

Abstract
Autosomal-dominant congenital stromal corneal dystrophy (CSCD) is a human genetic disease characterized by corneal opacities beginning shortly after birth. It is linked to a frameshift mutation in decorin, resulting in a C-terminal truncation lacking 33 amino acids that includes the “ear” repeat, a feature specific for small leucine-rich proteoglycans. Our goals are to elucidate the roles of the mutant decorin in CSCD pathophysiology and to decipher the mechanism whereby mutant decorin affects matrix assembly. A novel animal model that recapitulates human CSCD was generated. This transgenic mouse model targets expression of truncated decorin to keratocytes, thereby mimicking the human frameshift mutation. Mutant mice expressed both wild-type and mutant decorin. Corneal opacities were found throughout, with increased severity toward the posterior stroma. The architecture of the lamellae was disrupted with relatively normal lamellae separated by regions of abnormal fibril organization. Within abnormal zones, the interfibrillar spacing and the fibril diameters were increased. Truncated decorin negatively affected the expression of endogenous decorin, biglycan, lumican, and keratocan and positively affected fibromodulin. Our results provide a mechanistic explanation for the generation of corneal opacities in CSCD. Thus, truncated decorin acts in a dominant-negative manner to interfere dually with matrix assembly and binding to receptor tyrosine kinases, thereby causing abnormal expression of endogenous small leucine-rich proteoglycans leading to structural abnormalities within the cornea and vision loss.
Rigidly controlled fibrillogenesis and fibril organization provide the structural basis for corneal transparency and refractive stability.1 Small leucine-rich proteoglycans (SLRPs) are involved in these processes. Intriguingly, although SLRPs are widely distributed and modulate multiple signaling pathways,2 most of the genetic mutations in this family of proteoglycans are associated with ocular diseases. For instance, a missense mutation of keratocan is linked to cornea plana, and intronic variations in lumican and fibromodulin are associated with high myopia.3 Autosomal-dominant congenital stromal corneal dystrophy (CSCD) is a human disease associated with a mutant class I SLRP gene, decorin. Two independent families have been described harboring a single base pair deletion in the coding sequence, either at 941 (delC) or 967 (delT).4, 5, 6 Notably, both mutations cause a frameshift in the reading frame that leads to an identical truncated decorin protein core lacking the C-terminal 33 amino acids. Patients develop early-onset corneal opacities and a disrupted stromal structure, indicating a novel role for decorin in regulating fibril organization.4, 6
Decorin is the prototype SLRP and contains a protein core and a single chondroitin/dermatan sulfate side chain covalently attached to Ser7 near the N-terminus.7 The protein core has a central domain of tandem leucine-rich repeats (LRRs) flanked by conserved Cys-rich domains. Twelve LRRs give the decorin monomer a curved solenoid fold with an internal parallel β-sheet. LRR-XI extends laterally from the main body near the C-terminus and is referred to as the “ear” repeat, a distinctive feature of SLRPs.8 Decorin has a high-affinity binding site for collagen in LRRs 4 to 6 and decorates collagen fibrils, regulating collagen fibrillogenesis. Decorin also bridges collagen fibrils to other molecules such as collagens VI,9 XII,10 and XIV.11 All these interactions are possible in the corneal stroma.
Decorin is both a structural constituent as well as a signaling molecule involved in regulating cell growth, angiogenesis, and fibrosis.12 For example, decorin binds to epidermal growth factor receptor and ErB4 through the LRR7 domain and inhibits tumor cell growth.13, 14 The N-terminus binds to insulin-like growth factor-1 receptor, regulating cell proliferation and apoptosis.15 Decorin also suppresses tumor cell-mediated angiogenesis by inhibiting the endogenous secretion of vascular endothelial growth factor.16 Recent studies have also shown that decorin is an antagonistic ligand of the Met receptor.17, 18 Molecular modeling indicates that truncation of the decorin C-terminus in human CSCD affects the ear repeat, which is proposed to maintain the conformation of the protein and to regulate ligand binding.8 However, the dysfunctional role of mutant decorin in human autosomal-dominant CSCD and the pathophysiological basis of this inherited disease remains to be elucidated.
To provide an explanation of why heterozygous truncating mutations in the human decorin (DCN) gene lead to corneal clouding, we developed a novel mouse model in which the corneal stroma was targeted with a Cre-on approach, resulting in the concurrent expression of the decorin wild-type and the 952 delT alleles. We were able to reproduce the human CSCD phenotype in this mouse model. The mice expressing mutant decorin developed corneal opacities postnatally as in humans. The corneal opacities increased with age, and ultrastructural analysis showed altered fibrillogenesis and disrupted lamellar structure required for transparency. Unexpectedly, we discovered that the expression of the truncated decorin was associated with profound aberrant expression of endogenous SLRPs. Thus, we provide a mechanistic explanation for this rare genetic condition in which expression of a mutant decorin may act in a dominant-negative fashion to alter both matrix secretion at the cellular level and extracellular matrix assembly in the corneal microenvironment.
Materials and Methods
Generation of a 952 delT Decorin Transgenic Mouse
A pMSCV/Dec plasmid, which contains pMSCVpuro (Clontech, Mountain View CA) vector, and a full-length mouse decorin cDNA,19 was used as the template for making the mutant mouse decorin cDNA (952 delT) with the use of a Transformer Site-directed mutagenesis kit (Clontech). Mutant mouse decorin cDNA was subcloned into a plasmid pCAG-flox-CAT-flox vector.20 The construct backbone was linearized, and the pCAG-flox-CAT-flox-952delTdecorin DNA construct was used to generate the 952delTDcn transgenic mouse model in the Moffitt Cancer Center Mouse Models Core. The mice carrying 952 delT decorin were screened by PCR (DecDelT, 5′-CGAAAGGCTCCTACTCGGC-3′; DecF-3, 5′-TGGCACAGCATAAGTATATCC-3′; pvector-R, 5′-AACTTCAGGGTCAGCTTGC-3′) and crossed with Kera-Cre 4.2 mouse20 (KC-IRES426-452: 5′-GGCTCTCCTCAAGCGTATTCAACAAG-3′; Cre866-844: 5′-CCAGACCAGGCCAGGTATCTCTG-3′). Mice positive in both 952delT decorin and Kera-Cre PCR screening were used for the experiments described. All animal studies were performed in compliance with approved animal protocols from the University of South Florida Institutional Animal Care and Use Committee.
In Vivo Confocal Microscopy, Measurement of Corneal Haze and Thickness
In vivo corneal haze was analyzed with the Heidelberg Retinal Tomography HRT Rostock Cornea Module (HRT-III; Heidelberg Engineering Inc., Heidelberg, Germany). Briefly, four pairs of 2- to 3-month-old wild-type and 952delTDcn mice were anesthetized and restrained on an adapted stage. GenTeal Gel (Novartis Pharmaceuticals Corp., East Hanover, NJ) was applied to the corneal surface. Images of the apex of the cornea were obtained by mechanically or manually adjusting the CCD camera position. Series of images were sequentially collected from the surface of the epithelium to the endothelium as a z-axis scan at fixed intensity for all of the animals. Images were processed with Metamorph software (Premier Version 7.6.5.0, Molecular Devices, Sunnyvale, CA). A depth intensity profile from the scans was generated by plotting the average pixel intensity per plane as a function of corneal depth. Corneal haze (light scattering) was measured by calculating the total pixel intensity as measured by the area under the curve. Corneal thickness was determined by measuring the axial distance from the epithelium to endothelium.
