria lymphocytes in a mouse model of colitis. Proc Natl Acad Sci U S A. 2001;98:13936–13941. doi: 10.1073/pnas.241377298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Napoli C., Cicala C., Wallace J.L., de N.F., Santagada V., Caliendo G., Franconi F., Ignarro L.J., Cirino G. Protease-activated receptor-2 modulates myocardial ischemia-reperfusion injury in the rat heart. Proc Natl Acad Sci U S A. 2000;97:3678–3683. doi: 10.1073/pnas.97.7.3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Milia A.F., Salis M.B., Stacca T., Pinna A., Madeddu P., Trevisani M., Geppetti P., Emanueli C. Protease-activated receptor-2 stimulates angiogenesis and accelerates hemodynamic recovery in a mouse model of hindlimb ischemia. Circ Res. 2002;91:346–352. doi: 10.1161/01.res.0000031958.92781.9e. [DOI] [PubMed] [Google Scholar]
- 43.Dery O., Corvera C.U., Steinhoff M., Bunnett N.W. Proteinase-activated receptors: novel mechanisms of signaling by serine proteases [review] Am J Physiol. 1998;274:C1429–C1452. doi: 10.1152/ajpcell.1998.274.6.C1429. [DOI] [PubMed] [Google Scholar]
- 44.Ferrell W.R., Lockhart J.C., Kelso E.B., Dunning L., Plevin R., Meek S.E., Smith A.J., Hunter G.D., Mclean J.S., McGarry F., Ramage R., Jiang L., Kanke T., Kawagoe J. Essential role for proteinase-activated receptor-2 in arthritis. J Clin Invest. 2003;111:35–41. doi: 10.1172/JCI16913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O'Brien P.J., Prevost N., Molino M., Hollinger M.K., Woolkalis M.J., Woulfe D.S., Brass L.F. Thrombin responses in human endothelial cells-Contributions from receptors other than Par1 include the transactivation of Par2 by thrombin-cleaved Par1. J Biol Chem. 1994;275:13502–13509. doi: 10.1074/jbc.275.18.13502. [DOI] [PubMed] [Google Scholar]
- 46.Manning B.D., Cantley L.C. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Franke T.F., Yang S.I., Chan T.O., Datta K., Kazlauskas A., Morrison D.K., Kaplan D.R., Tsichlis P.N. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- 48.Peng X.D., Xu P.Z., Chen M.L., Hahn-Windgassen A., Skeen J., Jacobs J., Sundararajan D., Chen W.S., Crawford S.E., Coleman K.G., Hay N. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev. 2003;17:1352–1365. doi: 10.1101/gad.1089403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thrash B.R., Menges C.W., Pierce R.H., McCance D.J. AKT1 provides an essential survival signal required for differentiation and stratification of primary human keratinocytes. J Biol Chem. 2006;281:12155–12162. doi: 10.1074/jbc.M512116200. [DOI] [PubMed] [Google Scholar]
- 50.Branger J., van den Blink B., Weijer S., Madwed J., Bos C.L., Gupta A., Yong C.L., Polmar S.H., Olszyna D.P., Hack C.E., van Deventer S.J., Peppelenbosch M.P., van der Poll T. Anti-inflammatory effects of a p38 mitogen-activated protein kinase inhibitor during human endotoxemia. J Immunol. 2002;168:4070–4077. doi: 10.4049/jimmunol.168.8.4070. [DOI] [PubMed] [Google Scholar]
- 51.Song G.Y., Chung C.S., Chaudry I.H., Ayala A. MAPK p38 antagonism as a novel method of inhibiting lymphoid immune suppression in polymicrobial sepsis. Am J Physiol Cell Physiol. 2001;281:C662–C669. doi: 10.1152/ajpcell.2001.281.2.C662. [DOI] [PubMed] [Google Scholar]
- 52.Peng T., Lu X., Lei M., Moe G.W., Feng Q. Inhibition of p38 MAPK decreases myocardial TNF-alpha expression and improves myocardial function and survival in endotoxemia. Cardiovasc Res. 2003;59:893–900. doi: 10.1016/s0008-6363(03)00509-1. [DOI] [PubMed] [Google Scholar]
- 53.Choi I.J., Kim J.S., Kim J.M., Jung H.C., Song I.S. Effect of inhibition of extracellular signal-regulated kinase 1 and 2 pathway on apoptosis and bcl-2 expression in Helicobacter pylori-infected AGS cells. Infect Immun. 2003;71:830–837. doi: 10.1128/IAI.71.2.830-837.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Topically applied treatment 3 consecutive days was more effective than a single APC treatment in WT mice. Full-thickness 6-mm diameter wounds were treated topically with 10 μg of APC once a day for 1 day (black column) or 3 consecutive days (white column). Percentage of wound area/initial area was calculated from tracing the wounds, measured with Visitrak. Values are mean ± SD; n = 4 wounds.
Relative expression of EPCR (A) on WT, PAR-1 KO, and PAR-2 KO mice; PAR-2 (B) expression on WT and PAR-1 KO mice; and PAR-1 (C) expression on WT and PAR-2 KO mice. Expression of EPCR, PAR-1, and PAR-2 were detected by immunoblot analysis from homogenates of normal skin of WT, PAR-1 KO, and PAR-2 KO mice. β-Actin was used as an internal control.
A: Dose-dependent stimulation of PAR-2 in HaCat keratinocytes. Confluent cells were treated with or without APC for 24 hours, and PAR-2 was detected by immunoblot analysis. B: PAR-2 expression in human primary keratinocytes was assayed by immunoblot analysis after 48 hours of treatment with PAR-2 siRNA.