

Abstract
We have shown that substance P (SP) and its neurokinin-1 receptor (NK-1R) regulate intestinal angiogenesis by increasing expression of protein CYR61 (the cysteine-rich angiogenic inducer 61, or CCN1) in colonic epithelial cells. However, the mechanism involved in SP-induced CCN1 expression has not been studied, and the outcome of increased CCN1 expression in the development of colitis is not fully understood. Because histone deacetylase (HDAC) modulates transcription of several genes involved in inflammation, we investigated participation of HDAC in SP-induced CCN1 expression in human colonic epithelial NCM460 cells overexpressing NK-1R (NCM460-NK-1R) and in primary colonocytes. SP increased HDAC activity with deacetylation and dephosphorylation of nucleosome protein histone H3 in NCM460-NK-1R and/or primary colonocytes. Histone deacetylation and dephosphorylation was observed in colonic mucosa from irritable bowel disease patients. Similarly, colonic mucosal tissues from mice exposed to dextran sulfate sodium showed histone H3 deacetylation and dephosphorylation and increased HDAC activity that was reversed by the NK-1R antagonist CJ-12255. SP-induced increased CCN1 expression in NCM460-NK-1R cells was abolished by pharmacological HDAC inhibition. HDAC overexpression activated basal and SP-induced CCN1 promoter activity. Intracolonic CCN1 overexpression significantly ameliorated dextran sulfate sodium-induced colitis, with reduction of proinflammatory cytokine expression in mice. Thus, SP-mediated CCN1 expression in the inflamed human and mouse colon involves increased HDAC activity. Our results strongly suggest that increased CCN1 expression may be involved in mucosal healing during colitis.
Substance P (SP), an 11-amino-acid neuropeptide member of the tachykinin family isolated by Chang and Leeman,1 is localized in the central nervous system,2 enteric nerves,3 sensory neurons,4 and immune cells.5 SP binding to its high-affinity neurokinin-1 receptor (NK-1R) mediates important intestinal functions, including mucosal permeability,6 chloride secretion,7 motility,8 and inflammation.9 Interactions of SP and NK-1R promote inflammation via activation of cyclooxygenase-2 and secretion of PGE2 via JAK-STAT activation and by stimulating proinflammatory genes regulated by nuclear factor kappa B (NF-κB).10, 11, 12 Moreover, SP enhances mucosal healing by stimulating colonic epithelial cell proliferation that involves activation of metalloproteinases and transactivation of epithelial growth factor receptor (EGFR) and exerts anti-apoptotic effects via Akt phosphorylation in vivo and in vitro.13, 14, 15
We have previously reported that SP exerts proangiogenic effects by stimulating expression of CCN1 (recently redesignated as CYR61) in colonic epithelial cells.16 The CCN family consists of 30- to 40-kDa cysteine-rich proteins17 that stimulate cell proliferation, adhesion, apoptosis, extracellular matrix formation, angiogenesis, and tumor growth.18 CCN1 plays an essential role in vasculogenesis during embryogenesis.19, 20 Up-regulation of CCN1 is also evident in the colonic tissues of ulcerative colitis (UC) patients.16 However, the mechanism of CCN1 expression in colonic epithelial cells has never been investigated, and, except for angiogenesis, the outcome of its increased expression in the pathogenesis of colitis is not fully understood.
Histone deacetylases (HDACs) are a class of enzymes that remove acetyl groups from histone proteins.21 Their actions are opposite to those of histone acetyltransferase.21 Histones are found in nuclei of eukaryotic cells; they package DNA into nucleosomes and represent important components of chromatin.22 Histone H3 is a core histone that assembles DNA into nucleosomes.23 HDACs can regulate gene transcription through deacetylation of histone,24 indicating that histone H3 modifications are related to modulation of gene expression. Of note, previous results indicate that inhibition of HDAC results in amelioration of experimental colitis in mice,25 suggesting that HDAC may regulate expression of inflammation-related genes.
Given that SP is involved in colonic inflammation, we hypothesized that HDAC-related pathways may play a role in the SP-mediated colonic inflammation. Here, we report increased HDAC activity as well as histone H3 deacetylation and dephosphorylation in SP-exposed colonic epithelial cells, inflamed colon tissues of mice with experimental colitis, and colonic mucosa of patients with UC. HDAC activity in colonocytes is involved in SP-mediated CCN1 expression, and its overexpression in mouse colon reduces tissue damage in experimental colitis, implicating a healing role for CCN1 in the development of colitis.
Materials and Methods
Cell Cultures
NCM460 cells overexpressing NK-1R (NCM460-NK-1R), previously generated by us,10, 11, 14, 15 were cultured in M3D medium (INCELL, San Antonio, TX) containing 10% fetal calf serum and 1% penicillin/streptomycin (both from Invitrogen, Carlsbad, CA).14 Cells were treated with 0.1% trifluoroacetic acid as vehicle solution, SP, or human CCN1 protein (PeproTech, Rocky Hill, NJ) as indicated.16 Human primary colonic epithelial cells were isolated from colonic epithelial tissues of healthy subjects and from involved regions of UC patients after informed consent in accordance with procedures established by the Cedars-Sinai Institutional Review Board (IRB 3358). Epithelial cells were obtained as described previously.26 The top 0%/30% layer interface contained >90% pure epithelial cells. The primary cells were cultured in Dulbecco's modified Eagle's medium containing 10% fetal calf serum and 1% penicillin/streptomycin (both from Invitrogen). NCM460-NK-1R cells and human primary colonic epithelial cells were seeded on 24 well plates (2 × 106 cells/plate), unless otherwise specified. Cell viability had been checked by adherence to culture plates and exclusion of Trypan Blue dye.
Dextran Sodium Sulfate Colitis Model
Male 8- to 10-week-old C57BL/6 mice (n = 6/group) (Charles River Laboratories International, Wilmington, MA) were maintained at the animal facility under standard conditions. Animal studies were approved by the institutional animal care and use committees of Beth Israel Deaconess Medical Center and the University of California at Los Angeles. Mice received standard pelleted chow and tap water ad libitum, except that the colitis group received water containing dextran sulfate sodium (DSS) 5% (w/v), as described previously.10, 15 Groups of mice were also injected intraperitoneally with 200 μL PBS containing the NK-1R antagonist CJ-12255 (2.5 mg/kg/twice per day) or with PBS. After 5 days, mice were sacrificed by carbon dioxide euthanasia. Colon tissues were dissected and homogenized in immunoprecipitation buffer (Santa Cruz Biotechnology, Santa Cruz, CA), and equal amounts of protein (40 μg/lane) were subjected to Western blotting. Some colon tissues were also fixed in formalin and paraffin-embedded for immunohistochemistry.
Trinitrobenzene Sulfonic Acid Colitis Model
Mice were injected with either 30% ethanol in 100 μL as vehicle solution or with 100 mg/kg trinitrobenzene sulfonic acid (TNBS) in 30% ethanol intracolonically. A group of mice received sodium butyrate (0.3 mmol/kg of body weight) intraperitoneally every day for days 0, 1, and 2. After the end of day 2, colonic tissues were obtained for immunohistochemistry.
HDAC Activity Assay
HDAC activities in cell lysates or tissue homogenates in radioimmunoprecipitation assay buffer (∼50 μg/mL protein) were measured by colorimetric HDAC activity assay (BML-AK501; Enzo Life Sciences, Plymouth Meeting, PA) according to the manufacturer's instructions.
