

Abstract
Tissue inhibitor of matrix metalloproteinase–2 (TIMP-2) belongs to a small family of endogenous proteins that inhibits a group of enzymes, the matrix metalloproteinases (MMPs). TIMP-2 inhibits endothelial cell proliferation and migration in vitro and angiogenesis in vivo, through MMP-dependent and -independent mechanisms. However, little is known regarding the contribution of these mechanisms to the antitumor effects of TIMP-2. Using a retroviral delivery system, we stably overexpressed TIMP-2 and its mutant Ala+TIMP-2 (devoid of MMP inhibitory activity) in human adenocarcinoma A549 cells. Using real time PCR, and enzyme-linked immunosorbent assay (ELISA), we confirmed enhanced TIMP-2 expression and its MMP inhibitory activity by reverse zymography. In vitro, growth assays suggested that TIMP-2 and Ala+TIMP-2 did not alter basal cell proliferation rates, however, tumor cell migration and invasion were inhibited. In vivo, both TIMP-2 and Ala+TIMP-2 A549 xenografts exhibited reduced growth rate, CD31 immunostaining indicated decreased intratumoral microvascular density, and TUNEL demonstrated enhanced tumor cell apoptosis. Immunoblotting and immunohistochemical analyses of A549 xenograft tissues with either phospho-FAK (Tyr397) or phospho-AKT (Ser473) showed decreased activation in both TIMP-2 and Ala+TIMP-2 tumor cells. We conclude that TIMP-2-mediated inhibition of tumor growth occurs, at least in part, independently of MMP inhibition, and is a consequence of both direct effects of TIMP-2 on tumor cells and modulation of the tumor microenvironment.
Tissue inhibitor of matrix metalloproteinase–2 (TIMP-2) belongs to a family of multifunctional secreted proteins (TIMPs) that control the proteolytic activity of matrix metalloproteinases (MMPs). MMPs are the principal proteases implicated in promoting tumor angiogenesis, invasion, and metastasis of cancer cells.1, 2, 3 There are four TIMP family members, TIMP-1, -2, -3, and -4, that share significant sequence identity.4, 5 The N-terminus of TIMPs binds to the MMP catalytic domain and inhibits MMP activity, whereas their C-terminus interacts with the MMP hemopexin domain to stabilize complexes with both pro- and active MMPs.6, 7 TIMP-2 is the only TIMP member that specifically interacts on the cell membrane with both MT1-MMP (membrane bound) and pro-MMP-2 (latent zymogen) to facilitate the activation of pro-MMP-2.8, 9 It, therefore, functions both as an MMP inhibitor and activator.
During cancer progression, TIMP-2 is associated with inhibition of tumor growth, angiogenesis, invasion, and metastasis.10, 11, 12, 13, 14, 15, 16, 17, 18 Much evidence suggests that the TIMP-2 antiangiogenic effects are not only a consequence of MMP inhibition but also occur independent of MMP-mediated endothelial cell proteolysis.12, 19 Ala+TIMP-2 is a mutant form of TIMP-2 that contains the amino acid alanine appended to the N-terminus and makes the protein unable to inhibit MMP activity.20 Nevertheless, we were able to demonstrate that exogenous treatment with either TIMP-2 or Ala+TIMP-2 inhibited endothelial cell (EC) proliferation in vitro and angiogenesis in vivo, on vascular endothelial growth factor-A (VEGF-A) or fibroblast growth factor-2 (FGF-2)–induced growth. The antiangiogenic mechanism described involves binding of TIMP-2 or Ala+TIMP-2 to α3β1 integrin receptor on the ECs and, consequently, activating the SH2-containing protein tyrosine phosphatase-1 (SHP-1) to suppress the receptor tyrosine kinase (RTK) activation/phosphorylation, including VEGFR-2 and FGFR-1.12 More recently, TIMP-2 Loop 6, located at the C-terminus of the protein, was shown to inhibit angiogenesis in vivo by direct binding to the insulin-like growth factor receptor I (IGF-IR) on ECs and regulating IGF-IR downstream mitogenic signals.21 TIMP-2 regulates additional cellular activities including, inhibition of EC migration, myogenesis, and neuronal differentiation, all via an α3β1 integrin-dependent mechanism.22, 23, 24, 25 We have reported that TIMP-2 inhibits EC migration by inducing the expression of an MMP inhibitor, the reversion-inducing-cystein-rich protein with Kazal motif (RECK), leading to loss of endothelial cell migration.22, 26 TIMP-2 also interacts with α3β1 to inhibit growth and to promote neurite differentiation in vitro.23 TIMP-2 induces neurites to undergo G1 cell cycle arrest mediated by increased expression of the cyclin-dependent kinase inhibitor p21, reminiscent of TIMP-2-–mediated p27 induction, shown previously to occur in endothelial cells.27
TIMP-2 expression levels are decreased or absent in several human cancers, particularly in invasive and metastatic tumors, such as lymphoid, prostate, head and neck, and cervical cancers, either through epigenetic modifications such as hypermethylation of its promoter or genetic polymorphisms.28, 29, 30 Therefore, understanding, how TIMP-2 regulates tumor cell biology and the tumor microenvironment is critical in identifying new therapeutic interventions.
The purpose of this study was to determine the antitumor effects of TIMP-2 and Ala+TIMP-2 (independent of MMP proteolytic activity) on human A549 lung cancer cells in vitro and in vivo. A549 cells were chosen to stably overexpress TIMP-2 or Ala+TIMP-2 owing to the low endogenous expression of TIMP-2. Although A549 TIMP-2 and Ala+TIMP-2 stably overexpressing cells exhibited no difference in cell growth in vitro, significant down regulation of tumor cell migration and invasion were observed in both TIMP-2 and Ala+TIMP-2 A549 stable clones, indicative of MMP-independent mechanism(s). In vivo, A549 TIMP-2 and Ala+TIMP-2 xenograft tumors taken from two distinct murine models (nude and NOD-SCID), demonstrated significantly reduced tumor growth, accompanied by reduced angiogenesis and increased apoptosis. Reduced total levels and phosphorylated forms of focal adhesion kinase (FAK) and AKT in A549 TIMP-2 and Ala+TIMP-2 xenograft tumors suggest that TIMP-2 overexpression directly alters growth, apoptotic and migration pathways in tumor cells, in addition to its antiangiogenic effects.
Materials and Methods
Cell Culture and Stable Transfections
The A549 adenocarcinoma cell line (ATCC, catalog number CCL-185) was maintained in Dulbecco's Modified Eagle Medium (DMEM)/F-12 media 1:1 (Invitrogen, Carlsbad, CA) with 5% fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO) in a humidified incubator containing 5% CO2 at 37°C. Human TIMP-2 or Ala+TIMP-2 cDNA sequences were inserted into the pLXRN retrovirus vector (Clontech, Mountain View, CA). Using the Pantropic Retroviral Expression System (Clontech), infectious virus was produced from the GP2-293 packaging cells and used to infect A549 cells. Stable transfected A549 single clones were selected and the two highest TIMP-2 or Ala+TIMP-2 expressing clones, as determined by TIMP-2 ELISA and real-time quantitative RT-PCR analyses for TIMP-2, were pooled. Transfected cells were maintained under the selection of 400 μg/mL Geneticin. Cells were cultured up to 85% confluency before all experimental analyses. The designations used in hereafter are as follows: A549 wild-type (WT), empty vector control (EV), TIMP-2 (T2), and Ala+TIMP-2 (Ala+T2).
Cell Growth Assay
A total of 8 × 104 cells in 5 mL of DMEM/F-12 medium with 5% FBS were seeded into T25 cm2 tissue culture treated flasks (Corning, Corning, NY). Every day for 5 days, a set of three flasks from the A549 WT and stables were rinsed with PBS, trypsinized, and resuspended in complete media. Cell counts were obtained using the Z1 Coulter Particle Counter (Beckman Coulter, Brea CA). The average of three independent cell counts is plotted against time (days) to obtain a growth curve for each cell population.
Human TIMP-2 ELISA
TIMP-2 ELISA (R&D, Minneapolis, MN) was performed on the conditioned media (CM) prepared by replacing the complete media of 80% confluent cells with phenol free DMEM/F-12–containing 0.1% FBS for 48 hours. The final TIMP-2 concentration was adjusted to the number of cells. The assay was performed from five independent experiments according to the manufacturer's instructions.
