

Abstract
In flow-associated pulmonary arterial hypertension (PAH), increased pulmonary blood flow is an essential trigger for neointimal formation. Using microarray analysis, we recently found that the early growth response protein 1 (Egr-1) transcription factor is increased in experimental flow-associated end-stage PAH. Its role in PAH development is unknown. Here, we assessed the spatiotemporal expression of Egr-1 during neointimal development in flow-associated PAH. Flow-associated PAH was produced in rats by combining monocrotaline administration with an aortocaval shunt. Animals were sacrificed 1 day before or 1 day, 1 week, or 4 to 5 weeks after flow addition. Egr-1 expression was spatiotemporally assessed using laser microdissection, quantitative real-time PCR and immunohistochemistry. In addition, Egr-1 expression was assessed in a non-neointimal pulmonary hypertension model and in human PAH associated with congenital shunt. In 4 to 5 weeks, rats subjected to increased flow developed PAH with neointimal lesions. Egr-1 mRNA was increased 1 day after flow addition and in end-stage PAH, whereas monocrotaline only did not result in increased Egr-1 mRNA. Directly after flow addition, Egr-1 was expressed in endothelial cells. During disease development, Egr-1 protein expression increased and migrated throughout the vessel wall. In PAH patients, Egr-1 was expressed in vessels with media hypertrophy and neointimal lesions, including plexiform lesions. Thus, Egr-1 may be an important regulator in the development of pulmonary neointimal lesions induced by increased pulmonary blood flow.
Pulmonary arterial hypertension (PAH) is a vasoproliferative disorder in which vascular obstruction of the small pulmonary arteries leads to an increased pulmonary vascular resistance and eventually death.1 PAH is considered irreversible when pulmonary vascular remodeling is characterized by the development of unique neointimal lesions, including concentric laminar intima fibrosis and plexiform lesions.2, 3 These neointimal lesions cause intraluminal obstruction arising from proliferation of endothelial and smooth muscle cells, fibrosis, and inflammation.2, 4, 5
In congenital heart disease, increased pulmonary blood flow is an essential trigger for neointimal formation and disease development. Although neointimal development is well-described histopathologically, the pathogenesis of PAH and its typical vascular lesions is largely unknown. Several animal models have been described to investigate the pathogenesis of PAH.6 The toxic alkaloid monocrotaline model is a commonly used experimental model of pulmonary hypertension. However, in this model neointimal lesions, typical for irreversible PAH,3 are not seen. We and other investigators have shown that combining monocrotaline with an increased pulmonary blood flow results in pulmonary neointimal lesions in end-stage disease.7, 8, 9, 10 Little is known about the development of these lesions during disease progression.
In a rat model of flow-associated PAH, we recently identified the early growth response protein 1 (Egr-1) transcription factor, by microarray analysis, to be up-regulated in end-stage PAH only when increased pulmonary blood flow was added to monocrotaline administration.8 Egr-1 is expressed in a variety of cardiovascular processes in systemic vessels, such as intimal thickening, vascular proliferation, and vessel inflammation.11, 12, 13, 14 Egr-1 is poorly expressed in the normal arterial wall but can be enhanced by various stimuli, including fluid shear stress and hypoxia.11, 15, 16, 17 Once activated, Egr-1 is capable of activating or repressing the transcription of numerous genes, including genes known to play a role in the pathogenesis of PAH, such as transforming growth factor β, platelet-derived growth factor, and monocytes chemotactic protein 1 (MCP1).11
On the basis of these observations, we hypothesized that Egr-1 may be an important factor in the development of flow-induced neointimal lesions in PAH. In this study, we therefore investigated the spatiotemporal expression of Egr-1 during the development of pulmonary neointimal lesions in experimental flow-associated PAH and compared this expression pattern with that in a non-neointimal pulmonary hypertension model (monocrotaline only) and in human PAH associated with a congenital shunt.