Antibodies and Immunoblots
Rabbit anti-decorin (LF113; provided by Dr. Larry Fisher, National Institutes of Health, National Institute of Dental and Craniofacial Research, Bethesda, MD) was used at 1:250; both anti-biglycan (LF159, provided by Dr. Larry Fisher, National Institutes of Health, National Institute of Dental and Craniofacial Research) and anti-keratocan (provided by Dr. John Hassell, University of South Florida) were used at 1:200. Rabbit anti-lumican (provided by Dr. Ake Oldberg, Lund University, Lund, Sweden) was used at 1:1000. Rabbit anti-mouse fibromodulin provided by Dr. Larry Fisher, National Institutes of Health, National Institute of Dental and Craniofacial Research) was used at 1:200. Corneas were dissected and pooled followed by homogenization in a 20-fold excess (w/v) of extraction buffer (4 mol/L guanidine-HCl, 50 mmol/L sodium acetate, pH 5.8) with proteinase inhibitors (Thermo Scientific, Waltham, MA). SLRPs were extracted at 4°C for 48 hours with shaking. The extraction was clarified by centrifugation and then dialyzed against 150 mmol/L Tris-HCl, 150 mmol/L NaCl, pH 7.3. Samples were digested with Chondroitinase ABC or Endo-β-galactosidase (Seikagaku Corporation, Tokyo, Japan) for 24 hours at 37°C. To remove the N-glycans from decorin, samples were denatured and digested with N-Glycanase (PNGase F; ProZyme, San Leandro, CA), following the manufacturer's protocol. Total protein was determined with a BCA protein assay kit (Pierce, Rockford, IL). Proteins from each preparation were separated with 4% to 12% Bis-Tris gels (Invitrogen, Carlsbad, CA) and transferred to Hybond-C extra membranes (GE Health Care, Little Chalfont, Buckinghamshire, UK) for immunoblotting. Goat anti-rabbit IgG-peroxidase (GE Health Care) secondary antibodies were used at a 1:3000 dilution with an electrochemiluminescence (Pierce) detection system. Actin reactivity in each sample was detected with an antiactin antibody at a 1:200 dilution (Millipore, Billerica, MA). The volume of the bands was measured with a Gel Doc system and Quantity One software (Version 4.6.7, Bio-Rad, Hercules, CA).
Immunofluorescence and Transmission Electron Microscopy
Whole eyes, three to five per group, were embedded in OCT medium, frozen on dry ice, and stored at −80°C. Frozen sections (4-μm thick) were cut with a Microm HM505E cryostat (Leica, Wetzlar, Germany). The slides were blocked with 5% bovine serum albumin in PBS followed by the primary antibody. The secondary antibody was Alexa Fluor 568- or 488-conjugated goat anti-rabbit IgG (Molecular Probes, Eugene, OR) used at 1:100. Vectashield mounting solution with DAPI (Vector Laboratories, Inc., Burlingame, CA) was used as a nuclear marker. Images were captured with the use of a Leica CTR 5500 microscope and Leica DFC 340 FX camera. Identical conditions and set integration times were used to facilitate comparisons between samples. Cornea samples from wild-type and 952delTDcn transgenic mice were analyzed by transmission electron microscopy as previously described.21 Briefly, three corneas per group were dissected and fixed in 4% paraformaldehyde, 2.5% glutaraldehyde, 0.1 mol/L sodium cacodylate, pH 7.4, with 8.0 mmol/L CaCl2, postfixed with 1% OsO4. The corneas were dehydrated in graded ethanol series, followed by propylene oxide. The tissue samples were infiltrated and embedded in a mixture of Embed 812, nadic methyl anhydride, dodecenyl succinic anhydride, and DMP-30 (Electron Microscopy Sciences, Hatfield, PA). Thin sections (∼80 nm) were cut with a Leica ultramicrotome and poststained with 2% aqueous uranyl acetate and 1% phosphotungstic acid, pH 3.2. The sections were examined at 80 kV with a JEOL 1400 transmission electron microscope equipped with a Gatan Ultrascan US1000 2K digital camera.
Fibril Diameter Distribution
Three corneas from three different animals at postnatal day 30 were analyzed for both wild-type and 952delTDcn transgenic mice. Digital images (9/cornea) were taken from nonoverlapping regions in both the anterior and posterior stroma. Fibril diameters were measured with a RM Biometrics-Bioquant Image Analysis System (Nashville, TN) with the use of randomly chosen, masked digital images analyzed at a final magnification of ×48,000. For each of the groups, sample mean, sample median to first quartile distance, and sample third quartile distance to median were analyzed. The median indicates the location of the center of the distribution, whereas median to quartile distances reflect primarily the spread of the data. Distributions of the sample-to-sample means and both median to quartile distances for the anterior and posterior stroma in the transgenic cornea were compared with the wild-type group using a t-test.
Cuprolinic Blue Staining
Three corneas from both wild-type and 952delTDcn transgenic mice were fixed overnight in 2.5% glutaraldehyde in 25 mmol/L sodium acetate buffer, pH 5.7, containing 0.1 mol/L magnesium chloride and 0.05% cuprolinic blue (BDH Chemicals, London, UK). The samples were washed three times in 25 mmol/L sodium acetate buffer, pH 5.7, and then transferred to distilled water that contained 0.5% (w/v) sodium tungstate for 15 minutes, followed by 15 minutes in 50% ethanol containing 0.5% sodium tungstate. Specimens were then dehydrated in an ethanol series, followed by processing as described above.
Results
Creation of a Novel Mouse Model of Human CSCD
Only heterozygous patients carrying a normal and mutant decorin allele were found to have CSCD, suggesting that homozygosity may result in a lethal phenotype. We developed a transgenic mouse model of this disease with the use of a Cre-on approach to avoid embryonic lethality. A transgenic mouse line carrying the mutant decorin cDNA with a deleted T at 952, which is comparable to human 967delT mutation, was created (Figure 1A). We generated a vector harboring a ubiquitous CAG promoter followed by a loxP-flanked chloramphenicol acetyltransferase (CAT) gene that was followed by the mutant decorin cDNA (952delT) (Figure 1B). The transgenic mice were bred into a wild-type background (Dcn+/+). Cornea-specific 952delT decorin expression was turned on by breeding these mice with Kera-Cre mice which specifically deletes the LoxP-CAT, leading to mutant decorin expression by cornea stromal keratocytes. The activation of Kera at embryonic day 13.5 to 14.520 allows the expression of the truncated decorin allele because of the deletion of LoxP-flanked polyadenylated CAT gene by Cre-recombinase driven by keratocan promoter.
Figure 1.