Migration and Chemoinvasion Assays
Migration of A549 WT and stables was measured using the disposable 96-well cell migration ChemoTx System (NeuroProbe, Gaithersburg, MD) with an 8-μm pore, polycarbonate uncoated membrane. Cells were seeded in complete media for 24–48 hours, washed once with serum free media and cultured in 0.5% FBS DMEM/F-12 overnight. Next day, the lower compartment of the ChemoTx System was filled with 31.5 μL per well of DMEM/F-12 with no phenol red, supplemented with 5% FBS as chemoattractant. On the top, 25 μL of 30,000 cells resuspended in DMEM/F-12, with no phenol red and supplemented with 0.1% FBS, were placed on each of seven replicate wells and incubated for 5 hours at 37°C in a humidified incubator with 5% CO2. The membrane was fixed, nonmigrated cells were discarded from the top of the membrane and the migrated cells were stained using the Diff Quick protocol. Pictures of migrated cells were taken using the Olympus BX51 microscope from seven independent replicates using ×100 magnification. Migrated cells were counted using ImageJ software.31 Chemoinvasion was determined using the BioCoat Matrigel invasion 24-well chamber assay (BD Biosciences, Bedford, MA) with an 8-μm pore polycarbonate filter coated with Matrigel. Cells were cultured for 24 to 48 hours, detached and resuspended in serum-free media at 6 × 104 cells/mL. The lower compartment was filled with 0.7 mL prewarmed DMEM/F-12 with no phenol red, supplemented with 5% FBS as chemoattractant. In the upper compartment, 3 × 104 cells per well in serum-free media were placed in triplicate wells and incubated for 48 hours at 37°C in a humidified incubator with 5% CO2. The inserts were fixed in ice-cold methanol for 20 minutes and using a cotton-tipped swab the noninvasive cells were removed from the top of the membrane. After three washes in PBS cells were stained with Crystal Violet (0.5% in 20% methanol) for 20 minutes at room temperature. The membranes were finally thoroughly rinsed with dH2O before observed under the microscope. The number of invasive cells was determined from five different fields using ×200 objective magnification. The assays were repeated a minimum of three times.
Real-Time Quantitative RT-PCR
Cell pellets were collected for total RNA extraction (RNeasy kit; Qiagen, Gaithersburg, MD). RNA was spectrophotometrically quantified (ND-1000 spectrophotometer; Thermo Scientific Nanodrop, Rochester, NY), and 1 μg was reverse transcribed with the SuperScript First Strand Synthesis system (Invitrogen). Real-time PCR was performed in 96-well fast optical PCR plates (MicroAmp) using the 7500 Fast Real-Time PCR System (Applied Biosystems, Carlsbad, CA) in the presence of 12.5 μL of 2xSYBR Green PCR master mix (Applied Biosystems) 300 nmol/L forward and reverse primers, 1 μL cDNA (diluted 1:5) and dH20 up to 25 μL volume. The TIMP-2 primer sequences used are: forward, 5′-ACAGGCGTTTTGCAATGCA-3′ and reverse, 5′-GGGTTGCCATAAATGTCGTTTC-3′. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers were used to amplify the endogenous control: forward, 5′- GGAAGGTGAAGGTCGGAGTCA-3′ and reverse, 5′-GCAACAATATCCACTTTACCAGAGTTAA-3′. Mean Ct values for each gene were normalized to the Ct values of the endogenous control GAPDH. The mRNA gene expression analyzed versus GAPDH is expressed as fold 2(−Δct). The assays were repeated at least three times as independent experiments.
Immunoblotting and Immunoprecipitation
CM (see ELISA protocol for preparation) were used after concentrating the samples ∼15× using Amicon Ultra-15 (Billerica, MA) spin columns (Millipore), and the volumes were adjusted to the number of cells before loading on a 16% Novex Tris-Glycine gel (Invitrogen) followed by electrophoresis. Proteins were transferred onto a Protran nitrocellulose membrane (Whatman; GE Healthcare, Piscataway, NJ) and then blocked with 5% skim milk in 1× PBS or 4% BSA in 1xTBS + 0.1%Tween for 1 hour. The membrane was probed with the monoclonal antibody T2-101 for the detection of TIMP-2–secreted protein levels.32 Protein lysates from NOD-SCID–derived A549 xenograft homogenates were probed with anti-FAK (1:2000 dilution, clone 4.47; Millipore), anti-Phospho-FAK (Y397) (1:1000 dilution; BD Transduction Laboratories), anti-Akt1 (1:1000 dilution; Cell Signaling Technology, Danvers, MA), and anti-Phospho-Akt (Ser473) (1:1000 dilution; Cell Signaling Technology). Protein detection was performed with enhanced chemiluminescence, ECL Plus Western Blotting Detection System (GE Healthcare).
Immunoprecipitation (IP) was performed in the homogenates from the A549 xenograft tumors (see Reverse zymography). Supernatants were precleared by adding 3 μg of mouse IgG1 (the same species and isotype as the IP antibody to homogenates) along with 60 μL of Protein G PLUS-Agarose (Santa Cruz Biotechnology, Santa Cruz, CA). The mixtures were incubated for 1 hour on a shaker at 4°C followed by a centrifugation at 5000 rpm at 4°C for 5 minutes. T2-101 primary antibody (Abcam, Cambridge, MA) was added to the supernatant at a concentration of 2 μg/mg protein homogenate and incubated overnight at 4°C, under agitation. 100 μl of Protein G PLUS-Agarose (Santa Cruz), which had been washed twice with PBS, was added to supernatants and the homogenate-beads mixture was incubated at 4°C under rotary agitation for 2 hours. Samples were centrifuged to remove the supernatant and beads were washed 3 times in ice-cold PBS. Finally, the protein was eluted from the beads with 2× sample buffer and run on a 15% reverse zymogram (described below).
Gelatin Zymography
Cells were cultured in media containing 0.1% FBS for 48 hours. CM were collected and adjusted to number of cells before subsequent experimental analysis. To test whether MMP activity was changed in the stably transfected cells, MMP-2 enzymatic activities were measured by zymography.33 CM were mixed with 5× sample buffer and zymography was performed using precast gels (10% polyacrylamide, 0.1% gelatin), renaturing and developing buffers, according to the manufacturer's instructions (Invitrogen). Gelatinase activities were visualized by staining zymograms with DyeHard Coomassie Blue staining solution (Crystalgen, Commack, NY), and imaged using an Epson Perfection V750 Pro scanner. Densitometry was done to quantify band density subtracting background and normalizing to the control sample included on each gel (ImageQuant v. 5.2).
Reverse Zymography
Reverse gelatin zymography was performed to test how TIMP-2–associated MMP-2 inhibitory activity varied in the A549 stable transfected cells and in tumor xenografts, as previously described.34 CM from cultured cells were normalized to cell number. Each sample was mixed with 5X Tris-Glycine-SDS sample buffer. Recombinant human TIMP-1 and TIMP-2 (both R&D Systems) were used as controls. Reverse zymograms were incubated with the same renaturation and developing reagents used for gelatin zymography according to their manufacturer's instructions (Invitrogen). Gels were stained and imaged as described in gelatin zymography. Band density was measured after subtracting background and normalizing to the control sample (ImageQuant v. 5.2). For the analysis of tumor xenografts, tumors were excised, any nontumor tissue was removed, and weights were recorded. Tumor fragments were placed in ∼10 μL per mg tumor wet weight homogenization buffer [50 mmol/L Tris, pH 7.5, 200 mmol/L NaCl, 5 mmol/L CaCl2, 0.02% Brij3, protease inhibitor (Sigma-Aldrich) and phosphatase inhibitors (Roche, Indianapolis, IN)] and homogenized using the gentleMACS Dissociator (MACS Miltenyi Biotech, Auburn, CA). Immunoprecipitation of homogenates followed according to the protocol described above (see Immunoblotting and Immunoprecipitation). TIMP-1 and TIMP-2 activities were visualized and band densities were measured as described earlier.
Immunohistochemistry
Immunohistochemistry was performed using the avidin–biotin peroxidase method with Vectastain Elite ABC kits (Vector Laboratories, Burlingame, CA) according to the manufacturer's instructions with the following modifications. Slides were preheated at 60°C for 1 hour before deparaffinization, followed by antigen retrieval in 0.01mol/L Citrate buffer (pH 6) and incubation in 3% hydrogen peroxide to quench endogenous peroxidase. Intratumoral microvascular density was assessed using the rat anti-mouse CD31 antibody (PECAM-1, BD Biosciences) on zinc-fixed tumor tissue. Antibodies to focal adhesion kinase (FAK, Clone 4.47, Millipore), phospho-FAK (pY397, BD Transduction), Akt1 (C73H10, Cell Signaling), and phospho-AKT (Ser473, Cell Signaling) were performed on formalin-fixed, paraffin-embedded tumor tissue. All primary antibodies were incubated overnight at 4°C, and color development was achieved using 3,3′ diaminobenzidine (DAB, Sigma Chemical, St. Louis, MO).
TUNEL Assay
Apoptotic cell death was investigated using the terminal deoxynucleotidyl transferase (TdT)–mediated dUTP nick end-labeling (TUNEL) assay (Apoptag Plus Peroxidase In Situ Detection Kit; Chemicon) according to the vendor's instructions. Quantification of staining was assessed by calculating the mean number of TUNEL-positive cell nuclei within 20 high-power (×400) fields per animal group.
Nude and NOD-SCID A549 Xenografts
A549 EV, T2, Ala+T2 cells were counted and washed twice in 1× PBS. A total of 5 × 106 cells per 100 μL PBS were injected subcutaneously into the hind flank of 4- to 6-week-old female athymic nude (CD-1 Nude, n = 5 per group; Charles River) or NOD SCID mice (n = 20 per group NCI-DPT-FCRDC, Frederick, MD). All mice were housed under specific pathogen-free conditions under a 12-hour light/dark cycle with access to food and water ad libitum according to approved Institutional Animal Care and Use Committee protocol LP-003. Tumor size was measured biweekly by caliper measurement, and tumor volume was calculated using the following formula: tumor volume = (width2 × length) × 0.5. Mean tumor volume for each animal group was represented as the mean ± SEM. Xenografts from nude and NOD SCID mice were excised and weighed 38 and 21 days after inoculation, respectively.