Materials and Methods
Animals
Animal care and experiments were approved by the Institutional Animal Care and Use Committee. Seventy-one male Wistar rats (310 to 380 g) were used. Experimental flow-associated PAH was produced using a monocrotaline injection (60 mg/kg) followed by an abdominal aortocaval shunt 1 week later, as described previously.7
To investigate the evolution of neointimal lesions, rats were randomly assigned to the following groups: i) monocrotaline 7 (M 7): 1 day before flow (n = 12), 1 week after monocrotaline administration, sacrificed before flow addition; ii) monocrotaline plus flow 8 (M+F 8): early activation (n = 12), sacrificed after 1 day of flow addition; iii) monocrotaline plus flow 14 (M+F 14): early-stage PAH (n = 12), sacrificed after 1 week of flow addition; and iv) monocrotaline plus flow 30 (M+F 30): end-stage PAH (n = 15), sacrificed when clinical signs of heart failure occurred, as described previously.7 For each time point, 5 rats receiving either saline injection or saline injection and sham surgery served as the control. Because the sham groups showed no differences in hemodynamic measurements, histologic findings, or mRNA and protein expression, these groups were pooled as a control group.
Furthermore, as an additional control group, we investigated Egr-1 expression in a non–flow-associated, nonneointimal model of pulmonary hypertension. Rats were injected with monocrotaline only and assigned to three additional experimental groups: i) monocrotaline 8 (M 8) (n = 5), sacrificed 8 days after monocrotaline injection; ii) monocrotaline 14 (M 14) (n = 5), sacrificed 14 days after monocrotaline injection; and iii) monocrotaline 30 (M 30) (n = 5), sacrificed 30 days after monocrotaline injection. The time points of sacrifice for these groups were similar to the flow-associated PAH groups mentioned above.
Hemodynamic Measurements
Directly before sacrifice invasive hemodynamic measurements were performed using a closed chest technique that is routinely used in our laboratory.7, 8 Rats were anesthetized with 3% to 5% isoflurane inhalation. A fluid-filled pressure catheter was inserted into the right internal jugular vein and guided to the pulmonary artery under pressure waveform monitoring using a bedside monitor. Mean right atrial pressure, right ventricular systolic pressure. and pulmonary arterial pressures were measured.
Pathologic Analysis
After hemodynamic measurements, organs were harvested and prepared for further analysis. The left lateral liver lobe was excised for wet/dry weight ratio measurement as an indication for right ventricular failure. Heart and lungs were excised and weighed separately. The right lung was frozen in liquid nitrogen for molecular analysis. The left lung was fixated by filling the airways with 3.6% formalin and embedded in paraffin.
Pulmonary vascular morphology (n = 5 rats per experimental group; n = 2 per sham group) was qualitatively and quantitatively analyzed as described previously by van Suylen et al and our laboratory.7, 8, 18 Intra-acinar vessels without a clearly defined internal lamina elastica combined with luminal occlusion were defined as neointimal lesions. Intra-acinar vessels with a double lamina elastica for more than half of its circumference were defined as completely muscular. Intra-acinar vessels with a double lamina elastica for less than half of its circumference were defined as partially muscular. Normal, nonmuscular, intra-acinar vessels had a single lamina elastica and no luminal occlusion. Muscularization per time point was assessed and presented as the percentage of vessels per degree of muscularization (including neointimal lesions).
Laser Microdissection
Cryosections (9 μm) from rat lung tissue were mounted on membrane-covered slides (PEN membrane; PALM Microlaser Technologies, Bernried, Germany). Intra-acinar vessels (1 × 106 μm2 of tissue per rat) from the M+F 30 group (n = 5 rats) and control group (n = 2 rats) were dissected using the Laser Robot Microbeam System (LMD6000; Leica, Wetzlar, Germany). Intra-acinar vessels were identified based on size (<50 μm) and morphologic findings.
Real-Time RT-PCR
Whole Lung
For real-time RT-PCR of whole lung samples (n = 5 to 8 per group), commercially available kits were used. Total RNA was isolated from pulmonary tissue using TRIzol reagent (Invitrogen, Carlsbad, CA). Real-time RT-PCR experiments were performed on a CXF384 real-time system C1000 Thermal cycler (BioRad Laboratories, Veenendaal, The Netherlands). cDNA was synthesized using QuantiTect Reverse Transcription Kit (Qiagen, Benelux, Venlo, the Netherlands). Real-time RT-PCR was conducted using SYBR Green PCR Master Mix according to the manufacturer's instructions (Eurogentec, San Diego, CA). Primer sequences are available on request. Expression levels were obtained from a dilution standard curve and compared with those of cyclophilin or 36B4 mRNA to calculate the relative expression levels.