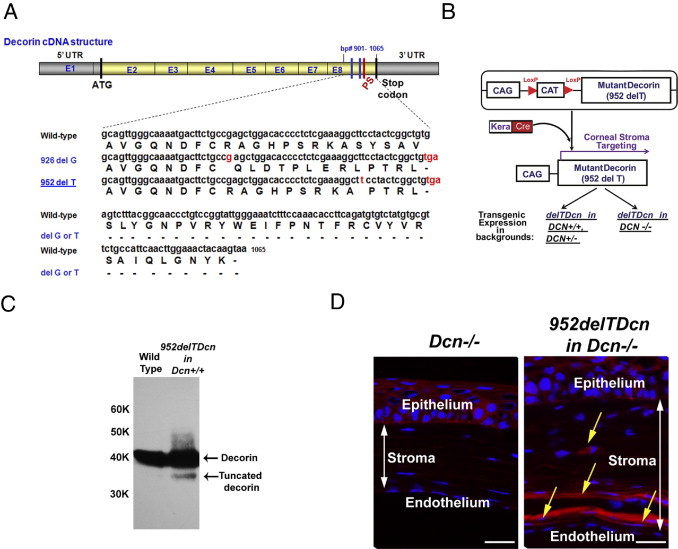
952delTDcn transgenic mice express both wild-type and mutant decorin. A: Two independent families with human CSCD have been reported. A single base pair deletion in the coding sequence at either 941 (delC) or 967 (delT) caused frameshift mutations and a truncation of the C-terminal 33 amino acids. In mice, a delG at 926 or a delT at 952 (highlighted in red) will result in a comparable frameshift mutation. The new stop codon TGA (highlighted in red) will yield a C-terminal truncated decorin comparable to that in human CSCD. B: A Cre-on approach was used to create a transgenic mouse carrying a mutant decorin cDNA with a deleted T at 952. A stop codon flanked with LoxP (red triangle) elements was inserted between a ubiquitous CAG promoter and the mutant decorin cDNA. Corneal stromal targeting of mutant decorin was generated through breeding with Kera-cre mouse. The mouse model was bred into different backgrounds, including decorin wild-type, heterozygous, and deficient backgrounds. C: Immunoblot analyses show that the transgenic mice in a decorin wild-type background expresses two decorin bands after sequential Chondroitinase ABC and PNGase F digestion, one migrating with the wild-type decorin core, the other migrating faster at ∼37 kDa, consistent with the C-terminal truncation. The level of mutant decorin was substantially less than the wild-type decorin. These qualitative immunoblots were overloaded/exposed to show the presence or absence of the truncated decorin core in mutant and wild-type corneas, respectively. The antibody used was generated against the N-terminal 17 amino acids of the decorin protein core and thus recognizes both the native and the C-terminal–truncated species. D: Breeding the mutant into a decorin-null background allowed the localization of the mutant protein core. Mutant decorin expression was identified by immunolocalization in a 952delTDcn mouse in decorin-deficient background (yellow arrow indicates the positive reactivity of mutant decorin in the corneal stroma). Scale bar = 25 μm.
Endogenous expression of wild-type and 952delT decorin was analyzed in the bi-transgenic animals. Extracts prepared from excised corneas were sequentially digested with chondroitinase ABC and N-glycanase. Immunoblot analysis with anti-decorin antibody showed a single band at ∼40 kDa in wild-type mice, whereas in mutant mice, there were two bands, one migrated with the wild type, the other one migrated faster, consistent with the 3- to 4-kDa truncation of the decorin core (Figure 1C). These data indicate that both normal and mutant decorin are concurrently expressed in the bi-transgenic mouse cornea, albeit the truncated decorin was expressed at lower levels (Figure 1C). The expression of the truncated decorin in the corneal stroma was further confirmed by breeding the 952delTDcn transgenic mice into a decorin-null background (Dcn−/−). Immunofluorescence analysis indicated that the mutant decorin was readily detectable and exhibited a higher immunoreactivity in the posterior stroma (Figure 1D).
Validation of the Mouse Model of Human CSCD
The 952delTDcn mice were fertile and generally normal with the exception of opaque corneal stromas in 952delTDcn compared with wild-type mice (Figure 2, A and B). Grossly, the extent of opacity in the cornea varied from hardly visualized cloudiness to overt opacity. Visualized opacities typically appeared at ∼1 month of postnatal life (postnatal day 30) with a slow progression of opacity afterward. The opacities were more severe in the central cornea. To further analyze the affected mutant corneas, we used in vivo confocal microscopy. In the mutant mice, the epithelium and endothelium were clear, comparable to that in the wild-type mice. However, opacities developed throughout the stromal layers in 952delTDcn mutant mice, with scattered regions of higher intensity in the posterior stroma (Figure 3A). The corneal haze in 952delTDcn mouse was significantly higher than in control mice (P = 0.05) (Figure 3B). However, no significant difference in corneal thickness was observed (Figure 3C). Moreover, in the 952delTDcn transgenic mice the lamellar structure was disrupted, and this was more severe in the posterior stroma (Figure 3D). Similar to the human CSCD disease, at the ultrastructural level, the corneal orthogonal lamellar structure was disrupted in the mouse model. Relatively normal lamellae were separated by abnormal zones in which collagen fibrils were irregularly packed and embedded in an electron-lucent substance (Figure 4, B and D) not observed in the normal controls (Figure 4, A and C). These abnormal zones were scattered throughout the corneal stroma, but they were consistently more severe in the posterior stromal layers. Collectively, these results indicate that our novel animal model recapitulates most of the macroscopic and ultrastructural abnormalities reported in human CSCD.
Figure 2.
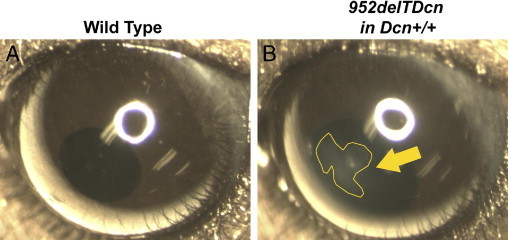
952delTDcn transgenic mice develop corneal opacities. Representative views of eyes from wild-type (A) and 952delTDcn transgenic (B) mice in a decorin wild-type background. Notice that the 952delTDcn transgenic mice develop cloudy corneas with the opacity more obvious in the central cornea (yellow arrow).
Figure 3.
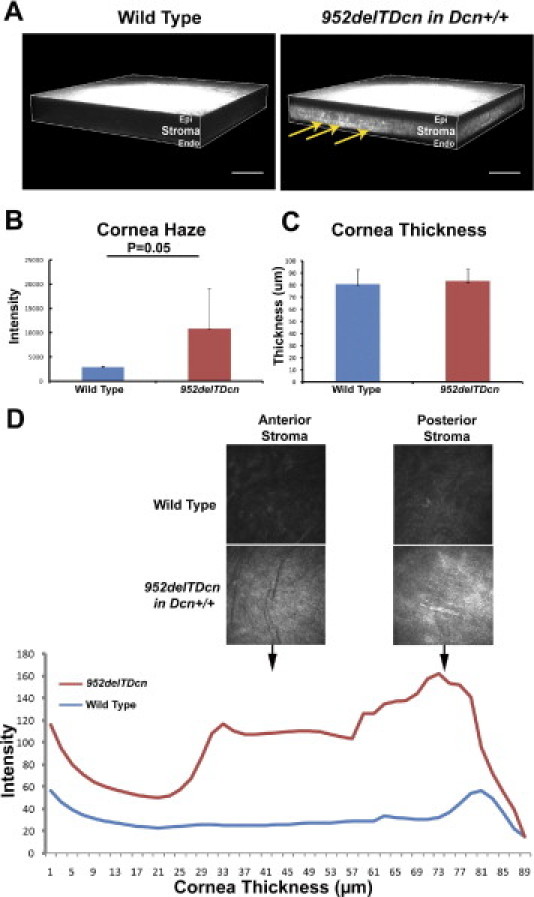
Location and severity of stromal haze in 952delTDcn transgenic mouse. A: Three-dimensional renderings of representative in vivo confocal z-stacks of scanning images show corneal opacity across the stroma with intact epithelium and endothelium in 952delTDcn transgenic mice. Notice the presence of scattered intensities, which are more severe in the posterior stroma (yellow arrows). B: Increased corneal haze in 952delTDcn transgenic mice than in control mice (P = 0.05). C: The cornea thickness was comparable between 952delTDcn transgenic mice and control mice. D: Little light scattering was observed in the epithelium and endothelium of wild-type and 952delTDcn transgenic mice; however, light scattering was increased in both the anterior and posterior stroma in 952delTDcn transgenic mice than in wild-type control mice. Stroma haze was present throughout the stroma but was more intense in posterior stroma as shown by the depth intensity profile. All 952delTDcn transgenic mice were in a Dcn+/+ background. Scale bar = 100 μm.