Statistical Analyses
Statistical significance in comparisons between two groups was determined by two-tailed Student's t-tests (GraphPad Prism). Two-way analysis of variance test was performed for comparisons between groups over time (in vivo growth assay). Calculations were done with the GraphPad Prism software. P value ≤0.05 was considered significant.
Results
Forced Expression of TIMP-2 and Ala+TIMP-2 in Human Adenocarcinoma A549 Cells
We generated retrovirally transfected A549 cell lines stably overexpressing TIMP-2 and Ala+TIMP-2 to address whether TIMP-2 affects tumor cell biology. Two clones that showed the highest level of expression for TIMP-2 and Ala+TIMP-2 were selected and pooled. TIMP-2 expression was evaluated by real time quantitative RT-PCR on cDNA and TIMP-2 ELISA on CM derived from the A549 stables (Figures 1, A and B). The empty vector (EV) control showed TIMP-2 mRNA levels similar to that of the parental (WT), whereas the T2 and Ala+T2 showed a four- to fivefold increase in mRNA levels compared with WT or EV control cells (Figure 1A). Similarly, there was no difference in TIMP-2 protein levels in CM derived from WT and EV control cells; however there was at least threefold increase in secreted TIMP-2 protein in T2 and Ala+T2 A549 cells.
Figure 1.
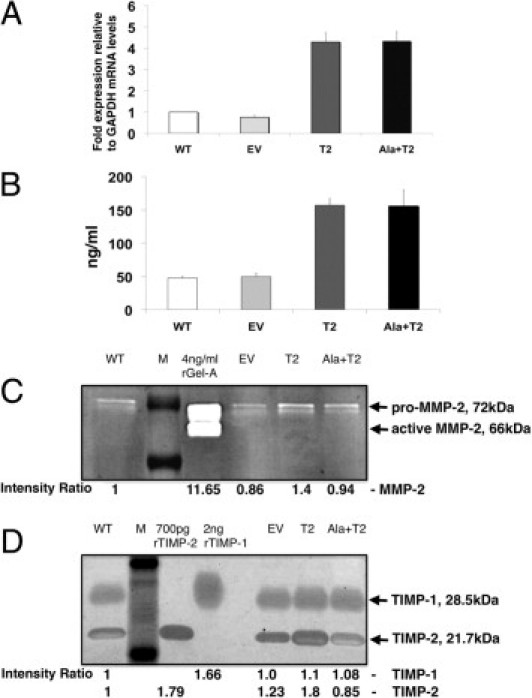
TIMP-2 expression levels, MMP-2 and TIMP-2 activities in A549 cell lines, A549 WT (wild-type, parental), EV (empty vector control), T2 (TIMP-2) and Ala+T2 (Ala+TIMP-2 mutant lacking MMP-2 inhibitory activity). A: Total RNA was extracted from the retrovirally transfected A549 cells and used to determine TIMP-2 mRNA levels using quantitative real-time RT-PCR. Fold expression mRNA levels are relative to those of the house-keeping gene GAPDH. Error bars represent the SEM from seven independent experiments. B: Supernatant from cultured A549 stable cells was collected 48 hours after culture and used to determine secreted TIMP-2 protein levels using a human TIMP-2 enzyme-linked immunosorbent assay (ELISA). TIMP-2 concentrations (ng/mL) were adjusted to the number of cultured cells. Error bars represent the SEM from five independent experiments. C: Conditioned media (CM), prepared as described above, were analyzed in gelatin zymography to determine MMP-2 (gelatinase A) activity in the A549 stables. MMP-2 shows two bands, the 72-kDa latent zymogen (pro-MMP-2) and the 66 kDa active enzyme (active MMP-2). A 4 ng quantity of recombinant gelatinase A (rGel-A) was run as a positive control. D: CM were analyzed in reverse zymography to determine TIMP-2 inhibitory activity in the A549 stables. Both TIMP-1 (28.5 kDa) and TIMP-2 (21.7 kDa) are present in the CM. Recombinant proteins (rTIMP-1 and rTIMP-2) were run as controls at the indicated amounts. For Figures 1C and 1D: Relative band intensities are shown below each figure as compared with A549 WT. Each zymogram is representative of at least three independent experiments. M = Molecular Weight Marker.
MMP and TIMP-2 Enzymatic Activities in A549 Cells
We next wanted to know whether the A549 stables had altered characteristics compared with the control cells regarding MMP-2 proteolytic activity. CM were collected and adjusted to number of cells before gelatin zymography (Figure 1C). As a control, recombinant MMP-2 (gelatinase A, rGel-A) was run in parallel with the CM from the A549 stables. The rGel-A shows two forms of MMP-2, the inactive latent pro-MMP-2 (72 kDa) and the activated MMP-2 (66 kDa). The stables show predominantly the latent form present in the CM. Although slight differences in MMP-2 activity were detected between WT and EV controls, as well as with the stables, none of these differences were significant. We also determined the TIMP-2 inhibitory activity by reverse zymography (Figure 1D). Both TIMP-1 and TIMP-2 are secreted in the CM from the A549 stables. Recombinant TIMP-1 (rTIMP-1, 28.5 kDa) and TIMP-2 (rTIMP-2, 21.7 kDa) were run in parallel with the samples to indicate protein size. According to the band intensities, there are no significant differences in the inhibitory activities of TIMP-1. There is, however, an almost 80% increase of TIMP-2 inhibitory activity compared with A549 WT and 60% compared with EV control, attributed to the overexpression TIMP-2 protein levels. On the contrary, although the TIMP-2 protein levels are similar between A549 T2 and Ala+T2, the inhibitory activity of Ala+T2 is almost similar to that of the A549 WT and twofold less than the A549 T2 stables. The above experiments suggest that the A549 stables overexpress TIMP-2 and Ala+TIMP-2 at similar mRNA and protein levels and the MMP-2 enzymatic activity is maintained at similar levels. However, the TIMP-2 inhibitory activity is increased in the A549 T2 but Ala+T2 does not inhibit MMP activity in the A549 stable cells.
TIMP-2–Forced Expression Does Not Alter A549 Cell Growth but Reduces Migration and Invasion of A549 Cells in Vitro
We then examined whether there was an effect on A549 cell growth in vitro. Each cell line was seeded at equal cell numbers (0.8 × 105) in three replicate flasks in DMEM/F12 with 5% FBS. Daily cell counts were performed for each cell line, however, no significant differences in the basal growth rates (not growth factor stimulated) were observed (Figure 2A). We then tested whether TIMP-2 overexpression affected migration of A549 cells (Figure 2, B and C). The migration assay revealed marked differences between the controls (WT and EV) and the overexpressing stables (T2 and Ala+T2) (Figure 2, B and C). Significantly lower migration rates, almost twofold, were observed in A549 T2 and Ala+T2 cells compared with the A549 EV control (**P = 0.0023 and **P = 0.0076 respectively). No-chemoattractant controls were also included and no migration was observed (data not shown). The assay was also performed using gelatin-coated membranes, which gave results similar to the uncoated membranes. We further determined the chemoinvasive abilities of the stable clones using a Matrigel in vitro invasion assay and 5% FBS as chemoattractant (Figure 2, D and E). As shown, there was a significant, almost 80%, decrease in invasion of both A549 T2 and Ala+T2 compared with EV control (***P < 0.0001 for both). These results suggest that TIMP-2 inhibits A549 cell migration and chemoinvasion independently of MMP-2 inhibition without affecting their growth in vitro.
Figure 2.
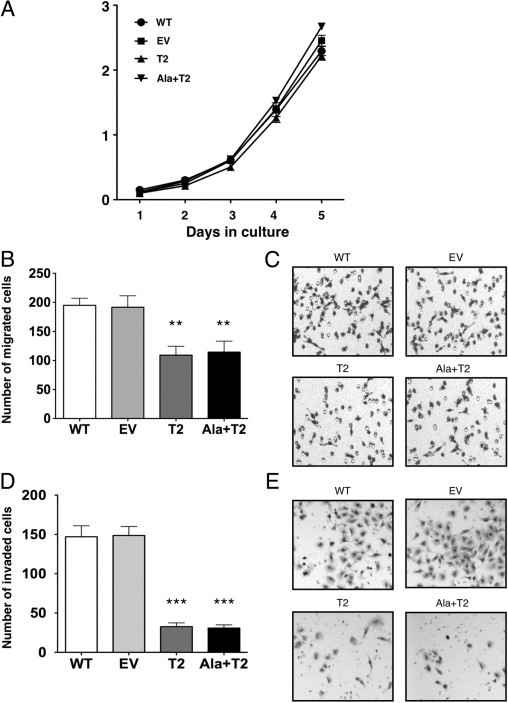
In vitro growth, migration and invasion assays of A549 stables. A:in vitro cell growth comparison of A549 WT, EV, T2 and Ala+T2 stables. Equal number of cells (0.8 × 105) were seeded in DMEM/F12 supplemented with 5% FBS for 5 days and cell numbers were quantified every day by cell counting. Error bars represent the SEM from three independent replicates. This experiment represents one from three independent experiments. B: Overexpression of TIMP-2 and Ala+TIMP-2 reduced migration potential of A549 cells. Using the disposable 96-well cell migration ChemoTx® System, cells were allowed to migrate toward 5% FBS for 5 hours. Cells were stained with the DiffQuick system. Numbers of migrated cells were determined using the average count from seven random microscopic fields at ×100 magnification. Values shown are means ± SEM from three independent experiments. **P value from comparisons with EV control: EV versus T2 P = 0.0023, EV versus Ala+T2 P = 0.0076. C: Microscopic images of migrated cells taken at ×100 magnification using the above system. D: Overexpression of TIMP-2 and Ala+TIMP-2 reduced invasive potential of A549 cells. The cells that invaded through Matrigel-coated transwell inserts toward the chemoattractant (5% FBS) were stained with crystal violet. Numbers of invaded cells were determined using the average count from five random microscopic fields at ×200 magnification. Values shown are means ± SEM from three independent replicates. ***P value from comparisons with EV control: EV versus T2 P < 0.0001, EV versus Ala+T2 P < 0.0001. This experiment is representative from two independent experiments. E: Microscopic images of invaded cells taken at ×100 magnification using the Matrigel-coated transwell inserts.