Enzyme-Linked Immunosorbent Assays
Immunoassays of colonic levels of mouse tumor necrosis factor α (TNFα) (DY410), IL-6 (DY406), and KC (redesignated as CXCL1) (DY453; R&D Systems, Minneapolis, MN) were performed according to the manufacturer's instructions.
Chromatin Immunoprecipitation
Serum-starved NCM460-NK-1R colonocytes were stimulated by vehicle 0.1% trifluoroacetic acid or SP (1 to 100 nmol/L) for 4 to 8 hours. Chromatin immunoprecipitation for determining the molecular association of histone H3 and CCN1 gene DNA was performed with a Pierce Agarose ChIP kit (no. 26156; Thermo Fisher Scientific, Rockford, IL). Cells were fixed and lysed by the reagents provided in the kit. Histone H3-CCN1 DNA complex was precipitated by anti-histone H3 polyclonal antibody (Pierce PA5-16183; Thermo Fisher Scientific). The precipitated protein was digested by proteinase K and the DNA was eluted. CCN1 DNA was detected by CCN1 primer (AJRR81V; Applied Biosystems, Foster City, CA) via real-time PCR. The CCN1 custom PCR primer set was automatically designed by Applied Biosystems based on the CCN1 genome sequence from 5′-CACAATGAGCCAGATTGCCC-3′ on chromosome 1, genome position from 86046664 to 86046803.
Intracolonic Overexpression of CCN1
CCN1 or an enhanced green fluorescent protein (eGFP)-overexpressing construct was transfected to colonic tissues in vivo, as described previously.16 Briefly, human CCN1 cDNA (clone TC310465; OriGene, Rockville, MD)-overexpressing construct mixed with a polyethyleneimine-based transfection reagent was intracolonically injected to anesthetized C57BL/6 mice. Mice were returned to consciousness and provided with 5% DSS in their drinking water or water alone for 5 days ad libitum. A separate group of mice were injected with the same amount of eGFP-expressing plasmid. The CCN1 transfection procedure was repeated on day 3, to increase transfection efficiency. Mice were monitored daily for body weight, stool consistency, and stool blood, with disease activity index DAI determined as described previously.27 Histological severity of colitis was determined by two investigators (H.W.K. and T.C.H.) in a blinded manner. Histology score measures combined parameters of crypt damage (score, 0 to 4), polymorphonuclear neutrophil (PMN) infiltrate (score, 0 to 3), edema (score, 0 to 3), erosion or ulceration (score, 0 to 3), and epithelial regeneration (score, 0 to 3), as described previously.28
Quantitative Real-Time PCR Analyses
Human irritable bowel disease (IBD) cDNA plates containing cDNA from 6 healthy subjects and 22 UC patients were obtained from OriGene (CCRT501). The PCR reactions were performed in an ABI Prism 7700 sequence detection system (Applied Biosystems, Carlsbad, CA) as described previously.16 The levels of mRNA were determined using the respective real-time primer set (HDAC1 Hs02621185_s1) obtained from Applied Biosystems according to the manufacturer's instructions. Levels were normalized to 18S mRNA using the respective primer set (Hs99999901_s1) and results were expressed as fold induction compared with their respective controls. All experiments were repeated three times.
Western Blot Analyses
Cells were lysed in 1× lysis buffer (no. 7722; Cell Signaling Technology, Danvers, MA). Equal amounts of cell extracts were fractioned by 10% SDS-PAGE, and proteins were transferred onto nitrocellulose membranes (Bio-Rad, Hercules, CA) with a 0.2 micron pore size at 400 mA for 2 hours at 4°C. Membranes were blocked in 5% nonfat milk in TBST (50 mmol/L Tris pH 7.5, 0.15 mol/L NaCl, 0.05% Tween 20), and then were incubated with antibodies against phospho-histone H3 Ser 10 (no. 9701; Cell Signaling Technology), acetyl-histone H3 Lys 9 (no. 9649S; Cell Signaling Technology), histone H3 (no. 9715; Cell Signaling Technology), CCN1 (sc-13100; Santa Cruz Biotechnology, Santa Cruz, CA), and β-actin (sc-47778; Santa Cruz Biotechnology). Horseradish peroxidase-labeled antibodies were detected by chemiluminescence (Fisher Scientific, Pittsburgh, PA), and signals were captured using an LAS4000 luminescent image analyzer (Fujifilm, Tokyo, Japan). In some experiments, Western blot bands were quantified by densitometry using Scion Image Analysis software version 1.1.
Promoter Activity of the CCN1 Gene
The DNA fragments of the CCN1 promoter (1.1 kb) upstream of the translational start site were cloned by PCR from human colonocyte genomic DNA and, after confirmation of the sequence identity, were subcloned into a pGL3 vector (pGL3-CCN1). NCM460-NK-1R cells were seeded in 12-well plates (2 × 106 cells/plate) overnight and transiently transfected with the pGL3-CCN1 construct together with an internal control pRL-TK (Promega, Madison, WI), as described previously.16 Transfected cells were serum-starved for 24 hours and then treated with SP for up to 24 hours. The relative promoter activity of CCN1 in equal amounts of cell extracts was measured using a dual luciferase reporter assay system (Promega). Empty pCMV6-XL5 vector, constructs overexpressing human HDAC1 (SC117054), human HDAC3 (SC112704), and human HDAC5 (SC124229) were purchased from OriGene. These constructs were transiently transfected to NCM460-NK-1R together with CCN1 promoter construct and pRL-TK control construct using Lipofectamine 2000 reagent (Invitrogen).
Immunohistochemistry
Human colonic tissue frozen sections were purchased from OriGene. Colon tissue sections were incubated with blocking buffer, followed by incubation with a rabbit polyclonal anti-phospho-histone H3 (Ser 10) antibody (no. 9701; Cell Signaling Technology) or acetyl histone H3 (Lys 9) antibody (no. 9649S; Cell Signaling Technology) overnight at 4°C. After a washing, sections were incubated with donkey anti-rabbit IgG and slides were stained with an ABC kit for color development (sc-2018; Santa Cruz Biotechnology). Immunohistochemistry experiments were performed with assistance of the Histology Core Facility of the University of California Los Angeles.
Measurement of Colitis Severity
The severity of DSS colitis was estimated by measuring body weight loss, stool consistency, and presence of fecal blood, leading to a clinical colitis score (scale, 0 to 4), as described previously.13, 15 The severity of TNBS colitis was estimated by measuring macroscopic damage score (scale, 0 to 10), as described previously.13
Statistical Analyses
Quantitative results are expressed as means ± SEM (as error bars within graphs). Results were analyzed using Prism professional statistics software program version 4 (GraphPad, La Jolla, CA). Student's t-tests were used for intergroup comparisons. P values of significant difference are reported within graphs.