Figure 4.
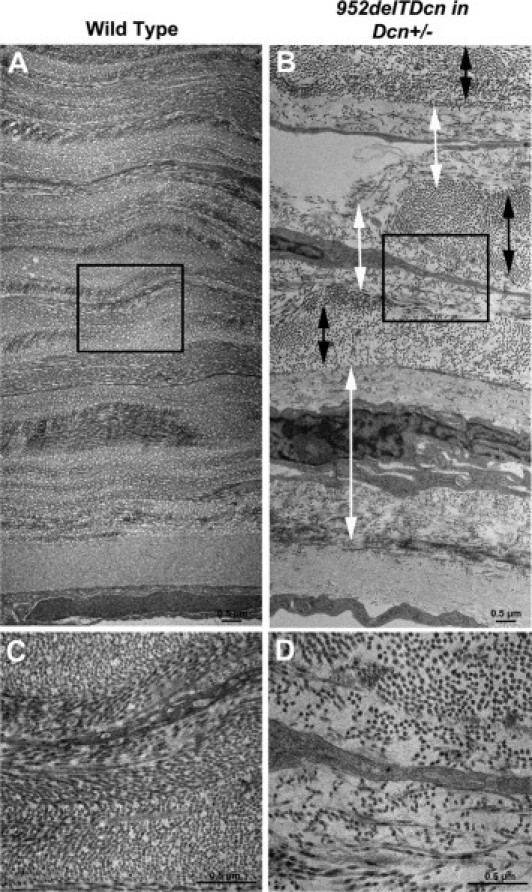
Disrupted corneal stromal structure in 952delTDcn transgenic mice. Compared with the wild-type cornea (A), in the 952delTDcn transgenic mouse, the orthogonal lamellar structure is disrupted (B). Relatively normal lamellae (black double-headed arrow) are separated by abnormal zones (white double-headed arrow). B: In these abnormal zones, collagen fibrils are irregularly packed and embedded in an electron-lucent substance, with increased interfibrillar spacing. Note that the abnormal zones are often adjacent to keratocytes and more pronounced in the posterior stroma. C: Higher magnification of boxed area in panel A. D: Higher magnification of boxed area in panel B. Scale bars = 0.5 μm (A–D).
Pathophysiological Mechanism of Dysfunctional Corneal Stroma in a Mouse Model of Human CSCD
Reduced proteoglycans are associated with disrupted fibril structure and organization in the 952delTDcn transgenic mouse. SLRPs not only regulate fibrillogenesis controlling fibril diameter but also are involved in control of interfibrillar spacing via the hydration of their glycosaminoglycan chains extending out into the interfibrillar space. The increased interfibril spacing in our mutant mice suggested that there may be accumulated SLRPs; however, with the use of cuprolinic blue histochemistry, we found that reduced sulfated proteoglycans were associated with disrupted fibrils. An accumulation or increase in proteoglycans was not observed in the electron lucent substance or associated with the abnormally small diameter fibrils (Figure 5B) in contrast to that seen in wild-type corneas (Figure 5A). The reduced histochemical reactivity for sulfated proteoglycans suggests a reduction in corneal stromal SLRPs.
Figure 5.
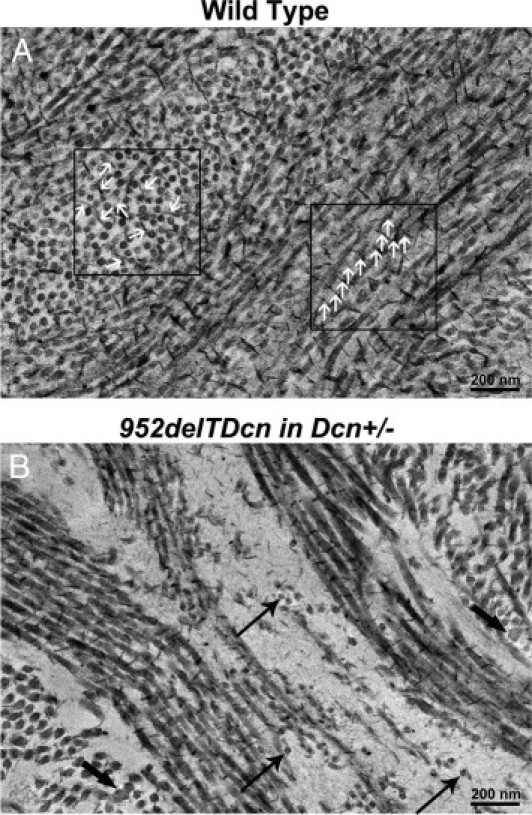
Reduced histochemical staining for proteoglycans associated with disrupted fibril structure and organization in the 952delTDcn transgenic mouse. A: In wild-type mice, cuprolinic blue reaction is associated with collagen fibrils. Note that the electron-dense deposits that label glycosaminoglycan side chains of the SLRPs are distributed along the fibril surface in a regular and periodic fashions. These proteoglycans extend into the interfibrillar space (white arrows). B: Marked reduction in cuprolinic blue-labeled SLRPs in 952delTDcn transgenic mouse corneas. Notice that the collagen fibrils exhibit decreased interaction relative to the control corneas. In addition, there is no accumulation of cuprolinic blue staining in the electron lucent substance indicative of proteoglycan sequestration. Thin arrows indicate relatively small-diameter fibrils; thick arrow indicates larger-diameter fibrils.
Altered Collagen Fibrillogenesis in 952delTDcn Transgenic Mice
Tightly regulated collagen fibrillogenesis is required for assembly of lamellae and stromal structure required for transparency.1 Our data show that fibrillogenesis was altered across the corneal stroma in our transgenic mouse model. In both the anterior and posterior stroma, collagen fibril diameters increased, and the fibril distribution shifted to larger diameters (Figure 6 and Table 1). The fibril distribution was heterogeneous in the mutant mice, and this alteration was more pronounced in the posterior stroma. Moreover, within the abnormal electron lucent zones, fibrils had abnormally small diameters. The present findings suggest that both regulation of fibril assembly and packing were disrupted in the mutant mouse corneas.
Figure 6.