Reduced Tumor Growth in Vivo of A549 Xenografts
Because both A549 WT and EV behaved similarly in the in vitro experiments, A549 WT was omitted from the in vivo assays and the A549 EV was considered to be the control. To address further the functional significance of TIMP-2 overexpression in A549 adenocarcinoma in vivo, we used two distinct murine models, the athymic nude and the NOD-SCID. Similar results were observed in both systems. A549 tumor cells overexpressing TIMP-2 and Ala+TIMP-2 were injected subcutaneously (5 × 106 cells per 100 μL PBS per cell line) into the hind flank of 4- to 6-week-old female athymic mice. A549 tumors were allowed to grow for 38 days and tumor growth rate was determined by measuring tumor sizes twice a week (Figure 3A). At the end of the experiment a 90% and 65% inhibition of A549 T2 and Ala+T2 tumor growth, respectively, was observed compared to mice injected with control A549 EV tumors (***P < 0.001 for both comparisons). We also looked at the proliferation rate of tumor cells by Ki-67 immunostaining and no difference was observed between the TIMP-2 transfected cells and the control (data not shown).
Figure 3.
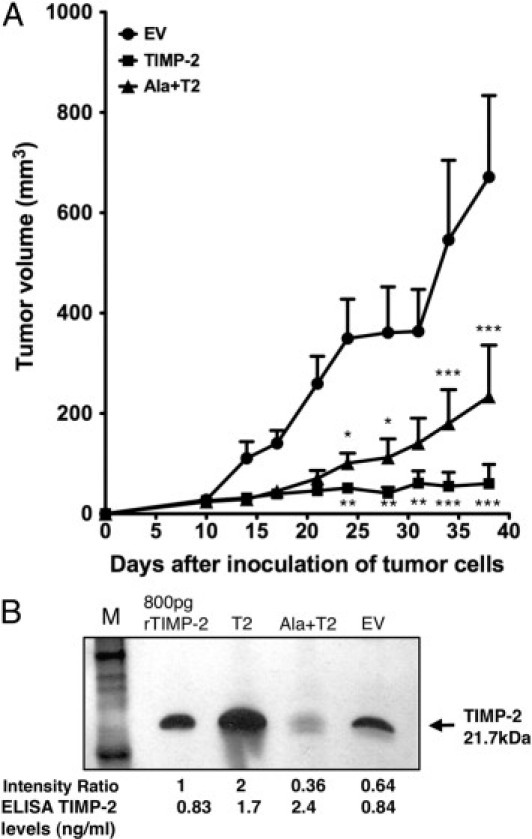
Evaluation of in vivo cell growth of A549 stables. A549 EV (control), T2 and Ala+T2 cells were cultured to 80% confluency, counted and washed twice with PBS. Equal number of cells (5 × 106), resuspended in 100 μL PBS per cell line, was injected subcutaneously into the hind flank of 4- to 6-week old female athymic nude (CD1 Nu) mice (n = 5 mice per group). A: Tumor volumes (mm3) were determined using caliper measurements twice a week starting at day 10 up to day 38 postinoculation. Graph is showing the overexpression of TIMP-2 and Ala+TIMP-2 effects on A549 xenograft tumor growth in athymic nude mice. Experiment was repeated at least three times in athymic nude mice and also in NOD-SCID mice giving similar results. At the end of the experiment it is shown that the tumor volumes of A549 T2 and Ala+T2 xenografts are significantly smaller than that of the EV control (two-way analysis of variance, ***P < 0.001, **P < 0.01 and *P < 0.05) whereas no significant difference is shown between A549 T2 and Ala+T2 xenografts. B: Reverse zymogram performed on TIMP-2 immunoprecipitates from equal total protein amounts of A549 tumor homogenates. Recombinant TIMP-2 protein was run as control at the indicated amount. Relative band intensities are shown below the figure compared with A549 EV control. ELISA for the determination of TIMP-2 levels in the tumor extracts was performed and the concentration shown in ng//mL. The zymogram is representative of two independent experiments. M = molecular weight marker.
When human TIMP-2 ELISA was performed in the IP (immunoprecipitated) tumor extracts, the total TIMP-2 protein levels were similar in A549 T2 and Ala+T2 (1.7 ng/mL and 2.4 ng/mL, respectively) and two- to threefold higher than the EV control (0.84 ng/mL). Performing a reverse zymography in TIMP-2 IP extracts from the A549 tumors, we observed a threefold increase and a 1.8-fold decrease of TIMP-2 inhibitory activity in A549 T2 and Ala+T2 tumors, respectively, compared with EV control (Figure 3B). The decreased TIMP-2 inhibitory activity below the EV control levels in the A549 Ala+TIMP-2 tumors may indicate that Ala+TIMP-2 protein may function in a dominant negative fashion to compete for endogenous TIMP-2 binding and inhibition of MMP activity. Overall, these results suggest that TIMP-2 and Ala+TIMP-2 inhibit A549 tumor growth in vivo (Figure 3, A and B) via both MMP-dependent and independent mechanisms.
Decreased Microvascular Density and Increased Apoptosis in A549 T2 and Ala+T2 Xenografts
Given that TIMP-2 has been shown to inhibit angiogenesis in vivo, we next determined if TIMP-2 and Ala+TIMP-2 inhibited tumor angiogenesis using the A549 xenograft model. Microvascular density (MVD) is used as a marker for tumor angiogenesis. We evaluated the MVD in the A549 xenograft tissue sections by determining the number of CD31 positive blood vessels in situ (Figure 4, A and B). The A549 EV control showed significantly higher average MVD than the T2 and Ala+T2 tumors. These differences were statistically significant (***P < 0.0001 for both comparisons). Interestingly, Ala+T2 tumors showed higher inhibition of angiogenesis compared with that in T2 tumors (***P = 0.0001). This result indicates that Ala+TIMP-2 might act more effectively as an angiogenesis inhibitor, possibly due to the fact that it does not bind to MMP active sites. We next used the TUNEL assay on the A549 tumor sections to evaluate whether the A549 stable xenografts exhibited higher apoptosis than the control (Figure 5, A and B). Indeed, a statistically significant increase in TUNEL positive (apoptotic) cells was observed in both A549 T2 and Ala+T2 tumor cells compared with the EV control (***P < 0.0001 for T2 and *P = 0.02 for Ala+T2). These data suggest that overexpression of TIMP-2 in A549 tumor cells leads to decreased tumor angiogenesis and a concomitant increase in apoptosis in an MMP-independent manner. These results confirm that the MMP-independent antiangiogenic and proapoptotic effects of TIMP-2 described previously contribute directly to the anti-tumor activity of TIMP-2 in vivo.
Figure 4.
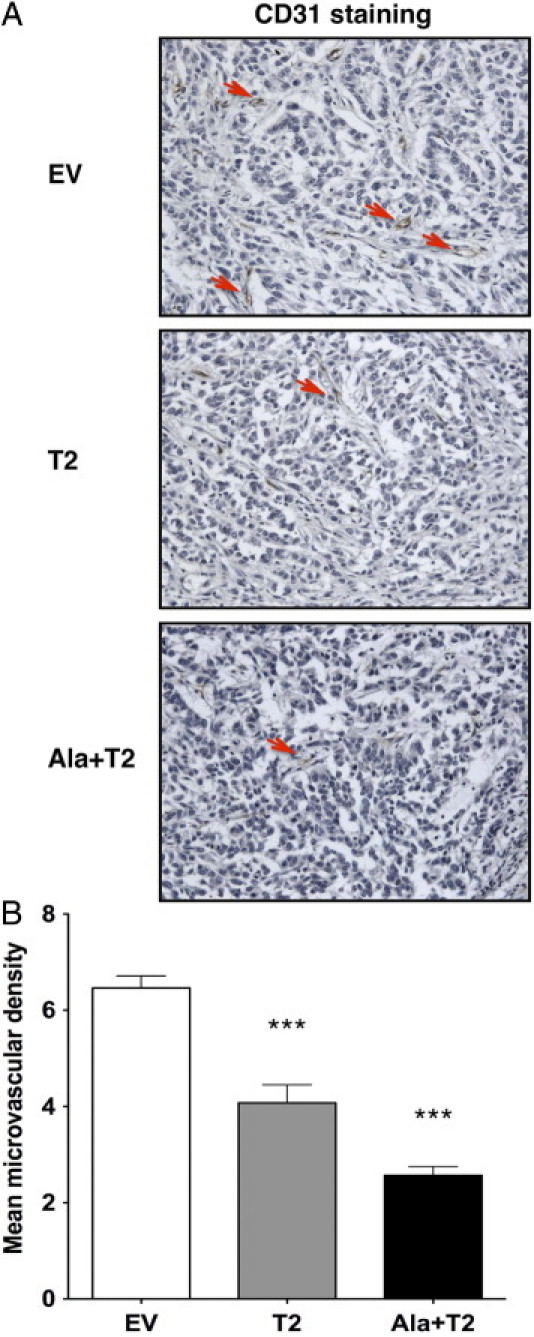
Increased TIMP-2 and Ala+TIMP-2 levels reduce intratumoral microvascular density (MVD). A: Tumor angiogenesis was measured by immunohistochemical staining of blood vessels with an anti-mouse CD31 antibody in tumor sections from A549 xenografts. Representative photographs shown are from 20 high-power fields taken at ×200 magnification. Red arrows indicate examples of CD31-positive blood vessels. B: The graph shows the average number of CD31-positive vessels from 20 high-power fields (×400) per group (EV, T2, Ala+T2). Error bars indicate the mean MVD from the measurement of at least 10 animals per group. ***P < 0.0001 as compared with EV control. Significant difference also is observed between A549 T2 and Ala+T2 (***P = 0.0001).