Results
SP Induces HDAC Activities in Human Primary Colonic Epithelial Cells and Colonic Biopsies from IBD Patients
HDAC has been proposed as a key factor in the mediation of the inflammation.29 Inhibition of HDAC activity by pharmacological agents such as short chain fatty acid butyrate and red grape-derived resveratrol results in reduced inflammatory responses.30, 31 Because SP modulates intestinal inflammation,9 we examined whether this neuropeptide can modulate HDAC activity in human primary colonic epithelial cells. SP (100 nmol/L) stimulated HDAC activities only in the human primary colonic epithelial cells from involved colonic regions, but not in cells from normal or uninvolved regions, of UC and Crohn's disease (CD) patients (Figure 1A). This trend correlates with significantly higher expression level of SP receptor NK-1R in human primary colonic epithelial cells from involved colonic regions of UC and CD patients, compared with healthy control subjects (Figure 1B). High expression of NK-1R in human primary colonic epithelial cells from IBD patients is consistent with our previous finding of elevated NK-1R mRNA expression in the colonic tissues of IBD patients,16, 32 making these primary cells suitable for studying SP-dependent pathways. The uninvolved colonic regions of UC and CD patients express low level of NK-1R (data not shown). Consistent with enhanced HDAC activity, we observed increased deacetylated and dephosphorylated histone H3 at the epithelial lining of the colonic biopsies obtained from UC and CD patients (Figure 1C). Cells below the epithelial lining of normal colon tissues remained acetylated and phosphorylated, indicating lower HDAC activity (Figure 1C). Moreover, the acetylation and phosphorylation state of cells in the lamina propria and submucosa were similar across all groups (Figure 1C).
Figure 1.
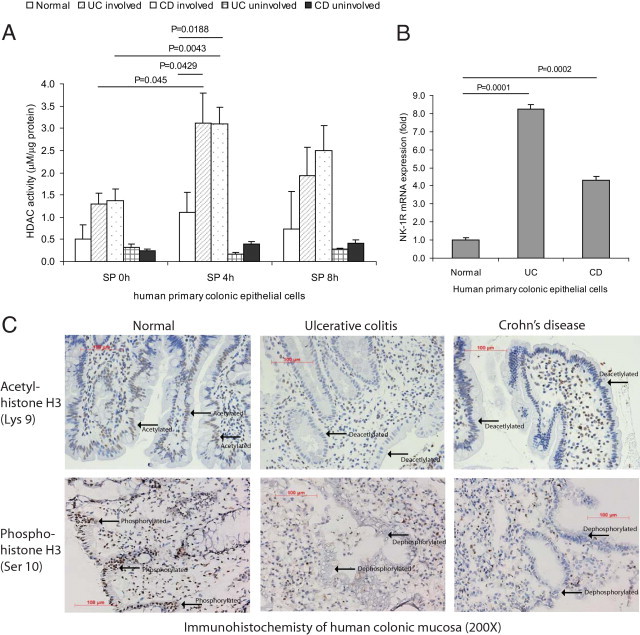
SP mediates histone deacetylase (HDAC) activity in human primary colonic epithelial cells and colons of IBD patients. A: Human primary colonic epithelial cells from normal, involved, and uninvolved regions of IBD patients were exposed to SP (100 nmol/L) for 0 to 8 hours. HDAC activities were measured. SP significantly stimulated HDAC activity of human primary colonic epithelial cells from involved regions of UC and CD patients; HDAC activities of normal and uninvolved regions of IBD patients remained low. B: Real-time RT-PCR showed that human primary colonic epithelial cells from UC and CD patients expressed significantly more NK-1R mRNA than those from healthy subjects. C: Immunohistochemistry of colonic biopsies from healthy subjects, UC patients, and CD patients was performed. The state of acetyl-histone H3 and phospho-H3 along the mucosal lining was observed (indicated by arrows) in samples from UC and CD patients, compared with colonic samples from healthy subjects. Results are representative of six patients per group. Original magnification, ×200. Scale bars = 100 μm.
SP-Dependent HDAC Activity in Mouse Colonic Mucosa with DSS-Induced Colitis
We next examined whether HDAC activity is dependent on the SP–NK-1R pathway, using a murine model of experimental DSS-induced colonic inflammation and an NK-1R specific antagonist (CJ-12255). As expected, DSS administration led to increase in colitis score, which was significantly reduced after CJ-12255 treatment (Figure 2B). Colonic levels of proinflammatory cytokines (TNFα, IL-6, and KC) in DSS-treated mice were significantly higher than those of water-treated mice, and they were significantly reduced by NK-1R antagonist CJ-12255 administration (Figure 2, D–F). Water-treated groups did not develop colitis, so their colitis scores are zero (Figure 2B) and the proinflammatory cytokine levels remain low (Figure 2, D–F). Similarly as in IBD patients, DSS-induced colitis in mice led to significantly higher colonic HDAC activity than was observed in the water-treated control group (Figure 2A). Administration of SP receptor antagonist (CJ-12255) significantly reduced colonic HDAC activity in the DSS-treated group (Figure 2A). CJ-12255 did not affect basal colonic HDAC activity among water-treated normal groups (Figure 2A). Deacetylation and dephosphorylation of histone H3 was also observed in the epithelial lining of DSS-exposed mouse colons, which were restored to an acetylated and phosphorylated state after CJ-12255 treatment (Figure 2C). The acetylation and phosphorylation states of the lamina propria and submucosal layer were similar across all groups. Colonic mucosal histone H3 of water-treated normal mice remained acetylated and phosphorylated (Figure 2C). CJ-12255 treatment did not alter the acetylation and phosphorylation states of histone H3, nor cytokine levels, in all water-treated control mice (Figure 2, C–F).
Figure 2.
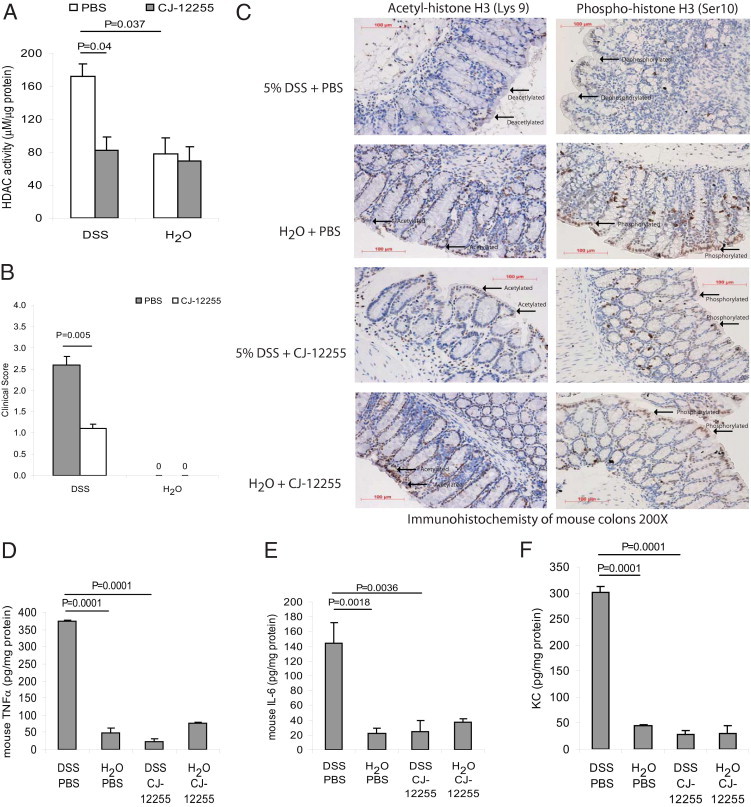
NK-1R mediates HDAC activity in colonic tissues of mice exposed to DSS. A: Mice were provided with water containing 5% DSS solution or water alone for 5 days. A separate group of mice were injected intraperitoneally with the NK-1R antagonist CJ-12255 or with PBS. HDAC activity in homogenized colonic tissues was measured. HDAC activity in inflamed colonic tissues is NK-1R-dependent. B: Clinical score of mice. Water-treated groups had no colitis, so the scores are zero. C: Acetylated and phosphorylated histone H3–positive cells were observed at the mucosa of normal colons exposed to water. Acetyl-histone H3-positive and phospho-histone H3-positive signals were reduced in inflamed colons of DSS-exposed mice, but were partially restored in DSS-exposed mice injected with NK-1R antagonist CJ-12255. This suggests that histone H3 deacetylation and dephosphorylation in colonic mucosa is NK-1R-dependent. Original magnification, ×200. Scale bars = 100 μm. D–F: Colonic levels of mouse proinflammatory cytokines TNFα (D), IL-6 (E), and KC (F). NK-1R antagonist CJ-12255 administration inhibited DSS colitis in mice. Results are representative of six mice per group.