Laser-Dissected Material
Total RNA was extracted from laser-dissected tissue using the RNAeasy Mini Kit (Qiagen, Leusden, Netherlands). DNA was removed using a RNase-free DNase set (Qiagen). RNA was quantified using the Nanodrop ND-1000 Spectrofotometer (Thermo Scientific, Waltham, MA). cDNA was synthesized using QuantiTect Rev. Transcription Kit (Qiagen). Primers and fluorogenic probes were purchased as assay-on-demand (Applied Biosystems Europe BV, Nieuwekerk aan de IJsel, Netherlands). Real-time RT-PCR was performed using an ABI 7900HT Sequence Detector (Applied Biosystems, Carlsbad, CA). RNA expression was normalized to the expression of GAPDH.
Immunohistochemistry
Paraffin-embedded lung sections were microwave heated for antigen retrieval. To reduce nonspecific staining, sections were preincubated in 0.3% hydrogen peroxide. Sections were then embedded for 1 hour at room temperature with primary antibodies for Egr-1 (rat: 1:50 dilution, Cell Signaling Technology Inc, Danvers, MA; human: 1:150 dilution, Abcam, Cambridge, MA), von Willebrand factor (rat: 1:250 dilution, Abcam), SMA (rat: 1:400 dilution, Dako, Carpinteria, CA), and CD68+ (1:300 dilution, AbD Serotec, Düsseldorf, Germany). Species-specific IgG coupled to peroxidase was used as a secondary antibody (1:100 dilution, Dako). The sections were stained with diaminobenzidine for 10 minutes and counterstained with hematoxylin. For immunofluorescence staining, the following secondary antibodies were used: Chromeo 488 IgG (1:400 dilution, Abcam) and Chromeo 546 IgG (1:50 dilution, Abcam). Nuclei were stained with DAPI (Vector Laboratories Inc, Burlingame, CA). For positive control for Egr-1 staining, prostate adenocarcinoma samples were used. Negative control (by replacing the primary antibody with PBS) showed no staining for all secondary antibodies.
Quantification of Immunohistochemical Analysis
Egr-1 staining was investigated (n = 5 rats per experimental group; n = 2 per sham group) to characterize spatiotemporal Egr-1 protein expression. Ten intra-acinar vessels were randomly selected per rat. Vessels were scored as followed: positive Egr-1 expression (0: no staining; 1: positive staining) and localization of Egr-1 expression [0: no staining; 1: mainly positive staining of endothelium; 2: mainly positive staining of media; and 3: diffuse positive staining (both) or perivascular staining]. Endothelium was defined as the cell layer bordering the vessel lumen. Media was defined as the layer between the outer elastic layer and the cell layer bordering the vessel lumen. In addition, the number of positive Egr-1 cells per 10 vessels was counted per experimental group.
For macrophage (CD68+) count, three rats per experimental group were analyzed. Ten intra-acinar vessels per rat were captured at ×400 magnification, and the number of macrophages was counted per field. For all analyses, slides were masked, making the experimental group unknown for the observers (M.G.D. and H.S.).
Patient Material
Human lung tissue samples were obtained from explanted lungs from five patients with PAH associated with congenital cardiac shunts that underwent lung transplantation. For control, lung tissue from two patients with disease-free lung parenchyma was obtained from autopsy.
Statistical Analysis
Data are presented as mean ± SEM. Differences between groups were determined by one-way analysis of variance with the Bonferroni post hoc test. For data not normally distributed, the Mann-Whitney U-test was performed. P < 0.05 was considered to be significant. Statistical analyses were performed using SPSS statistical software, version 16 (SPSS Inc, Chicago, IL).
Results
Development of Experimental Flow-Associated PAH
Surgery was performed successfully in all but four rats, which died due to perioperative complications: one rat assigned to the M+F 14 (early-stage PAH) group and three rats assigned to the M+F 30 (end-stage PAH) group. In addition, in the M+F 30 group three spontaneous deaths were seen in week 2 after increased flow induction. Necropsy revealed a considerable amount of pleural fluid in all three rats, which was presumed to be the cause of death.
In rats that received monocrotaline administration and an aortocaval shunt (M+F), increased mean pulmonary arterial pressure and peak systolic right ventricular pressure did not occur at M+F 8 (early activation; 1 day after flow induction) but were first seen at M+F 14 (early-stage PAH; 1 week after flow induction) (Table 1). Mean pulmonary arterial pressure and systolic right ventricular pressure further increased at M+F 30 (end-stage PAH) (Table 1). Increased right ventricular hypertrophy (Fulton index), right atrial pressure, and liver congestion (indications for right ventricular failure) were only seen at M+F 30 (end-stage PAH) (Table 1). The duration for rats in the M+F 30 group to develop severe, symptomatic PAH leading to sacrifice was 4 to 5 weeks (30 ± 3 days).