Figure 5.
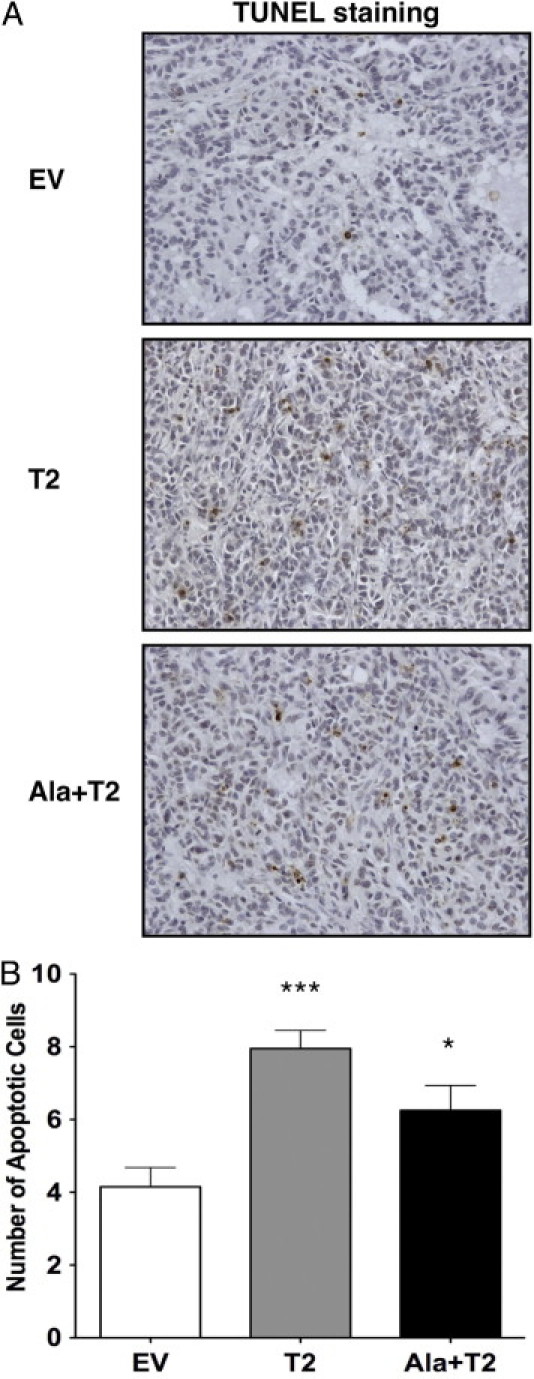
Increased TIMP-2 and Ala+TIMP-2 levels enhance tumor cell apoptosis. A: Apoptosis in A549 xenograft sections was measured by IHC using a TUNEL assay. Representative photographs shown are from 20 high-power fields taken at ×200 magnification. Brown-colored nuclei indicate apoptotic tumor cells. B: Graph shows the average number of apoptotic cells from 20 high-power fields (×400) per group (EV, T2, Ala+T2). Error bars indicate the mean number of apoptotic cells from the measurement of at least five animals per group. ***P < 0.0001, *P = 0.02 as compared with EV control.
Decreased Focal Adhesion Kinase and AKT1 Expression and Activation in A549 T2 and Ala+T2 Xenografts
The focal adhesion kinase (FAK) signaling pathway has been associated with cell migration and FAK expression correlates with tumor cell invasion and metastasis. Similarly, the PI3K/AKT (AKT1) pathway has been implicated in regulating survival and proliferation of a variety of cancers. We showed above that cell migration is significantly decreased in the A549 T2 and Ala+T2 transfectants and tumor cell growth is inhibited in vivo. Subsequently, A549 tumor cell lysates were analyzed by immunoblotting and tumor tissue was examined by immunohistochemistry (IHC) for total and activated FAK and AKT1 protein levels (Figure 6).
Figure 6.
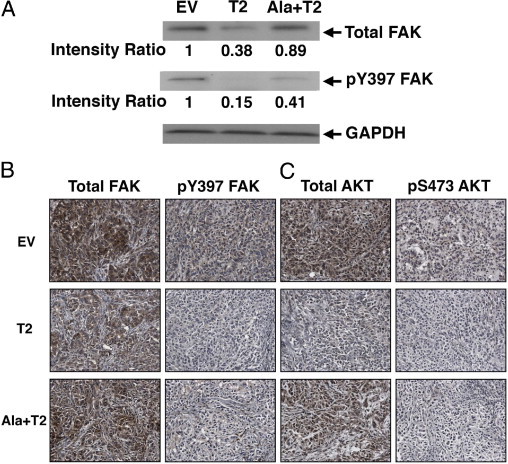
FAK and AKT protein levels and activation are decreased in A549 tumors by immunoblotting and immunoperoxidase staining of A549 xenograft tumor cells. A: FAK Western Blotting in NOD-SCID derived A549 xenograft tumor cell lysates. Total FAK protein levels are reduced in A549 T2 and Ala+T2 tumors. Similarly, the phosphorylation of FAK Y397 is significantly decreased in both A549 T2 and Ala+T2 tumors compared with the EV control. Relative band intensities are shown below each blot. GAPDH is the loading control. B and C: Total FAK (B, left panels) and phospho Y397 FAK (B, right panels) and C: total AKT1 (C, left panels) and phospho AKT Ser473 (C, right panels) by IHC showing a decrease in their levels in both A549 T2 and Ala+T2 compared with EV control. The reduced levels of total FAK and AKT are shown predominantly in A549 T2, whereas, pFAK and pAKT are diminished in both T2 and Ala+T2 tumors. Magnification, ×200.
Western blot analysis of tumor lysates prepared from NOD-SCID A549 xenografts showed a decrease in total and activated FAK (phospho Y397) protein levels in T2 and Ala+T2 tumors compared with the EV control (Figure 6A). More specifically, total FAK protein was reduced over 60% and 10% respectively in A549 T2 and Ala+T2 tumors. In addition, the major autophosphorylation site of FAK pY397 that regulates downstream cellular signaling events was significantly hypophosphorylated in A549 T2 and Ala+T2 tumors (85% and 60% decrease of phosphorylation) compared with EV control. We next analyzed the total levels and activation of AKT in the xenograft tumors, however, we could not see obvious changes by Western blot, possibly due to lack of suitable antibody (data not shown) although there were prominent differences by IHC (see below). These results indicate that TIMP-2 regulates FAK signaling in vivo via an MMP-independent manner.
To confirm the above data, we next examined the expression of total and phosphorylated forms of FAK and AKT1 by IHC in sections from the A549 EV, T2, and Ala+T2 tumor cells injected in nude mice (Figure 6, B and C). Similar to the Western blot results, a reduction in total FAK expression was observed in both A549 T2 and Ala+T2 xenografts compared with the EV control (Figure 6B, left panel). Moreover, the levels of the activated form of FAK (pY397 FAK) were dramatically diminished in both A549 T2 and Ala+T2 pFAK (Figure 6B, right panel). Again, these findings support the notion that TIMP-2 disrupts tumor cell migration in vitro and in vivo and is mediated by an MMP-independent mechanism.
Similarly, the A549 EV control shows the strongest staining of total AKT in tumor cells, whereas the levels of expression diminished in A549 T2 (Figure 6C, left panel). A549 Ala+T2 showed an intermediate level of total AKT expression. When we stained for pAKT, the levels were dramatically reduced in the tumor cells in both A549 T2 and Ala+T2 xenografts (Figure 6B, right panel). The findings suggest that in addition to suppressing directly tumor-induced angiogenesis via an MMP-independent mechanism, both TIMP-2 and Ala+TIMP-2 may also directly influence tumor cell survival/growth and migration signaling pathways in vivo.