We also found histone H3 deacetylation and dephosphorylation at the inflamed colonic epithelial lining of TNBS-exposed mice (see Supplemental Figure S1 at http://ajp.amjpathol.org). Administration of the HDAC inhibitor sodium butyrate partially reversed TNBS colitis and histone H3 to acetylated and phosphorylated states. These results are consistent with previous findings25 and indicate that colonic inflammation involves HDAC activity, which can be reduced by an HDAC inhibitor.
SP Induces HDAC Activity in Human Colonocytes
In addition to primary colonic epithelial cells, we also measured HDAC activity in nontransformed human colonocytes overexpressing NK-1R (NCM460-NK-1R). SP (10 nmol/L) significantly increased HDAC activity of NCM460-NK-1R cells between 4 and 8 hours; activity then returned to basal level (Figure 3A). Also, starting from 1 nmol/L, SP significantly induced HDAC activity in a concentration-dependent manner (Figure 3B). SP-dependent HDAC activity resulted in a concentration-dependent dephosphorylation and deacetylation of histone H3 in NCM460-NK-1R cells. At 100 nmol/L, SP almost abolished histone H3 phosphorylation and significantly reduced histone acetylation (Figure 3, C and D). SP-induced HDAC activity was blocked by both of the HDAC inhibitors, trichostatin A and sodium butyrate, indicating HDAC involvement in SP-mediated HDAC activity (Figure 3E). Neither HDAC inhibitor appeared to affect basal HDAC activity (Figure 3E).
Figure 3.
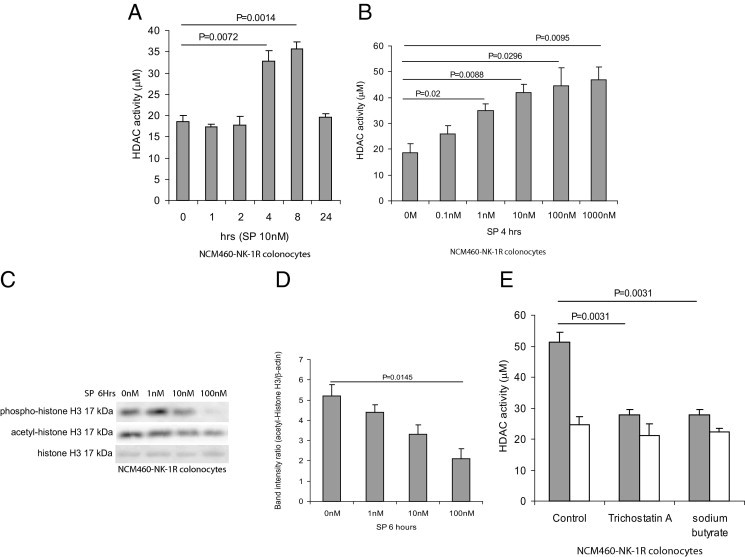
SP induces HDAC activities in human colonic epithelial cells. A: NCM460-NK-1R colonocytes were exposed to 10 nmol/L of SP for 0 to 24 hours. B: NCM460-NK-1R colonocytes were exposed to 0 to 1000 nmol/L of SP for 4 hours. HDAC activity in cell lysates was measured. SP induces HDAC activity in colonocytes. C: NCM460-NK-1R colonocytes were exposed to 0 to 100 nmol/L of SP for 6 hours. Phospho-histone H3, acetyl-histone H3, and total histone H3 were detected by Western blot analyses. D: Quantification of acetyl-histone signal showed that SP (100 nmol/L) significantly reduced histone H3 acetylation. Results are representative of three independent experiments. SP stimulates histone H3 dephosphorylation in colonocytes. E: NCM460-NK-1R colonocytes were treated with 1 μmol/L trichostatin A or 5 mmol/L sodium butyrate for 30 minutes, followed by SP (10 nmol/L) or 0.1% trifluoroacetic acid for 8 hours. HDAC activities in cell lysates were measured. The HDAC inhibitors trichostatin A and sodium butyrate significantly inhibit SP-mediated HDAC activity. Results are representative of three independent experiments.
SP Induces CCN1 Expression in Human Colonocytes via an HDAC-Dependent Mechanism
Similar to our previous findings in NCM460-NK-1R colonocytes,16 human primary colonic epithelial cells from UC and CD patients expressed significantly higher CCN1 mRNA, compared with cells from healthy subjects (Figure 4A). Additionally, we found that primary colonic epithelial cells from both UC and CD patients, but not from healthy individuals, expressed CCN1 mRNA in response to SP (Figure 4A).
Figure 4.
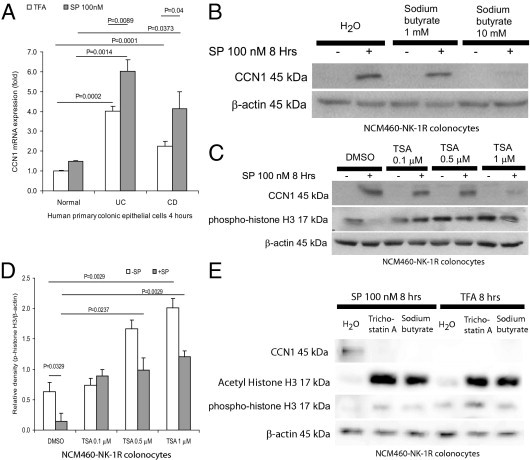
SP induces CCN1 expression in colonocytes via HDAC mechanism. A: Human primary colonic epithelial cells were exposed to trifluoroacetic acid or SP (100 nmol/L) for 4 hours. CCN1 mRNA levels were measured by real-time RT-PCR. Basal and SP-induced CCN1 expression levels from UC and CD patients were significantly higher than those from healthy subjects. B: NCM460-NK-1R cells were pretreated with 0 to 10 mmol/L of the HDAC inhibitor sodium butyrate for 30 minutes, followed by 100 nmol/L of SP exposure for 8 hours. Sodium butyrate treatment inhibited SP-induced CCN1 protein expression in colonocytes, as detected by Western blot analyses. C: NCM460-NK-1R cells were pretreated with 0 to 1 μmol/L of the HDAC inhibitor trichostatin A (TSA) or DMSO (vehicle) for 30 minutes, followed by 100 nmol/L of SP exposure for 8 hours. The levels of CCN1 and/or phospho-histone H3 were detected by Western blot analyses. Trichostatin A inhibits SP-induced CCN1 expression. D: Densitometric analyses of Figure 4C with phospho-histone H3 normalized to β-actin. Trichostatin A increases basal histone H3 phosphorylation and reverses SP-mediated histone H3 dephosphorylation. E: NCM460-NK-1R cells were pretreated with DMSO (vehicle), trichostatin A (1 μmol/L), or sodium butyrate (10 mmol/L) for 30 minutes, followed by 100 nmol/L of SP for 8 hours. SP-induced CCN1 expression was inhibited by both trichostatin A and sodium butyrate. Both inhibitors acetylated and phosphorylated histone H3. Results are representative of four independent experiments.