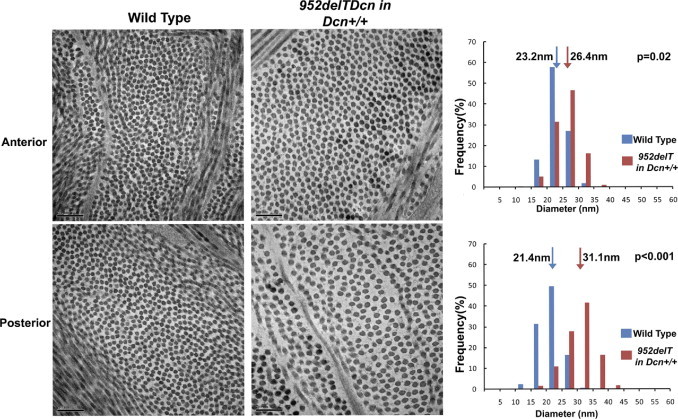
Altered collagen fibrillogenesis in 952delTDcn transgenic mice. Fibrillogenesis is altered across the corneal stroma in the 952delTDcn transgenic mouse model. Note that in both the anterior and posterior stroma, collagen fibril diameter increases with the fibril distribution shifted to the right. The fibril distribution is heterogeneous in the mutant mice, and this feature is more pronounced in the posterior stroma. Scale bar = 200 nm; arrow indicates the position of the median in fibril diameter distribution.
Table 1.
Altered Collagen Fibril Diameter Distribution in 952delTDcn Transgenic Mice
| Anterior stroma |
Posterior stroma |
|||||
|---|---|---|---|---|---|---|
| Wild-type (nm) | 952delTDcn in Dcn+/+ background (nm) | P | Wild-type (nm) | 952delTDcn in Dcn+/+ background (nm) | P | |
| Mean | 23.3 | 25.81 | 0.02 | 21.8 | 28.8 | <0.001 |
| Range | 14.0–32.9 | 15.0–37.5 | 10.7–34.8 | 10.2–44.9 | ||
| Median | 23.2 | 26.4 | 0.02 | 21.4 | 31.1 | <0.001 |
| M–Q1 | 1.9 | 2.7 | 0.009 | 1.9 | 3.2 | <0.001 |
| Q3–M | 2.0 | 2.5 | 0.008 | 1.9 | 3.6 | 0.002 |
M, median; Q1, 25th percentile; Q3, 75th percentile.
Deregulated Expression of SLRPs in the Corneal Stroma of 952delTDcn Transgenic Mice
SLRPs are involved in regulating lateral fibril growth from immature protofibrils.1, 22 Blocking the lateral growth is critical for the maintenance of homogenous small diameter fibrils required for corneal transparency. The corneal stroma is enriched in SLRPs, which cooperate in regulating fibrillogenesis. For instance, in decorin-deficient mice, biglycan is up-regulated to compensate for the loss of decorin.23 Therefore, we analyzed the expression levels of several SLRPs in our mutant mouse corneas. Immunoblot analyses showed a marked down-regulation of endogenous decorin (fourfold to eightfold) and biglycan (threefold to sevenfold) (Figure 7A) and a >10-fold decrease of lumican and keratocan (Figure 7B). In addition, significant fibromodulin expression was observed in the mutant corneas with a virtual absence in the wild-type mice. The immunoblot data were further corroborated by immunofluorescence analysis of the mutant corneas. These data show an overall expression pattern for the various SLRPs, consistent with the immunoblot analyses (Figure 8). In these analyses, some variation in SLRPs between samples was observed that may be related to the varying degrees of opacity observed.
Figure 7.
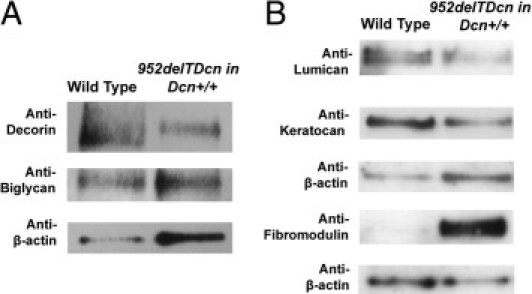
Dysregulation of SLRP expression in the corneal stroma of 952delTDcn transgenic mice. A: Expression of class I SLRPs, decorin and biglycan, was decreased in 952delTDcn transgenic mice compared with wild-type controls. Semiquantitative immunoblot analyses show a fourfold to eightfold decrease in decorin and a threefold to sevenfold decrease in biglycan expression relative to β-actin. B: Class II SLRPs also show altered expression in the 952delTDcn transgenic mice. The expression of lumican and keratocan is decreased >10-fold. Fibromodulin, which is virtually absent in wild-type corneas, showed consistent and significant expression in the mutant corneas.
Figure 8.
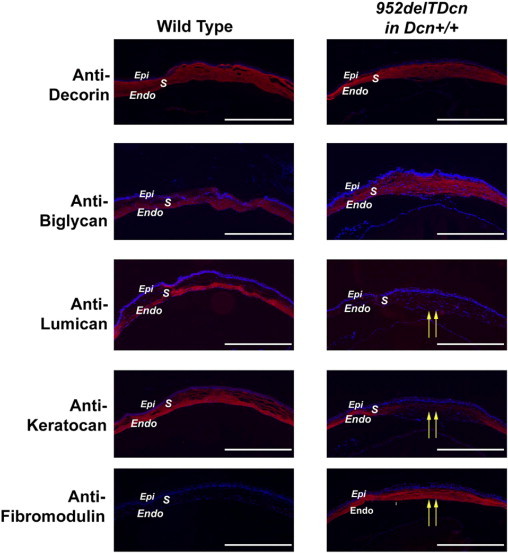
Altered immunolocalization of SLRPs in the corneal stroma of 952delTDcn transgenic mice. The immunolocalization of class I SLRPs, decorin and biglycan, is comparable in wild-type and transgenic mice. The decreased expression observed (Figure 7) is not obvious as decreased reactivity in these studies. All class II SLRPs show altered immunolocalization. Both lumican and keratocan reactivity are absent from the central cornea. Keratocan reactivity is present in the peripheral cornea of transgenic animals. In contrast, fibromodulin reactivity is absent from wild-type controls but present in the transgenic corneal stroma. Endo, endothelium; Epi, epithelium; S, stroma. Yellow arrows indicate altered class II SLRPs in central corneal stroma. Scale bar = 500 μm.
Collectively, our findings indicate that de novo expression of a C-terminal–truncated decorin affects the synthesis and secretion of various members of the SLRP family, all of which contribute to corneal transparency and homeostasis. The extensive alterations in SLRP expression suggest that the presence of the mutant decorin may act in a dominant-negative fashion and affect general receptor tyrosine kinases such as epidermal growth factor receptor, Met, or insulin-like growth factor-1 receptor, which would alter corneal morphogenesis and homeostasis.