Discussion
There is evidence to suggest that TIMP-2 plays a major role in regulation of the tumor microenvironment and in particular, inhibition of tumor angiogenesis. However, our understanding of the biological role of TIMP-2 in tumor growth in vivo is primarily limited to inhibition of MMP activity. In the current study, we stably overexpressed TIMP-2 and the mutant Ala+TIMP-2 (with no MMP inhibitory activity) in A549 human lung adenocarcinoma cells and investigated the biological effects on tumor cells in vitro and in vivo. We report, here, for the first time that overexpression of TIMP-2 or Ala+TIMP-2 is associated with in vitro inhibition of A549 tumor cell migration and invasion despite unchanged proliferative potential. These results indicate that TIMP-2 has direct anti-migratory and anti-invasive activities that are apparently independent of MMP inhibition. Our results also demonstrate that, in vivo, A549 T2 and, for the first time, Ala+T2 tumor xenografts showed significantly lower tumor growth compared with control A549 EV. In addition, TIMP-2 or Ala+TIMP-2 expressing A549 xenografts showed evidence of reduced tumor angiogenesis and a concomitant increase in tumor cells apoptosis. These findings are consistent with previous data showing that TIMP-2 and Ala+TIMP-2 inhibit angiogenesis in vivo, and are in agreement with the known association of antiangiogenic therapy and increased tumor cell apoptosis.35, 36 Finally, we demonstrate loss of activity of two important cell signaling pathways commonly hyperactivated during tumor progression, FAK and AKT. Our data suggest that TIMP-2, in addition to modulating the tumor microenvironment, may also directly modulate tumor cell migration and growth leading to diminished tumor progression and enhanced apoptosis.
TIMPs have been implicated in both promoting and inhibiting cell growth.4, 37 Early studies showed TIMP-1 and TIMP-2 having erythroid potentiating activity and over the years their growth stimulatory properties were also observed in various cell lines including fibroblasts, keratinocytes, and tumor cells. In addition, many studies suggested that the growth stimulation by TIMPs was independent of its ability to inhibit MMP activity. This was demonstrated by the use of growth promoting TIMPs lacking MMP inhibitory activity either by reductive alkylation or sequence mutation. Furthermore, several reports describe growth inhibitory activities for TIMP-1, TIMP-2, and TIMP-3.38, 39, 40 TIMP-2 has been shown to inhibit the growth of several cell types including (endothelial, neuronal, and tumor cells). More specifically, we have previously shown that exogenous treatment with TIMP-2, but not TIMP-1 or the potent synthetic MMP inhibitor BB-94, inhibited EC proliferation stimulated by basic fibroblast growth factor (bFGF).41 The contradictory results on TIMP-2 regulation of cell proliferation may be attributable to the different methodologies (exogenous treatment with TIMP-2 versus TIMP-2 overexpressing transfected cells, in the presence or absence of serum) and TIMP concentrations (picomolar versus nanomolar) used in several in vitro model systems (fibroblast, endothelial, tumor cells) in these studies. In the current study, we have shown that overexpressing TIMP-2 or Ala+TIMP-2 in A549 cells has no effect on basal level of tumor cell proliferation in vitro. We have previously shown that exogenous treatment of A549 cells with TIMP-2 and Ala+TIMP-2 in serum free conditions inhibits cell growth on stimulation with epithelial growth factor (EGF).42 In the present study, we did not use a growth factor to stimulate growth as cell proliferation was monitored in the presence of serum. Our result agrees with previous data derived from MC38 murine colon adenocarcinoma cells retrovirally transfected with TIMP-2.43 Similarly to our results, MC38 cells expressing TIMP-2 were cultured in complete media in the presence of serum and showed no change in basal proliferation. The effects of TIMP-2 previously observed on growth factor–stimulated proliferation in vitro could be masked by the presence of multiple growth factors in the serum.
In our previous studies we showed that exogenous treatment with TIMP-2 results in inhibition of EC migration toward bFGF as a chemoattractant (in a Boyden chamber), or after stimulation with VEGF (in a wound healing assay).22, 41 Both TIMP-2 and Ala+TIMP-2 were able to inhibit EC migration and this could be explained by dephosphorylation and suppression of VEGFR-2 signaling.12 Using a different in vitro system, TIMP-2 overexpression inhibited H-ras–transformed MCF10A breast epithelial cell migration and invasion, however, these effects were attributed to TIMP-2 modulating MMP-2 activity.16 In addition, Albini et al showed the ability of TIMP-2 to inhibit invasion of human fibrosarcoma HT1080 cells.10 In our study we show for the first time that TIMP-2 and Ala+TIMP-2 can inhibit migration and invasion of A549 tumor cells, suggesting that this effect may be mediated independently of MMP inhibitory activity; however, the possible mechanisms of this effect await further investigation.
During a 5-day in vitro proliferation assay we did not detect significant differences in growth. In contrast, subcutaneous injection of the A549 T2 or Ala+T2 cells into nude or NOD-SCID mice showed significant (90% and 65%, respectively) inhibition of tumor growth. We posit that host-tumor interaction in the tumor microenvironment is critical for TIMP-2 and Ala+TIMP-2 antitumoral effects. This is based on the observations that tumor growth inhibition occurs in vivo with no observed inhibition of tumor cell growth in vitro, and the proliferation index of tumor cells in the xenografts remained the same, as determined by Ki-67 staining (data not shown). Previous studies have shown in different in vivo mouse syngeneic systems11, 18, 44 and human xenografts45, 46 that TIMP-2 inhibits tumor growth, however, it was mainly attributed to inhibition of MMP activity. Our study shows that tumor growth is significantly inhibited in both A549 TIMP-2 and Ala+TIMP-2 xenografts suggesting that additional mechanisms are involved, which are independent of MMP inhibition.
In addition to being a potent MMP inhibitor and, therefore, restraining tumor cell spreading through matrix degradation, TIMP-2 has antiangiogenic effects that are mediated independently of MMP inhibitory activity.12, 19, 21 Indeed, we show that intratumoral angiogenesis is significantly reduced in A549 stables. Because TIMP-2 and Ala+TIMP-2 are overexpressed in A549 tumors we expect that the antiangiogenic effect might be caused by paracrine mechanisms involving TIMP-2 secretion and interaction with the surface receptors on the ECs. Another angiostatic agent, endorepellin, shares similar biological properties with TIMP-2; they both bind beta1-containing integrin, induce SHP-1 to dephosphorylate VEGFR-2, inhibit EC migration and tumor growth in vivo.47, 48 It would be of great interest to address whether endorepellin would also function not only as antiangiogenic but directly as antitumoral agent. Our findings clearly demonstrate that the MMP-independent antiangiogenic effects of TIMP-2, previously reported by several groups, contribute to the tumor suppressing activity of this member of the TIMP family.
Although TIMP-2 effects on programed cell death have not been well characterized, we assessed in our in vivo mouse model whether tumor growth inhibition also involves tumor cell apoptosis. Indeed, we found enhanced apoptosis in A549 T2 and Ala+T2 tumor cells compared with control cells. However, we did not see differences in apoptosis in A549 stables in vitro when we measured caspase activity (data not shown). These results suggest that TIMP-2 blocks tumor growth through its regulation of tumor angiogenesis with apparent related pro-apoptotic effects,35, 36 although a direct effect on tumor cell apoptosis cannot be excluded (vide infra).
FAK is a key regulator of cell adhesion and motility in normal and tumor cells and transduces biochemical signals on integrin-ligand interaction. FAK is frequently overexpressed in several tumors and its expression correlates with tumor progression and metastatic potential.49 In our current study we demonstrated that TIMP-2 inhibits migration and invasion of A549 tumor cells independent of MMP inhibition in vitro. We, therefore, examined the levels and activation of FAK in the A549 tumor xenografts and show that both FAK total protein levels and activation are significantly impaired in both TIMP-2 and Ala+TIMP-2 overexpressing A549 tumor cells. As FAK signaling occurs upstream of PI3K/AKT signaling pathway, it is possible that TIMP-2 mediated deregulation of FAK results in the downstream inhibition of AKT signaling that would eventually impact tumor cell growth and apoptosis.
The proto-oncogene serine-threonine kinase AKT is constitutively activated in many types of cancer as it critically regulates diverse cellular processes including cell proliferation, survival and glucose metabolism.50, 51 In particular, AKT regulates cell growth through its inhibitory effects on tumor suppressor genes including p21 and p27, whereas its pro-survival effects are due to its direct inhibitory signals on pro-apoptotic factors including Bad and Bax. It was recently reported that TIMP-2 Loop 6 directly binds to the insulin-like growth factor receptor I (IGF-IR) on ECs and regulates IGF-IR downstream mitogenic signals.21 As a result, AKT phosphorylation (Ser473) levels were reduced and the downstream receptor signaling was disrupted in ECs. We have also previously shown that Ala+TIMP-2 inhibits VEGF-R2 phosphorylation at tyrosine 1175, a phosphorylation site responsible for AKT signaling activation. Consequently AKT phosphorylation (Ser473) was inhibited in ECs, which is implicated in endothelial nitric oxide synthase phosphorylation.52 Despite the evidence of decreased tumor angiogenesis and the known link with increased tumor cell apoptosis, we examined whether inhibition of A549 tumor growth was directly affected by TIMP-2 overexpression. To this end we examined AKT1 and phospho-AKT expression in the A549 tumor xenografts. Total and activated AKT (Ser473) levels were clearly reduced in both A549 TIMP-2 and Ala+TIMP-2 tumors suggesting that TIMP-2 involvement in inhibition of tumor growth appears to involve additional signaling pathways, in addition to inhibition of tumor angiogenesis. These findings suggest that TIMP-2 may directly regulate differential gene expression in tumor cells. This hypothesis is currently under investigation in our laboratory using microarray analysis.