We next used the HDAC inhibitor sodium butyrate followed by SP exposure to determine whether SP-induced CCN1 expression is mediated by HDAC. At 10 mmol/L but not 1 mmol/L, sodium butyrate abolished SP-induced CCN1 expression in human colonocytes (Figure 4B). Another HDAC inhibitor, trichostatin A (1 μmol/L), reduced SP-induced CCN1 expression in NCM460-NK-1R (Figure 4C). At this concentration (1 μmol/L), trichostatin A reversed SP-mediated dephosphorylation and significantly phosphorylated histone H3 (Figure 4, C and D). Both trichostatin A and sodium butyrate abolished SP-induced CCN1 expression, but acetylated and phosphorylated histone H3 protein, indicating that both inhibitors greatly reduced HDAC activity in the cells (Figure 4E). Moreover, both of these HDAC inhibitors significantly reduced SP-induced and basal CCN1 promoter activity (Figure 5A).
Figure 5.
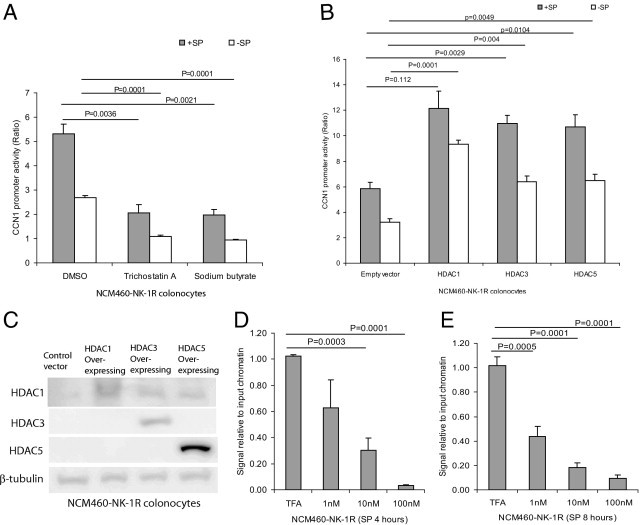
HDAC mediates CCN1 transcriptional activity in colonocytes. A: NCM460-NK-1R cells were transiently transfected with CCN1 promoter construct, followed by pretreatment with 1 μmol/L of trichostatin A, 10 mmol/L of sodium butyrate, or DMSO (vehicle) for 30 minutes and then exposure to SP (10 nmol/L) for 8 hours. CCN1 promoter activity was measured by luciferase assays. The HDAC inhibitors inhibited SP-induced CCN1 promoter activity. B: NCM460-NK-1R cells were transfected with empty vector or various isoforms of HDAC 1-, 3-, and 5-overexpressing constructs and CCN1 promoter construct, followed by exposure to SP (10 nmol/L) for 8 hours. CCN1 promoter activity was determined by luciferase assays. CCN1 promoter activity is mediated by multiple HDAC isoforms. C: HDAC 1, 3, and 5 protein expression after transient transfection of HDAC-overexpressing constructs into NCM460-NK-1R colonocytes. D and E: NCM460-NK-1R cells were exposed to SP (0 to 100 nmol/L) for 4 and 8 hours. Histone H3-DNA complex was immunoprecipitated and CCN1 DNA after immunoprecipitation was detected by real-time PCR. SP reduces association of histone H3 and CCN1 gene in colonocytes, providing access for CCN1 transcription. Results are representative of three independent experiments.
We also overexpressed HDAC isoforms 1, 3, and 5 via transient transfection of DNA constructs and examined CCN1 promoter activity in NCM460-NK-1R colonocytes. Overexpression of HDAC constructs significantly increased basal and SP-induced CCN1 promoter activity (Figure 5B), indicating that multiple HDAC isoforms mediate SP-induced CCN1 expression in colonocytes. Protein overexpression of HDAC 1, 3, and 5 isoforms was verified by Western blot analysis (Figure 5C).
To test the hypothesis that SP increases CCN1 transcription via increased HDAC activity and subsequent histone H3 modification, we used chromatin immunoprecipitation to determine the molecular association of histone H3 and the CCN1 gene (CYR61). Exposure of NCM460-NK-1R to SP for 4 and 8 hours (Figure 5, D and E) significantly decreased the CCN1 DNA signal, relative to input chromatin, after histone H3 immunoprecipitation in a concentration-dependent manner. This finding suggests that the histone H3 protein disassociated from the CCN1 gene on exposure to SP. The histone H3 disassociation from the CCN1 gene facilitates access of RNA polymerase and other transcription factors for CCN1 gene transcription.33
CCN1 Overexpression Reduces Colonic Mucosal Damage in DSS-Exposed Mice
We have previously reported that intracolonic CCN1 overexpression stimulates colonic angiogenesis during colitis in vivo.16 It is not known, however, whether increased CCN1 expression modulates the development of colitis. To directly examine the role of increased CCN1 expression in the colon, we transfected CCN1 or eGFP-overexpressing control constructs into mouse colon in vivo and then exposed animals to 5% DSS for 5 days. In vivo overexpression of CCN1 had been verified previously.16 Intracolonic CCN1 overexpression reduced tissue damage in DSS-exposed mice, as evidenced by a significant reduction of the colitis histology score (Figure 6, A and C) and DAI score of DSS-exposed mice (Figure 6B), both reflecting reduction of colitis severity. Moreover, CCN1 overexpression significantly reduced colonic levels of the proinflammatory cytokines TNFα, IL-6, and KC (Figure 6, D–F). Taken together, our data show that increased intracolonic CCN1 reduced tissue damage and inflammation in experimental DSS-induced colitis.
Figure 6.
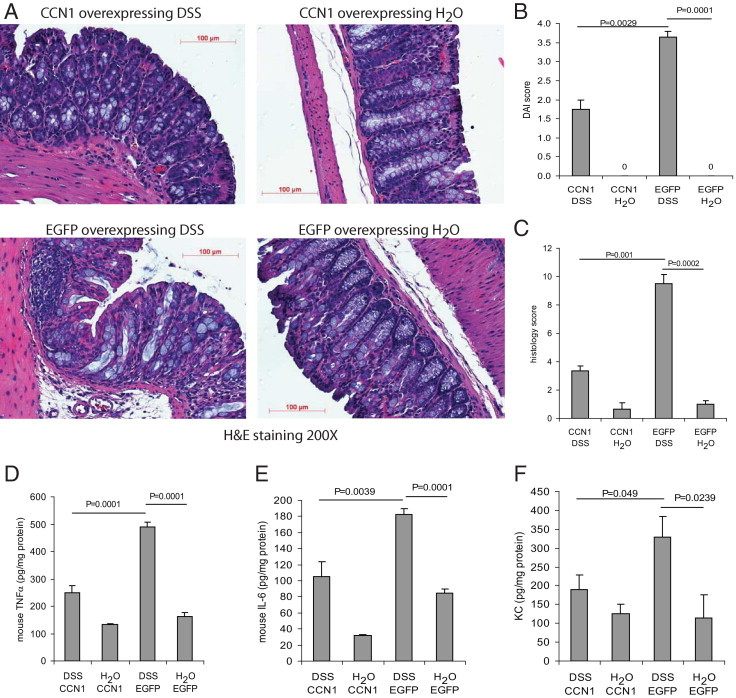
CCN1 reduces inflammation and mediates colonic healing in mice. Mice (C57BL/6, n = 4 per group) were transfected intracolonically with constructs overexpressing CCN1 and eGFP and then were treated with 5% DSS in their drinking water or water alone for 5 days. A: H&E staining shows that DSS colitis-induced tissue damage was reduced by CCN1 overexpression. Original magnification, ×200. Scale bars = 100 μm. B: Disease activity index (DAI) score and (C) histology score reflect reduction of inflammation and colonic tissue damage after CCN1 overexpression during colitis. Water-treated groups had no colitis, so the scores are zero. D–F: Colonic levels of mouse proinflammatory cytokines TNFα (D), IL-6 (E), and KC (F). CCN1 overexpression ameliorated DSS colitis and reduced colonic levels of proinflammatory cytokines. Results are representative of six mice.