Discussion
In this work, we have developed a novel transgenic mouse model expressing truncated decorin and have validated it as a model of human CSCD at the gross level as well as molecular and ultrastructural levels. With the use of this model, the pathophysiological mechanisms underlying CSCD were investigated. The data indicate that the presence of C-terminal–truncated decorin interferes with normal function of the native decorin, potentially by altering the interaction of decorin with various signaling receptors on the surface of keratocytes. This could occur directly or indirectly through other matrix constituents. We hypothesize that receptor tyrosine kinases such as the epidermal growth factor receptor, Met, or insulin-like growth factor-1 receptor might be directly involved in this loop.13, 14, 15, 16, 17, 18 The altered signaling has profound consequences insofar as the expression of endogenous decorin, biglycan, lumican, and keratocan is markedly down-regulated, whereas that of fibromodulin is induced. These changes would be expected to alter the precise regulation involving coordinated expression of SLRPs essential for the architecture of stromal extracellular matrix. Thus, the developing corneal stroma is not under the proper temporal regulation by SLRPs leading to corneal opacities. We believe that we have uncovered a novel mechanism of action for a rare disease such as CSCD in which a mutant decorin acts as a dominant-negative factor to dramatically affect the synthesis and secretion of key members of the SLRP gene family leading to structural abnormalities in the corneal stroma. The end result of this dysfunctional SLRP expression is abnormal protein-protein interaction in the stroma that leads to a dysregulated control of fibrillogenesis and fibril organization, thus the aberrant lattice structure in the cornea stroma (Figure 9). The data not only indicate the importance of the C-terminus in decorin but also provide novel evidence for interactions involving different classes of SLRPs in finely modulating microenvironment, thus contributing to the corneal stromal-specific structure.
Figure 9.
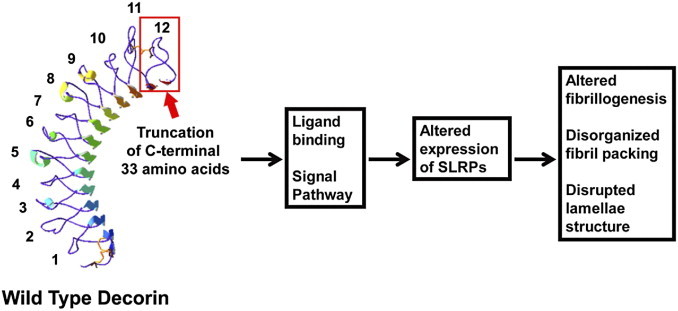
Pathophysiological mechanisms of dysfunctional matrix assembly in human CSCD. Swiss model (automated mode) of wild-type decorin core protein. The LRR domains are numbered from N-terminus to C-terminus (1 to 12). Red box indicates the truncation of C-terminal 33 amino acids in our mouse model, which deletes part of LRR11 and the entire LRR12 containing the ear repeat. The ear repeat has been implicated in regulating conformational stability and influencing ligand binding.8 Therefore, we suggest that loss of the C-terminus alters decorin binding to keratocytes, either directly or indirectly through other extracellular matrix components. Therefore, truncated decorin could act as a dominant-negative molecule that interferes with binding to receptor tyrosine kinases, thereby causing abnormal synthesis and secretion of endogenous SLRPs. In addition, the truncated decorin could act in a dominant-negative manner to interfere with decorin interaction with collagen necessary for regulation of fibrillogenesis. We propose that this dual interference results in altered fibrillogenesis, disorganized packing, and disrupted lamellae structure, which is the pathophysiological mechanism underlying human CSCD.
SLRPs have co-evolved with collagens primarily because of their ability to regulate collagen fibrillogenesis, a process required for the generation of an orthogonal lattice structure composed of homogenously smaller diameter fibrils, decreasing light scattering, thus resulting in corneal transparency.24 A unique feature of the corneal stroma is its enrichment in class I SLRPs (eg, decorin and biglycan) and class II SLRPs (eg, lumican, fibromodulin, and keratocan). SLRPs are involved in corneal stromal development, wound healing, and repair/remodeling and have been linked to several ocular abnormalities such as myopia25 and cornea plana,26 as well as CSCD.4, 5 Notably, decorin-null mice have clear corneas probably because of a functional compensation from up-regulated biglycan. However, the double-mutant mice lacking both Dcn and Bgn genes show a severe stromal phenotype, which document decorin's central role in regulating fibrillogenesis in the corneal stroma.23 Interestingly, the phenotype in our 952delT decorin transgenic mouse model was different from Dcn−/−;Bgn−/− mice, suggesting that C-terminus–truncated decorin caused a gain of function.
All of the SLRPs bind collagen fibrils through LRR domains; however, multiple different binding regions have been documented. Class I and II SLRPs were shown to bind different sites on the collagen fibril surface, eg, class I SLRPs, decorin and biglycan, bind collagen fibrils at the “b” and “d” zones,27, 28 whereas class II lumican and fibromodulin bind fibrils at the “a” and “c” zones.29, 30 On the one hand, analysis with the use of rotary shadow electron microscopy and photoaffinity labeling mapped the decorin binding site at the c zone, a main intermolecular cross-linking site of collagen heterotrimers.31 On the other hand, multiple other collagen binding sites have been reported in SLRPs.30, 32 The balance of competition, cooperation, and compensation among SLRPs and different binding sites results in highly specified regulatory functions in collagen fibrillogenesis.
An emerging concept is that SLRPs affect collagen fibrillogenesis and matrix assembly not only through physical binding to the collagen fibrils but also through regulation of their own expression. For example, in Dcn−/− mice, biglycan expression is increased to compensate for the loss of decorin,23 whereas in Fmod−/− mice lumican is up-regulated.33 In addition, the absence of lumican results in decreased keratocan expression.34 Another example of a coregulatory mechanism is provided by the lumican and keratocan genes, which are clustered in cis within one chromosome (chromosome 12 in human, chromosome 10 in mouse). In addition, part of the distal region of the keratocan promoter shares 89% homology with the lumican promoter, both of which contain NF-1 transcription factor-binding sites serving as responsive elements for many signal pathways.35 For example, in Tgfβ2−/− mice, both Lum and Kera transcription were decreased.36 In our transgenic mouse model, we observed that all SLRPs were affected, however; there was some variation between samples. This could be because of the differing expression levels of mutant decorin in the mice but may also be complicated by the interactions between SLRPs. The C-terminal truncation affects the decorin ear repeat, which was proposed to be implicated in ligand binding and maintenance of conformation.8 A recent study by Vial et al37 showed that LRR 12 can neutralize CCN2-mediated activity on C2C12 cells in vitro, suggesting a role for the C-terminus of decorin in this signal pathway.
The regulatory functions of SLRPs in corneal stromal collagen fibrillogenesis are complicated in a tissue-specific temporal-spatial manner. During development, both decorin and biglycan are distributed across the whole cornea; however, biglycan decreases after birth. Lumican is initially homogenous in both the anterior and posterior stroma, but it is then restricted to the posterior layers in the adult animal. Keratocan has a constant, homogeneous temporal and spatial expression pattern in corneal development. Fibromodulin was originally thought not to be a molecular component of the cornea; however, we observed that its expression extends into the central cornea in early postnatal development.33 In our 952delTDcn transgenic mouse model, the structural alterations and functional deficiency are present throughout the corneal stroma but more severe in posterior stroma, which is consistent with the depletion of posterior stroma-restricted lumican.38 These data suggest that the CSCD phenotype is partially a result of dysregulation of lumican-mediated fibril diameter regulation, and ultrastructural analysis shows structural alterations in the 952delTDcn transgenic mouse model consistent with the lumican-null phenotype (see Supplemental Figure S1 at http://ajp.amjpathol.org). In addition, the overall decrease in decorin and biglycan would be expected to affect the phenotype and may be exacerbated by dominant-negative influences of the mutant decorin influencing fibril as well as cell interactions. A distinguishing phenotype in both the human disease and our mouse model is the relatively small diameter fibrils embedded in an electron lucent substance separated by regions of relatively normal stromal structure. These abnormal zones are often adjacent to keratocytes and suggest regional disruption of matrix assembly mechanisms. The “simple” orthogonal packing of homogenously smaller diameter fibrils in the corneal stroma results from complex regulatory mechanisms. Different extracellular matrix molecules and multiple interactions between them and keratocytes are ultimately responsible for providing corneal transparency. An investigation into the reason this mutant decorin phenotype mainly affects corneal structure and function and how SLRPs cooperate in a cornea-specific temporal manner is important for a better understanding of corneal repair, regeneration, and pathophysiology.