In conclusion, we demonstrate that TIMP-2 plays an important role in controlling tumor development and progression via both MMP-dependent and independent mechanisms. We observed for the first time that TIMP-2 and Ala+TIMP-2 directly inhibit tumor cell migration and invasion. We confirmed that TIMP-2 mediates inhibition of tumor angiogenesis in vivo resulting in reduced tumor growth and concomitant induction of tumor cell apoptosis. Ala+TIMP-2 expressing tumor xenografts are clearly shown to have reduced MMP inhibitory activity, demonstrating that the antitumor effects are at least partially independent of MMP inhibitory activity. These data emphasize the in vivo antitumoral role of TIMP-2, similarly to other angiogenesis inhibitors that act also as inducers of apoptosis. Moreover, signaling pathways frequently hyperactivated during tumor progression and metastasis, such as those involving FAK and AKT, are also modulated in tumor cells overexpressing TIMP-2, implicating additional, unidentified mechanisms worthy of further investigation. Finally, our results indicate that the preclinical development of TIMP-2 as a novel biological agent for cancer therapy is warranted.
Acknowledgments
We thank the members of the Extracellular Matrix Pathology Section, Radiation Oncology Branch, NCI for critical discussions and reading of the manuscript. We also thank members of the NIH Division of Veterinary Resources for their technical assistance.
Footnotes
Supported by National Cancer Institute Center for Cancer Research grant #ZIA SC009179-21.
References
- 1.Stetler-Stevenson W.G., Yu A.E. Proteases in invasion: matrix metalloproteinases. Semin Cancer Biol. 2001;11:143–152. doi: 10.1006/scbi.2000.0365. [DOI] [PubMed] [Google Scholar]
- 2.Kessenbrock K., Plaks V., Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67. doi: 10.1016/j.cell.2010.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nguyen D.X., Bos P.D., Massague J. Metastasis: from dissemination to organ-specific colonization. Nat Rev Cancer. 2009;9:274–284. doi: 10.1038/nrc2622. [DOI] [PubMed] [Google Scholar]
- 4.Brew K., Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta. 2010;1803:55–71. doi: 10.1016/j.bbamcr.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baker A.H., Edwards D.R., Murphy G. Metalloproteinase inhibitors: biological actions and therapeutic opportunities. J Cell Sci. 2002;115:3719–3727. doi: 10.1242/jcs.00063. [DOI] [PubMed] [Google Scholar]
- 6.Fernandez-Catalan C., Bode W., Huber R., Turk D., Calvete J.J., Lichte A., Tschesche H., Maskos K. Crystal structure of the complex formed by the membrane type 1-matrix metalloproteinase with the tissue inhibitor of metalloproteinases-2, the soluble progelatinase A receptor. EMBO J. 1998;17:5238–5248. doi: 10.1093/emboj/17.17.5238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maskos K., Lang R., Tschesche H., Bode W. Flexibility and variability of TIMP binding: x-ray structure of the complex between collagenase-3/MMP-13 and TIMP-2. J Mol Biol. 2007;366:1222–1231. doi: 10.1016/j.jmb.2006.11.072. [DOI] [PubMed] [Google Scholar]
- 8.Hernandez-Barrantes S., Bernardo M., Toth M., Fridman R. Regulation of membrane type-matrix metalloproteinases. Semin Cancer Biol. 2002;12:131–138. doi: 10.1006/scbi.2001.0421. [DOI] [PubMed] [Google Scholar]
- 9.Strongin A.Y., Collier I., Bannikov G., Marmer B.L., Grant G.A., Goldberg G.I. Mechanism of cell surface activation of 72-kDa type IV collagenase: Isolation of the activated form of the membrane metalloprotease. J Biol Chem. 1995;270:5331–5338. doi: 10.1074/jbc.270.10.5331. [DOI] [PubMed] [Google Scholar]
- 10.Albini A., Melchiori A., Santi L., Liotta L.A., Brown P.D., Stetler-Stevenson W.G. Tumor cell invasion inhibited by TIMP-2. J Natl Cancer Inst. 1991;83:775–779. doi: 10.1093/jnci/83.11.775. [DOI] [PubMed] [Google Scholar]
- 11.Imren S., Kohn D.B., Shimada H., Blavier L., DeClerck Y.A. Overexpression of tissue inhibitor of metalloproteinases-2 retroviral-mediated gene transfer in vivo inhibits tumor growth and invasion. Cancer Res. 1996;56:2891–2895. [PubMed] [Google Scholar]
- 12.Seo D.W., Li H., Guedez L., Wingfield P.T., Diaz T., Salloum R., Wei B.Y., Stetler-Stevenson W.G. TIMP-2 mediated inhibition of angiogenesis: an MMP-independent mechanism. Cell. 2003;114:171–180. doi: 10.1016/s0092-8674(03)00551-8. [DOI] [PubMed] [Google Scholar]
- 13.Johansson E., Komuro A., Iwata C., Hagiwara A., Fuse Y., Watanabe A., Morishita Y., Aburatani H., Funa K., Kano M.R., Miyazono K. Exogenous introduction of tissue inhibitor of metalloproteinase 2 reduces accelerated growth of TGF-beta-disrupted diffuse-type gastric carcinoma. Cancer Sci. 2010;101:2398–2403. doi: 10.1111/j.1349-7006.2010.01688.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang S.W., Cody J.J., Rivera A.A., Waehler R., Wang M., Kimball K.J., Alvarez R.A., Siegal G.P., Douglas J.T., Ponnazhagan S. Conditionally replicating adenovirus expressing TIMP2 for ovarian cancer therapy. Clin Cancer Res. 2011;17:538–549. doi: 10.1158/1078-0432.CCR-10-1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Waleh N.S., Murphy B.J., Zaveri N.T. Increase in tissue inhibitor of metalloproteinase-2 (TIMP-2) levels and inhibition of MMP-2 activity in a metastatic breast cancer cell line by an anti-invasive small molecule SR13179. Cancer Lett. 2010;289:111–118. doi: 10.1016/j.canlet.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 16.Ahn S.M., Jeong S.J., Kim Y.S., Sohn Y., Moon A. Retroviral delivery of TIMP-2 inhibits H-ras-induced migration and invasion in MCF10A human breast epithelial cells. Cancer Lett. 2004;207:49–57. doi: 10.1016/j.canlet.2003.11.025. [DOI] [PubMed] [Google Scholar]
- 17.Lee Y.K., So I.S., Lee S.C., Lee J.H., Lee C.W., Kim W.M., Park M.K., Lee S.T., Park D.Y., Shin D.Y., Park C.U., Kim Y.S. Suppression of distant pulmonary metastasis of MDA-MB 435 human breast carcinoma established in mammary fat pads of nude mice by retroviral-mediated TIMP-2 gene transfer. J Gene Med. 2005;7:145–157. doi: 10.1002/jgm.645. [DOI] [PubMed] [Google Scholar]
- 18.DeClerck Y.A., Perez N., Shimada H., Boone T.C., Langley K.E., Taylor S.M. Inhibition of invasion and metastasis in cells transfected with an inhibitor of metalloproteinases. Cancer Res. 1992;52:701–708. [PubMed] [Google Scholar]
- 19.Fernandez C.A., Butterfield C., Jackson G., Moses M.A. Structural and functional uncoupling of the enzymatic and angiogenic inhibitory activities of tissue inhibitor of metalloproteinase-2 (TIMP-2): loop 6 is a novel angiogenesis inhibitor. J Biol Chem. 2003;278:40989–40995. doi: 10.1074/jbc.M306176200. [DOI] [PubMed] [Google Scholar]
- 20.Wingfield P.T., Sax J.K., Stahl S.J., Kaufman J., Palmer I., Chung V., Corcoran M.L., Kleiner D.E., Stetler-Stevenson W.G. Biophysical and functional characterization of full-length, recombinant human tissue inhibitor of metalloproteinases-2 (TIMP-2) produced in Escherichia coli: Comparison of wild type and amino-terminal alanine appended variant with implications for the mechanism of TIMP functions. J Biol Chem. 1999;274:21362–21368. doi: 10.1074/jbc.274.30.21362. [DOI] [PubMed] [Google Scholar]
- 21.Fernandez C.A., Roy R., Lee S., Yang J., Panigrahy D., Van Vliet K.J., Moses M.A. The anti-angiogenic peptide, loop 6, binds insulin-like growth factor-1 receptor. J Biol Chem. 2010;285:41886–41895. doi: 10.1074/jbc.M110.166439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oh J., Seo D.W., Diaz T., Wei B., Ward Y., Ray J.M., Morioka Y., Shi S., Kitayama H., Takahashi C., Noda M., Stetler-Stevenson W.G. Tissue inhibitors of metalloproteinase 2 inhibits endothelial cell migration through increased expression of RECK. Cancer Res. 2004;64:9062–9069. doi: 10.1158/0008-5472.CAN-04-1981. [DOI] [PubMed] [Google Scholar]