Discussion
We previously reported that CCN1 expression is up-regulated in both murine colitis and in the inflamed colonic mucosa of IBD patients.16 This is mediated in part by its interaction with the SP–NK-1R signaling pathway.16 In the present study, we examined the mechanism by which SP stimulates CCN1 expression in human colonic epithelial cells and studied the consequences of increased colonic CCN1 expression in the development of experimental colitis. We found that SP via NK-1R induces CCN1 expression in colonocytes, at least in part by increasing HDAC activity. To our knowledge, increased HDAC activity with histone H3 deacetylation/dephosphorylation by the SP–NK1R signaling pathway has not been described previously. Moreover, our results showing that increased intracolonic CCN1 expression modulates experimental murine colitis indicate that CCN1 may be a novel therapeutic target for IBD.
Studies from our laboratory and others showed increased NK-1R in IBD mucosa, including colonic tissues from UC patients.16, 32 Our present results provide in vitro and in vivo evidence for increased HDAC activity with histone H3 deacetylation and dephosphorylation in response to SP–NK-1R interactions (Figure 3, Figure 4). Elevated HDAC activity may play a role in gut mucosal inflammation, because we found increased histone H3 deacetylation and dephosphorylation in the inflamed mucosa of UC and CD patients, as well as in experimental DSS colitis in mice (Figure 1C and Figure 2, B and C; see also Supplemental Figure S1 at http://ajp.amjpathol.org). HDAC inhibitors, including sodium butyrate (a bacterial metabolite short chain fatty acid), broccoli-derived sulforaphane, and red grape-derived resveratrol, may modulate inflammation and inhibit inflammation-associated dysplasia.34, 35, 36 Oral administration of two other HDAC inhibitors, valproic acid and suberoylanilide hydroxamic acid, results in hyperacetylation of histone H3 at the site of colonic inflammation and amelioration of DSS-induced and TNBS-induced colitis in mice.25 We also show that sodium butyrate treatment inhibited TNBS-induced colitis in mice and partially reversed histone H3 deacetylation and dephosphorylation at the epithelium (see Supplemental Figure S1 at http://ajp.amjpathol.org). Along these lines, antagonism of NK-1R inhibited HDAC activity and in turn reduced DSS-induced colitis (Figure 2), suggesting an important novel pathway for the therapeutic application of NK-1R antagonists in the development of colitis.
We show that HDAC isoforms 1, 3, and 5 may contribute to CCN1 expression (Figure 5). Of note, HDAC1 mRNA levels are significantly reduced in colonic cDNA from UC patients (see Supplemental Figure S2 at http://ajp.amjpathol.org). Together, histone H3 deacetylation and dephosphorylation in the inflamed colon of IBD patients may be caused by increased HDAC enzymatic activity rather than increased gene expression. Given that increased HDAC activity led to increased SP-induced and basal CCN1 promoter activity (Figure 5), the present study further addressed the molecular mechanism of HDAC-mediated histone modification in the transcription of CCN1. Our chromatin immunoprecipitation experiments showed SP-dependent dissociation of histone H3 protein from the CCN1 gene (Figure 5), suggesting that an SP-HDAC mechanism facilitates chromatin decondensation and subsequent gene transcription.
Given the mitogenic functions of HDAC activity, SP-mediated HDAC activity may contribute to healing during colitis. Increased HDAC activity had also been shown to stimulate gene transcription, including cyclin D1.37 Inhibition of HDAC activity suppresses gene transcription by interfering RNA polymerase II recruitment,38 and often leads to cell death.39 For the same reason, HDAC activity is necessary for gene transcription and cell survival. This finding is consistent with previous reports of SP-mediated protective function in colitis,13 and SP and CCN1 have been shown to mediate cell proliferation in astrocytoma cell.40, 41 With the present study, we have demonstrated that CCN1 overexpression in vivo can significantly reduce severity of DSS colitis and colonic tissue damage with reduction of cytokine levels (Figure 6). SP-mediated CCN1 expression may therefore promote wound healing and accelerated recovery from colitis.
Our previous studies indicate that SP–NK-1R interactions lead to both proinflammatory and mucosal healing intestinal responses by stimulating both proinflammatory (NF-κB and Stat5) and cell proliferative/anti-apoptotic pathways (EGFR, Akt). Although partial inhibition of NK-1R by CJ-12255 effectively inhibits murine model of colonic inflammation, complete NK-1R deficiency actually leads to severe colitis.13, 42, 43 Moreover, administration of CJ-12255 in the healing phase of DSS-induced colitis worsens histological and clinical signs of colitis and enhances colonic apoptosis.15 We show here for the first time that SP–NK-1R modulates expression of CCN1 through an HDAC-histone H3 pathway. Thus, CNN1, along with our previously identified EGFR and Akt signaling,14, 15 may participate in the prohealing responses to SP. The prohealing function of CCN1 is supported by our data showing that overexpression of CCN1 lowers histology score as well as proinflammatory cytokine levels during experimental DSS colitis (Figure 6).
In conclusion, SP mediates HDAC activity with histone H3 deacetylation and dephosphorylation in colonic epithelial cells colonocytes. This epigenetic modulation of transcriptional activity mediates proangiogenic and colonocyte growth factor CCN1 expression and in turn stimulates colonic angiogenesis16 and colonic healing. A schematic summary of the SP–CCN1 expression pathway is given in Figure 7.
Figure 7.
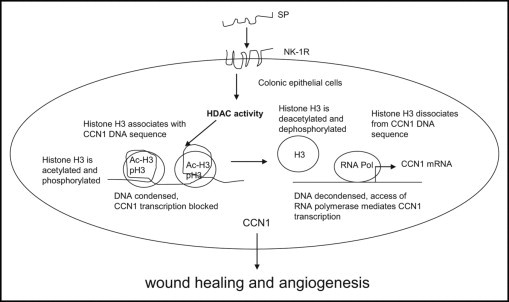
Schematic of SP-mediated HDAC pathway. SP activates HDAC activity, which in turn deacetylates and possibly dephosphorylates histone H3 in colonic epithelial cells. Under normal conditions, histone H3 is associated with chromatin and blocks CCN1 transcription. On exposure to SP, histone H3 dissociates from chromatin and opens up to access of transcriptional factors and RNA polymerases. This facilitates CCN1 transcription. CCN1 mediates wound healing and angiogenesis.
Acknowledgments
We thank Pfizer, Inc., for generously providing the NK-1R antagonist CJ-12255 and the Translational Pathology Core Laboratory (Department of Pathology, UCLA) for assistance in some of the histological staining procedures.
Footnotes
Supported in whole or in part by the NIH (grant DK47343 to C.P.), the Eli and Edythe Broad Chair (C.P.), a Research Fellowship Award and Career Development Award from the Crohn's and Colitis Foundation of America (H.W.K.), and the Blinder Research Foundation for Crohn's Disease (H.W.K.). The NK-1R antagonist CJ-12255 was provided by Pfizer, Inc.