Acknowledgments
We thank Sheila Adams for expert technical assistance with transmission electron microscopy and Qingmei Yao with the maintenance of the mouse lines.
Footnotes
Supported by the NIHEY05129 (D.E.B.), EY011845 (R.V.I.), and CA39481 (R.V.I.); Research to Prevent Blindness, Inc.; Ohio Lions Eye Research Foundation (W.W.K.); and 2010 American Association of Anatomists, 2010 Fight For Sight Postdoctoral Fellowship Awards (S.C.).
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.07.026.
A guest editor acted as editor-in-chief for the manuscript. No person at Thomas Jefferson University was involved in the peer review process or final disposition of this article.
Current address of X.M., Department of Pathology, Pennsylvania Hospital of University of Pennsylvania Health System, Philadelphia, PA.
Supplementary data
952delDcn mice exhibit characteristics of a lumican-deficient phenotype. Increased incidence of large-diameter fibrils with irregular cross-sectional profiles (thick black arrow) in the posterior stroma were observed in the 952delTDcn transgenic mouse model. Lumican has a critical role in regulating collagen fibrillogenesis in the corneal stroma.38 Lumican-null mice exhibit an increased incidence of large-diameter fibrils with irregular contours. This phenotype is more severe in posterior stroma where lumican localization is restricted in the mature cornea. Interestingly, we also observed a similar “lumican-deficient” phenotype in our 952delTDcn transgenic mouse localized to the posterior stroma. This is consistent with the depletion of posterior stromal-restricted lumican in our 952delTDcn transgenic mouse model. However, the phenotype in our model was more severe than in the lumican-null cornea, indicating a compound phenotype that results from dysfunctional expression of the normal corneal stroma SLRPs.
References
- 1.Birk D.E., Bruckner P. In: Mecham R.P., editor. Springer-Verlag; Berlin: 2011. Collagens, Suprastructures, and Collagen Fibril Assembly; pp. 77–115. [Google Scholar]
- 2.Iozzo R.V., Schaefer L. Proteoglycans in health and disease: novel regulatory signaling mechanisms evoked by the small leucine-rich proteoglycans. FEBS J. 2010;277:3864–3875. doi: 10.1111/j.1742-4658.2010.07797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaefer L., Iozzo R.V. Biological functions of the small leucine-rich proteoglycans: from genetics to signal transduction. J Biol Chem. 2008;283:21305–21309. doi: 10.1074/jbc.R800020200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bredrup C., Knappskog P.M., Majewski J., Rodahl E., Boman H. Congenital stromal dystrophy of the cornea caused by a mutation in the decorin gene. Invest Ophthalmol Vis Sci. 2005;46:420–426. doi: 10.1167/iovs.04-0804. [DOI] [PubMed] [Google Scholar]
- 5.Rodahl E., Van Ginderdeuren R., Knappskog P.M., Bredrup C., Boman H. A second decorin frame shift mutation in a family with congenital stromal corneal dystrophy. Am J Ophthalmol. 2006;142:520–521. doi: 10.1016/j.ajo.2006.03.064. [DOI] [PubMed] [Google Scholar]
- 6.Van Ginderdeuren R., De Vos R., Casteels I., Foets B. Report of a new family with dominant congenital heredity stromal dystrophy of the cornea. Cornea. 2002;21:118–120. doi: 10.1097/00003226-200201000-00025. [DOI] [PubMed] [Google Scholar]
- 7.Iozzo R.V. The biology of the small leucine-rich proteoglycans: Functional network of interactive proteins. J Biol Chem. 1999;274:18843–18846. doi: 10.1074/jbc.274.27.18843. [DOI] [PubMed] [Google Scholar]
- 8.McEwan P.A., Scott P.G., Bishop P.N., Bella J. Structural correlations in the family of small leucine-rich repeat proteins and proteoglycans. J Struct Biol. 2006;155:294–305. doi: 10.1016/j.jsb.2006.01.016. [DOI] [PubMed] [Google Scholar]
- 9.Bidanset D.J., Guidry C., Rosenberg L.C., Choi H.U., Timpl R., Hook M. Binding of the proteoglycan decorin to collagen type VI. J Biol Chem. 1992;267:5250–5256. [PubMed] [Google Scholar]
- 10.Font B., Eichenberger D., Rosenberg L.M., van der Rest M. Characterization of the interactions of type XII collagen with two small proteoglycans from fetal bovine tendon, decorin and fibromodulin. Matrix Biol. 1996;15:341–348. doi: 10.1016/s0945-053x(96)90137-7. [DOI] [PubMed] [Google Scholar]
- 11.Ehnis T., Dieterich W., Bauer M., Kresse H., Schuppan D. Localization of a binding site for the proteoglycan decorin on collagen XIV (undulin) J Biol Chem. 1997;272:20414–20419. doi: 10.1074/jbc.272.33.20414. [DOI] [PubMed] [Google Scholar]
- 12.Iozzo R.V., Goldoni S., Berendsen A., Young M. In: Mecham R.P., editor. Springer-Verlag; Berlin: 2011. Small leucine-rich proteoglycans; pp. 197–232. [Google Scholar]
- 13.Santra M., Reed C.C., Iozzo R.V. Decorin binds to a narrow region of the epidermal growth factor (EGF) receptor, partially overlapping but distinct from the EGF-binding epitope. J Biol Chem. 2002;277:35671–35681. doi: 10.1074/jbc.M205317200. [DOI] [PubMed] [Google Scholar]
- 14.Tralhao J.G., Schaefer L., Micegova M., Evaristo C., Schonherr E., Kayal S., Veiga-Fernandes H., Danel C., Iozzo R.V., Kresse H., Lemarchand P. In vivo selective and distant killing of cancer cells using adenovirus-mediated decorin gene transfer. FASEB J. 2003;17:464–466. doi: 10.1096/fj.02-0534fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schonherr E., Sunderkotter C., Iozzo R.V., Schaefer L. Decorin, a novel player in the insulin-like growth factor system. J Biol Chem. 2005;280:15767–15772. doi: 10.1074/jbc.M500451200. [DOI] [PubMed] [Google Scholar]
- 16.Grant D.S., Yenisey C., Rose R.W., Tootell M., Santra M., Iozzo R.V. Decorin suppresses tumor cell-mediated angiogenesis. Oncogene. 2002;21:4765–4777. doi: 10.1038/sj.onc.1205595. [DOI] [PubMed] [Google Scholar]