- 23.Perez-Martinez L., Jaworski D.M. Tissue inhibitor of metalloproteinase-2 promotes neuronal differentiation by acting as an anti-mitogenic signal. J Neurosci. 2005;25:4917–4929. doi: 10.1523/JNEUROSCI.5066-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lluri G., Langlois G.D., McClellan B., Soloway P.D., Jaworski D.M. Tissue inhibitor of metalloproteinase-2 (TIMP-2) regulates neuromuscular junction development via a beta1 integrin-mediated mechanism. J Neurobiol. 2006;66:1365–1377. doi: 10.1002/neu.20315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lluri G., Langlois G.D., Soloway P.D., Jaworski D.M. Tissue inhibitor of metalloproteinase-2 (TIMP-2) regulates myogenesis and beta1 integrin expression in vitro. Exp Cell Res. 2008;314:11–24. doi: 10.1016/j.yexcr.2007.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oh J., Diaz T., Wei B., Chang H., Noda M., Stetler-Stevenson W.G. TIMP-2 upregulates RECK expression via dephosphorylation of paxillin tyrosine residues 31 and 118. Oncogene. 2006;25:4230–4234. doi: 10.1038/sj.onc.1209444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Seo D.W., Li H., Qu C.K., Oh J., Kim Y.S., Diaz T., Wei B., Han J.W., Stetler-Stevenson W.G. Shp-1 mediates the antiproliferative activity of tissue inhibitor of metalloproteinase-2 in human microvascular endothelial cells. J Biol Chem. 2006;281:3711–3721. doi: 10.1074/jbc.M509932200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Galm O., Suzuki H., Akiyama Y., Esteller M., Brock M.V., Osieka R., Baylin S.B., Herman J.G. Inactivation of the tissue inhibitor of metalloproteinases-2 gene by promoter hypermethylation in lymphoid malignancies. Oncogene. 2005;24:4799–4805. doi: 10.1038/sj.onc.1208599. [DOI] [PubMed] [Google Scholar]
- 29.Pulukuri S.M., Patibandla S., Patel J., Estes N., Rao J.S. Epigenetic inactivation of the tissue inhibitor of metalloproteinase-2 (TIMP-2) gene in human prostate tumors. Oncogene. 2007;26:5229–5237. doi: 10.1038/sj.onc.1210329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ivanova T., Vinokurova S., Petrenko A., Eshilev E., Solovyova N., Kisseljov F., Kisseljova N. Frequent hypermethylation of 5′ flanking region of TIMP-2 gene in cervical cancer. Int J Cancer. 2004;108:882–886. doi: 10.1002/ijc.11652. [DOI] [PubMed] [Google Scholar]
- 31.Abramoff M.D., Magelhaes P.J., Ram S.J. Image Processing with ImageJ. Biophoton Int. 2004;11:36–42. [Google Scholar]
- 32.Hoyhtya M., Fridman R., Komarek D., Porter-Jordan K., Stetler-Stevenson W.G., Liotta L.A., Liang C.M. Immunohistochemical localization of matrix metalloproteinase 2 and its specific inhibitor TIMP-2 in neoplastic tissues with monoclonal antibodies. Int J Cancer. 1994;56:500–505. doi: 10.1002/ijc.2910560408. [DOI] [PubMed] [Google Scholar]
- 33.Kleiner D.E., Stetler-Stevenson W.G. Quantitative zymography: detection of picogram quantities of gelatinases. Anal Biochem. 1994;218:325–329. doi: 10.1006/abio.1994.1186. [DOI] [PubMed] [Google Scholar]
- 34.Oliver G.W., Leferson J.D., Stetler-Stevenson W.G., Kleiner D.E. Quantitative reverse zymography: analysis of picogram amounts of metalloproteinase inhibitors using gelatinase A and B reverse zymograms. Anal Biochem. 1997;244:161–166. doi: 10.1006/abio.1996.9895. [DOI] [PubMed] [Google Scholar]
- 35.Folkman J. Angiogenesis and apoptosis. Semin Cancer Biol. 2003;13:159–167. doi: 10.1016/s1044-579x(02)00133-5. [DOI] [PubMed] [Google Scholar]
- 36.Streit M., Riccardi L., Velasco P., Brown L.F., Hawighorst T., Bornstein P., Detmar M. Thrombospondin-2: a potent endogenous inhibitor of tumor growth and angiogenesis. Proc Natl Acad Sci USA. 1999;96:14888–14893. doi: 10.1073/pnas.96.26.14888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stetler-Stevenson W.G. Tissue inhibitors of metalloproteinases in cell signaling: metalloproteinase-independent biological activities. Sci Signal. 2008;1:re6. doi: 10.1126/scisignal.127re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taube M.E., Liu X.W., Fridman R., Kim H.R. TIMP-1 regulation of cell cycle in human breast epithelial cells via stabilization of p27(KIP1) protein. Oncogene. 2006;25:3041–3048. doi: 10.1038/sj.onc.1209336. [DOI] [PubMed] [Google Scholar]
- 39.Kang K.H., Park S.Y., Rho S.B., Lee J.H. Tissue inhibitor of metalloproteinases-3 interacts with angiotensin II type 2 receptor and additively inhibits angiogenesis. Cardiovasc Res. 2008;79:150–160. doi: 10.1093/cvr/cvn072. [DOI] [PubMed] [Google Scholar]
- 40.Qi J.H., Ebrahem Q., Moore N., Murphy G., Claesson-Welsh L., Bond M., Baker A., Anand-Apte B. A novel function for tissue inhibitor of metalloproteinases-3 (TIMP3): inhibition of angiogenesis by blockage of VEGF binding to VEGF receptor-2. Nat Med. 2003;9:407–415. doi: 10.1038/nm846. [DOI] [PubMed] [Google Scholar]
- 41.Murphy A.N., Unsworth E.J., Stetler-Stevenson W.G. Tissue inhibitor of metalloproteinases-2 inhibits bFGF-induced human microvascular endothelial cell proliferation. J Cell Physiol. 1993;157:351–358. doi: 10.1002/jcp.1041570219. [DOI] [PubMed] [Google Scholar]
- 42.Hoegy S.E., Oh H.R., Corcoran M.L., Stetler-Stevenson W.G. Tissue inhibitor of metalloproteinases-2 (TIMP-2) suppresses TKR-growth factor signaling independent of metalloproteinase inhibition. J Biol Chem. 2001;276:3203–3214. doi: 10.1074/jbc.M008157200. [DOI] [PubMed] [Google Scholar]
- 43.Feldman A.L., Stetler-Stevenson W.G., Costouros N.G., Knezevic V., Baibakov G., Alexander H.R., Jr., Lorang D., Hewitt S.M., Seo D.W., Miller M.S., O'Connor S., Libutti S.K. Modulation of tumor-host interactions, angiogenesis, and tumor growth by tissue inhibitor of metalloproteinase 2 via a novel mechanism. Cancer Res. 2004;64:4481–4486. doi: 10.1158/0008-5472.CAN-03-2929. [DOI] [PubMed] [Google Scholar]
- 44.Valente P., Fassina G., Melchiori A., Masiello L., Cilli M., Vacca A., Onisto M., Santi L., Stetler-Stevenson W.G., Albini A. TIMP-2 over-expression reduces invasion and angiogenesis and protects B16F10 melanoma cells from apoptosis. Int J Cancer. 1998;75:246–253. doi: 10.1002/(sici)1097-0215(19980119)75:2<246::aid-ijc13>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 45.Albini A., Fontanini G., Masiello L., Tacchetti C., Bigini D., Luzzi P., Noonan D.M., Stetler-Stevenson W.G. Angiogenic potential in vivo by Kaposi's sarcoma cell-free supernatants and HIV-1 tat product: inhibition of KS-like lesions by tissue inhibitor of metalloproteinase-2. AIDS. 1994;8:1237–1244. doi: 10.1097/00002030-199409000-00004. [DOI] [PubMed] [Google Scholar]
- 46.Montgomery A.M., Mueller B.M., Reisfeld R.A., Taylor S.M., DeClerck Y.A. Effect of tissue inhibitor of the matrix metalloproteinases-2 expression on the growth and spontaneous metastasis of a human melanoma cell line. Cancer Res. 1994;54:5467–5473. [PubMed] [Google Scholar]
- 47.Goyal A., Pal N., Concannon M., Paul M., Doran M., Poluzzi C., Sekiguchi K., Whitelock J.M., Neill T., Iozzo R.V. Endorepellin, perlecan angiostatic module, interacts with both the α2β1 integrin and VEGFreceptor 2: A dual receptor antagonism. J Biol Chem. 2011;286:25947–25962. doi: 10.1074/jbc.M111.243626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nystrom A., Shaik Z.P., Gullberg D., Krieg T., Eckes B., Zent R., Pozzi A., Iozzo R.V. Role of tyrosine phosphatase SHP-1 in the mechanism of endorepellin angiostatic activity. Blood. 2009;114:4897–4906. doi: 10.1182/blood-2009-02-207134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McLean G.W., Carragher N.O., Avizienyte E., Evans J., Brunton V.G., Frame M.C. The role of focal-adhesion kinase in cancer—a new therapeutic opportunity. Nat Rev Cancer. 2005;5:505–515. doi: 10.1038/nrc1647. [DOI] [PubMed] [Google Scholar]
- 50.Engelman J.A. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–562. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 51.Manning B.D., Cantley L.C. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee S.J., Tsang P.S., Diaz T.M., Wei B.Y., Stetler-Stevenson W.G. TIMP-2 modulates VEGFR-2 phosphorylation and enhances phosphodiesterase activity in endothelial cells. Lab Invest. 2010;90:374–382. doi: 10.1038/labinvest.2009.136. [DOI] [PMC free article] [PubMed] [Google Scholar]