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.07.038.
Supplementary data
A: Immunohistochemistry of acetyl-histone H3 and phospho-histone H3 of the trinitrobenzene sulfonic acid (TNBS) model. Compared with the control group, TNBS-exposed inflamed colons had deacetylation and dephosphorylation at the epithelium which was reversed by sodium butyrate treatment. Scale bars = 100 μm. B: Macroscopic damage score shows significant amelioration of colitis by butyrate treatment in TNBS-exposed mice. Results are representative of six mice per group.
Real-time RT-PCR of HDAC1 mRNA of colon biopsies from healthy subjects and ulcerative colitis patients. HDAC1 mRNA expression is lower in colon tissues of ulcerative colitis and Crohn's disease patients, compared with those of healthy subjects. Results are representative of three independent experiments.
References
- 1.Chang M.M., Leeman S.E. Isolation of a sialogogic peptide from bovine hypothalamic tissue and its characterization as substance P. J Biol Chem. 1970;245:4784–4790. [PubMed] [Google Scholar]
- 2.Mantyh P.W. Neurobiology of substance P and the NK1 receptor. J Clin Psychiatry. 2002;63(Suppl 11):6–10. [PubMed] [Google Scholar]
- 3.Costa M., Furness J.B., Franco R., Llewellyn-Smith I., Murphy R., Beardsley A.M. Substance P in nerve tissue in the gut. Ciba Found Symp. 1982:129–144. doi: 10.1002/9780470720738.ch8. [DOI] [PubMed] [Google Scholar]
- 4.Maggi C.A. Capsaicin-sensitive nerves in the gastrointestinal tract. Arch Int Pharmacodyn Ther. 1990;303:157–166. [PubMed] [Google Scholar]
- 5.Bost K.L. Quantification of macrophage-derived substance P receptor mRNA using competitive polymerase chain reaction. Adv Exp Med Biol. 1995;373:219–223. doi: 10.1007/978-1-4615-1951-5_30. [DOI] [PubMed] [Google Scholar]
- 6.Pothoulakis C., Castagliuolo I., LaMont J.T., Jaffer A., O'Keane J.C., Snider R.M., Leeman S.E. CP-96,345, a substance P antagonist, inhibits rat intestinal responses to Clostridium difficile toxin A but not cholera toxin. Proc Natl Acad Sci USA. 1994;91:947–951. doi: 10.1073/pnas.91.3.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Riegler M., Castagliuolo I., So P.T., Lotz M., Wang C., Wlk M., Sogukoglu T., Cosentini E., Bischof G., Hamilton G., Teleky B., Wenzl E., Matthews J.B., Pothoulakis C. Effects of substance P on human colonic mucosa in vitro. Am J Physiol. 1999;276:G1473–G1483. doi: 10.1152/ajpgi.1999.276.6.G1473. [DOI] [PubMed] [Google Scholar]
- 8.Holzer P., Holzer-Petsche U. Tachykinins in the gut: Part I. Expression, release and motor function. Pharmacol Ther. 1997;73:173–217. doi: 10.1016/s0163-7258(96)00195-7. [DOI] [PubMed] [Google Scholar]
- 9.Koon H.W., Pothoulakis C. Immunomodulatory properties of substance P: the gastrointestinal system as a model. Ann N Y Acad Sci. 2006;1088:23–40. doi: 10.1196/annals.1366.024. [DOI] [PubMed] [Google Scholar]
- 10.Koon H.W., Zhao D., Zhan Y., Rhee S.H., Moyer M.P., Pothoulakis C. Substance P stimulates cyclooxygenase-2 and prostaglandin E2 expression through JAK-STAT activation in human colonic epithelial cells. J Immunol. 2006;176:5050–5059. doi: 10.4049/jimmunol.176.8.5050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koon H.W., Zhao D., Zhan Y., Simeonidis S., Moyer M.P., Pothoulakis C. Substance P-stimulated interleukin-8 expression in human colonic epithelial cells involves protein kinase Cdelta activation. J Pharmacol Exp Ther. 2005;314:1393–1400. doi: 10.1124/jpet.105.088013. [DOI] [PubMed] [Google Scholar]
- 12.Zhao D., Kuhnt-Moore S., Zeng H., Pan A., Wu J.S., Simeonidis S., Moyer M.P., Pothoulakis C. Substance P-stimulated interleukin-8 expression in human colonic epithelial cells involves Rho family small GTPases. Biochem J. 2002;368:665–672. doi: 10.1042/BJ20020950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castagliuolo I., Morteau O., Keates A.C., Valenick L., Wang C.C., Zacks J., Lu B., Gerard N.P., Pothoulakis C. Protective effects of neurokinin-1 receptor during colitis in mice: role of the epidermal growth factor receptor. Br J Pharmacol. 2002;136:271–279. doi: 10.1038/sj.bjp.0704697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koon H.W., Zhao D., Na X., Moyer M.P., Pothoulakis C. Metalloproteinases and transforming growth factor-alpha mediate substance P-induced mitogen-activated protein kinase activation and proliferation in human colonocytes. J Biol Chem. 2004;279:45519–45527. doi: 10.1074/jbc.M408523200. [DOI] [PubMed] [Google Scholar]
- 15.Koon H.W., Zhao D., Zhan Y., Moyer M.P., Pothoulakis C. Substance P mediates antiapoptotic responses in human colonocytes by Akt activation. Proc Natl Acad Sci USA. 2007;104:2013–2018. doi: 10.1073/pnas.0610664104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koon H.W., Zhao D., Xu H., Bowe C., Moss A., Moyer M.P., Pothoulakis C. Substance P-mediated expression of the pro-angiogenic factor CCN1 modulates the course of colitis. Am J Pathol. 2008;173:400–410. doi: 10.2353/ajpath.2008.080222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brigstock D.R. The CCN family: a new stimulus package. J Endocrinol. 2003;178:169–175. doi: 10.1677/joe.0.1780169. [DOI] [PubMed] [Google Scholar]
- 18.Moussad E.E., Brigstock D.R. Connective tissue growth factor: what's in a name? Mol Genet Metab. 2000;71:276–292. doi: 10.1006/mgme.2000.3059. [DOI] [PubMed] [Google Scholar]
- 19.Brigstock D.R. Regulation of angiogenesis and endothelial cell function by connective tissue growth factor (CTGF) and cysteine-rich 61 (CYR61) Angiogenesis. 2002;5:153–165. doi: 10.1023/a:1023823803510. [DOI] [PubMed] [Google Scholar]
- 20.Mo F.E., Muntean A.G., Chen C.C., Stolz D.B., Watkins S.C., Lau L.F. CYR61 (CCN1) is essential for placental development and vascular integrity. Mol Cell Biol. 2002;22:8709–8720. doi: 10.1128/MCB.22.24.8709-8720.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y., Fang H., Jiao J., Xu W. The structure and function of histone deacetylases: the target for anti-cancer therapy. Curr Med Chem. 2008;15:2840–2849. doi: 10.2174/092986708786242796. [DOI] [PubMed] [Google Scholar]
- 22.Miremadi A., Oestergaard M.Z., Pharoah P.D., Caldas C. Cancer genetics of epigenetic genes. Hum Mol Genet. 2007;16(Spec No 1):R28–R49. doi: 10.1093/hmg/ddm021. [DOI] [PubMed] [Google Scholar]