- 17.Buraschi S., Pal N., Tyler-Rubinstein N., Owens R.T., Neill T., Iozzo R.V. Decorin antagonizes Met receptor activity and down-regulates {beta}-catenin and Myc levels. J Biol Chem. 2010;285:42075–42085. doi: 10.1074/jbc.M110.172841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goldoni S., Humphries A., Nystrom A., Sattar S., Owens R.T., McQuillan D.J., Ireton K., Iozzo R.V. Decorin is a novel antagonistic ligand of the Met receptor. J Cell Biol. 2009;185:743–754. doi: 10.1083/jcb.200901129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scholzen T., Solursh M., Suzuki S., Reiter R., Morgan J.L., Buchberg A.M., Siracusa L.D., Iozzo R.V. The murine decorin: Complete cDNA cloning, genomic organization, chromosomal assignment, and expression during organogenesis and tissue differentiation. J Biol Chem. 1994;269:28270–28281. [PubMed] [Google Scholar]
- 20.Kao W.W. Ocular surface tissue morphogenesis in normal and disease states revealed by genetically modified mice. Cornea. 2006;25:S7–S19. doi: 10.1097/01.ico.0000247207.55520.a4. [DOI] [PubMed] [Google Scholar]
- 21.Birk D.E., Trelstad R.L. Extracellular compartments in matrix morphogenesis: collagen fibril, bundle, and lamellar formation by corneal fibroblasts. J Cell Biol. 1984;99:2024–2033. doi: 10.1083/jcb.99.6.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reed C.C., Iozzo R.V. The role of decorin in collagen fibrillogenesis and skin homeostasis. Glycoconj J. 2002;19:249–255. doi: 10.1023/A:1025383913444. [DOI] [PubMed] [Google Scholar]
- 23.Zhang G., Chen S., Goldoni S., Calder B.W., Simpson H.C., Owens R.T., McQuillan D.J., Young M.F., Iozzo R.V., Birk D.E. Genetic evidence for the coordinated regulation of collagen fibrillogenesis in the cornea by decorin and biglycan. J Biol Chem. 2009;284:8888–8897. doi: 10.1074/jbc.M806590200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maurice D.M. The structure and transparency of the cornea. J Physiol. 1957;136:263–286. doi: 10.1113/jphysiol.1957.sp005758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cornuet P.K., Blochberger T.C., Hassell J.R. Molecular polymorphism of lumican during corneal development. Invest Ophthalmol Vis Sci. 1994;35:870–877. [PubMed] [Google Scholar]
- 26.Pellegata N.S., Dieguez-Lucena J.L., Joensuu T., Lau S., Montgomery K.T., Krahe R., Kivela T., Kucherlapati R., Forsius H., de la Chapelle A. Mutations in KERA, encoding keratocan, cause cornea plana. Nat Genet. 2000;25:91–95. doi: 10.1038/75664. [DOI] [PubMed] [Google Scholar]
- 27.Pringle G.A., Dodd C.M. Immunoelectron microscopic localization of the core protein of decorin near the d and e bands of tendon collagen fibrils by use of monoclonal antibodies. J Histochem Cytochem. 1990;38:1405–1411. doi: 10.1177/38.10.1698203. [DOI] [PubMed] [Google Scholar]
- 28.Schonherr E., Witsch-Prehm P., Harrach B., Robenek H., Rauterberg J., Kresse H. Interaction of biglycan with type I collagen. J Biol Chem. 1995;270:2776–2783. doi: 10.1074/jbc.270.6.2776. [DOI] [PubMed] [Google Scholar]
- 29.Hedlund H., Mengarelli-Widholm S., Heinegard D., Reinholt F.P., Svensson O. Fibromodulin distribution and association with collagen. Matrix Biol. 1994;14:227–232. doi: 10.1016/0945-053x(94)90186-4. [DOI] [PubMed] [Google Scholar]
- 30.Kalamajski S., Oldberg A. Homologous sequence in lumican and fibromodulin leucine-rich repeat 5–7 competes for collagen binding. J Biol Chem. 2009;284:534–539. doi: 10.1074/jbc.M805721200. [DOI] [PubMed] [Google Scholar]
- 31.Keene D.R., San Antonio J.D., Mayne R., McQuillan D.J., Sarris G., Santoro S.A., Iozzo R.V. Decorin binds near the C terminus of type I collagen. J Biol Chem. 2000;275:21801–21804. doi: 10.1074/jbc.C000278200. [DOI] [PubMed] [Google Scholar]
- 32.Svensson L., Heinegard D., Oldberg A. Decorin-binding sites for collagen type I are mainly located in leucine-rich repeats 4–5. J Biol Chem. 1995;270:20712–20716. doi: 10.1074/jbc.270.35.20712. [DOI] [PubMed] [Google Scholar]
- 33.Chen S, Oldberg A, Chakravarti S, Birk DE: Fibromodulin regulates collagen fibrillogenesis during peripheral corneal development. Dev Dyn 239:844–854 [DOI] [PMC free article] [PubMed]
- 34.Carlson E.C., Liu C.Y., Chikama T., Hayashi Y., Kao C.W., Birk D.E., Funderburgh J.L., Jester J.V., Kao W.W. Keratocan, a cornea-specific keratan sulfate proteoglycan, is regulated by lumican. J Biol Chem. 2005;280:25541–25547. doi: 10.1074/jbc.M500249200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kao W.W., Liu C.Y. Roles of lumican and keratocan on corneal transparency. Glycoconj J. 2002;19:275–285. doi: 10.1023/A:1025396316169. [DOI] [PubMed] [Google Scholar]
- 36.Saika S., Liu C.Y., Azhar M., Sanford L.P., Doetschman T., Gendron R.L., Kao C.W., Kao W.W. TGFbeta2 in corneal morphogenesis during mouse embryonic development. Dev Biol. 2001;240:419–432. doi: 10.1006/dbio.2001.0480. [DOI] [PubMed] [Google Scholar]
- 37.Gutierrez J., Vial C., Santander C., Cabrera D., Brandan E. Decorin interacts with CTGF/CCN2 through LRR12 inhibiting its biological activity. J Biol Chem. 2011;286:24242–24252. doi: 10.1074/jbc.M110.189365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chakravarti S., Zhang G., Chervoneva I., Roberts L., Birk D.E. Collagen fibril assembly during postnatal development and dysfunctional regulation in the lumican-deficient murine cornea. Dev Dyn. 2006;235:2493–2506. doi: 10.1002/dvdy.20868. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
952delDcn mice exhibit characteristics of a lumican-deficient phenotype. Increased incidence of large-diameter fibrils with irregular cross-sectional profiles (thick black arrow) in the posterior stroma were observed in the 952delTDcn transgenic mouse model. Lumican has a critical role in regulating collagen fibrillogenesis in the corneal stroma.38 Lumican-null mice exhibit an increased incidence of large-diameter fibrils with irregular contours. This phenotype is more severe in posterior stroma where lumican localization is restricted in the mature cornea. Interestingly, we also observed a similar “lumican-deficient” phenotype in our 952delTDcn transgenic mouse localized to the posterior stroma. This is consistent with the depletion of posterior stromal-restricted lumican in our 952delTDcn transgenic mouse model. However, the phenotype in our model was more severe than in the lumican-null cornea, indicating a compound phenotype that results from dysfunctional expression of the normal corneal stroma SLRPs.