- 23.Campos E.I., Reinberg D. Histones: annotating chromatin. Annu Rev Genet. 2009;43:559–599. doi: 10.1146/annurev.genet.032608.103928. [DOI] [PubMed] [Google Scholar]
- 24.Perez-Cadahia B., Drobic B., Davie J.R. H3 phosphorylation: dual role in mitosis and interphase. Biochem Cell Biol. 2009;87:695–709. doi: 10.1139/O09-053. [DOI] [PubMed] [Google Scholar]
- 25.Glauben R., Batra A., Fedke I., Zeitz M., Lehr H.A., Leoni F., Mascagni P., Fantuzzi G., Dinarello C.A., Siegmund B. Histone hyperacetylation is associated with amelioration of experimental colitis in mice. J Immunol. 2006;176:5015–5022. doi: 10.4049/jimmunol.176.8.5015. [DOI] [PubMed] [Google Scholar]
- 26.Mayer L., Shlien R. Evidence for function of Ia molecules on gut epithelial cells in man. J Exp Med. 1987;166:1471–1483. doi: 10.1084/jem.166.5.1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cooper H.S., Murthy S.N., Shah R.S., Sedergran D.J. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–249. [PubMed] [Google Scholar]
- 28.Ungaro R., Fukata M., Hsu D., Hernandez Y., Breglio K., Chen A., Xu R., Sotolongo J., Espana C., Zaias J., Elson G., Mayer L., Kosco-Vilbois M., Abreu M.T. A novel Toll-like receptor 4 antagonist antibody ameliorates inflammation but impairs mucosal healing in murine colitis. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1167–G1179. doi: 10.1152/ajpgi.90496.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Halili M.A., Andrews M.R., Sweet M.J., Fairlie D.P. Histone deacetylase inhibitors in inflammatory disease. Curr Top Med Chem. 2009;9:309–319. doi: 10.2174/156802609788085250. [DOI] [PubMed] [Google Scholar]
- 30.Zhang L., Jin S., Wang C., Jiang R., Wan J. Histone deacetylase inhibitors attenuate acute lung injury during cecal ligation and puncture-induced polymicrobial sepsis. World J Surg. 2010;34:1676–1683. doi: 10.1007/s00268-010-0493-5. [DOI] [PubMed] [Google Scholar]
- 31.Rahman I., Marwick J., Kirkham P. Redox modulation of chromatin remodeling: impact on histone acetylation and deacetylation, NF-kappaB and pro-inflammatory gene expression. Biochem Pharmacol. 2004;68:1255–1267. doi: 10.1016/j.bcp.2004.05.042. [DOI] [PubMed] [Google Scholar]
- 32.Goode T., O'Connell J., Anton P., Wong H., Reeve J., O'Sullivan G.C., Collins J.K., Shanahan F. Neurokinin-1 receptor expression in inflammatory bowel disease: molecular quantitation and localisation. Gut. 2000;47:387–396. doi: 10.1136/gut.47.3.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wegel E., Shaw P. Gene activation and deactivation related changes in the three-dimensional structure of chromatin. Chromosoma. 2005;114:331–337. doi: 10.1007/s00412-005-0015-7. [DOI] [PubMed] [Google Scholar]
- 34.Chen Z., Clark S., Birkeland M., Sung C.M., Lago A., Liu R., Kirkpatrick R., Johanson K., Winkler J.D., Hu E. Induction and superinduction of growth arrest and DNA damage gene 45 (GADD45) alpha and beta messenger RNAs by histone deacetylase inhibitors trichostatin A (TSA) and butyrate in SW620 human colon carcinoma cells. Cancer Lett. 2002;188:127–140. doi: 10.1016/s0304-3835(02)00322-1. [DOI] [PubMed] [Google Scholar]
- 35.Nian H., Delage B., Ho E., Dashwood R.H. Modulation of histone deacetylase activity by dietary isothiocyanates and allyl sulfides: studies with sulforaphane and garlic organosulfur compounds. Environ Mol Mutagen. 2009;50:213–221. doi: 10.1002/em.20454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kai L, Samuel SK, Levenson AS: Resveratrol enhances p53 acetylation and apoptosis in prostate cancer by inhibiting MTA1/NuRD complex. Int J Cancer 126:1538-1548 [DOI] [PubMed]
- 37.Hu J., Colburn N.H. Histone deacetylase inhibition down-regulates cyclin D1 transcription by inhibiting nuclear factor-kappaB/p65 DNA binding. Mol Cancer Res. 2005;3:100–109. doi: 10.1158/1541-7786.MCR-04-0070. [DOI] [PubMed] [Google Scholar]
- 38.Furumai R., Ito A., Ogawa K., Maeda S., Saito A., Nishino N., Horinouchi S., Yoshida M. Histone deacetylase inhibitors block nuclear factor-kappaB-dependent transcription by interfering with RNA polymerase II recruitment. Cancer Sci. 2011;102:1081–1087. doi: 10.1111/j.1349-7006.2011.01904.x. [DOI] [PubMed] [Google Scholar]
- 39.Dashwood R.H., Myzak M.C., Ho E. Dietary HDAC inhibitors: time to rethink weak ligands in cancer chemoprevention? Carcinogenesis. 2006;27:344–349. doi: 10.1093/carcin/bgi253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O'Kelly J., Chung A., Lemp N., Chumakova K., Yin D., Wang H.J., Said J., Gui D., Miller C.W., Karlan B.Y., Koeffler H.P. Functional domains of CCN1 (Cyr61) regulate breast cancer progression. Int J Oncol. 2008;33:59–67. [PubMed] [Google Scholar]
- 41.Castagliuolo I., Valenick L., Liu J., Pothoulakis C. Epidermal growth factor receptor transactivation mediates substance P-induced mitogenic responses in U-373 MG cells. J Biol Chem. 2000;275:26545–26550. doi: 10.1074/jbc.M003990200. [DOI] [PubMed] [Google Scholar]
- 42.Stucchi A.F., Shebani K.O., Leeman S.E., Wang C.C., Reed K.L., Fruin A.B., Gower A.C., McClung J.P., Andry C.D., O'Brien M.J., Pothoulakis C., Becker J.M. A neurokinin 1 receptor antagonist reduces an ongoing ileal pouch inflammation and the response to a subsequent inflammatory stimulus. Am J Physiol Gastrointest Liver Physiol. 2003;285:G1259–G1267. doi: 10.1152/ajpgi.00063.2003. [DOI] [PubMed] [Google Scholar]
- 43.Koon H.W., Shih D., Karagiannides I., Zhao D., Fazelbhoy Z., Hing T., Xu H., Lu B., Gerard N., Pothoulakis C. Substance P modulates colitis-associated fibrosis. Am J Pathol. 2010;177:2300–2309. doi: 10.2353/ajpath.2010.100314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A: Immunohistochemistry of acetyl-histone H3 and phospho-histone H3 of the trinitrobenzene sulfonic acid (TNBS) model. Compared with the control group, TNBS-exposed inflamed colons had deacetylation and dephosphorylation at the epithelium which was reversed by sodium butyrate treatment. Scale bars = 100 μm. B: Macroscopic damage score shows significant amelioration of colitis by butyrate treatment in TNBS-exposed mice. Results are representative of six mice per group.
Real-time RT-PCR of HDAC1 mRNA of colon biopsies from healthy subjects and ulcerative colitis patients. HDAC1 mRNA expression is lower in colon tissues of ulcerative colitis and Crohn's disease patients, compared with those of healthy subjects. Results are representative of three independent experiments.