Table 1.
Animal Characteristics
| Parameter | Controls | M 7 | M+F 8 | M+F 14 | M+F 30 |
|---|---|---|---|---|---|
| Hemodynamics | |||||
| Mean RAP, mmHg | 3 ± 1 | 3 ± 1 | 3 ± 1 | 3 ± 1 | 9 ± 2⁎ |
| sRVP, mmHg | 25 ± 4 | 23 ± 2 | 23 ± 4 | 33 ± 3⁎ | 64 ± 12⁎ |
| sPAP, mmHg | 25 ± 4 | 22 ± 5 | 22 ± 3 | 32 ± 3⁎ | 60 ± 10⁎ |
| dPAP, mmHg | 11 ± 4 | 12 ± 4 | 14 ± 5 | 17 ± 4⁎ | 29 ± 7⁎ |
| mPAP, mmHg | 18 ± 3 | 17 ± 3 | 18 ± 2 | 25 ± 2⁎ | 42 ± 6⁎ |
| Heart rate/min | 343 ± 60 | 317 ± 40 | 341 ± 46 | 351 ± 20 | 246 ± 35⁎ |
| Pathology | |||||
| Body weight at day 1, g | 346 ± 20 | 382 ± 10 | 336 ± 13 | 332 ± 12 | 330 ± 16 |
| Body weight at sacrifice, g | 373 ± 24 | 379 ± 7 | 338 ± 14 | 327 ± 35 | 380 ± 35 |
| RV weight, g | 0.22 ± 0.03 | 0.21 ± 0.03 | 0.22 ± 0.03 | 0.26 ± 0.09 | 0.48 ± 0.04⁎ |
| Fulton index [RV/(IVS/LV)] | 0.29 ± 0.04 | 0.26 ± 0.04 | 0.3 ± 0.02 | 0.33 ± 0.02 | 0.57 ± 0.08⁎ |
| Liver wet/dry weight ratio | 2.8 ± 0.3 | 3.0 ± 0.2 | 3.1 ± 0.1 | 3.0 ± 0.2 | 3.4 ± 0.1⁎ |
| Pulmonary vascular remodeling | |||||
| Intra-acinar vessels (<50 μm) | |||||
| Wall thickness, μm | 0.1 ± 0.1 | 0.5 ± 0.2 | 1.0 ± 0.2 | 2.6 ± 1.0 | 9.3 ± 2.1⁎ |
| Wall/lumen ratio | 0.003 ± 0.002 | 0.022 ± 0.010 | 0.030 ± 0.015 | 0.112 ± 0.067 | 1.806 ± 1.562⁎ |
| Muscularization | |||||
| Nonmuscularized, % of vessels | 91.8 ± 5.9 | 65.5 ± 5.9⁎ | 44.7 ± 10.3⁎ | 22.8 ± 11.1⁎ | 0.0 ± 0.0⁎ |
| Partially muscularized, % of vessels | 7.4 ± 5.2 | 23.7 ± 6.5 | 32.9 ± 9.0⁎ | 27.4 ± 3.5⁎ | 2.5 ± 4.3 |
| Totally muscularized, % of vessels | 0.8 ± 1.1 | 6.9 ± 2.8 | 18.0 ± 7.0 | 43.0 ± 5.5⁎ | 22.4 ± 9.9⁎ |
| Neointima, % of vessels | 0.0 ± 0.0 | 0.5 ± 1.1 | 0.5 ± 1.1 | 4.7 ± 4.3 | 75.1 ± 11.0⁎ |
| Preacinar vessels (>50 μm) | |||||
| Wall thickness, μm | 7.1 ± 2.4 | 11.1 ± 2.6 | 10.3 ± 1.5 | 11.0 ± 3.2 | 19.6 ± 11.5 |
| Wall/lumen ratio | 0.07 ± 0.02 | 0.14 ± 0.03 | 0.11 ± 0.05 | 0.13 ± 0.06 | 0.19 ± 0.08⁎ |
Muscularization per experimental group is presented as the percentage of intra-acinar vessels per degree of muscularization (including neointimal lesions). Data are presented as mean ± SD from 5 to 10 rats per group.
dPAP, diastolic pulmonary arterial pressure; IVS, intraventricular septum; LV, left ventricle; mPAP, mean pulmonary arterial pressure; RAP, right atrial pressure; RV, right ventricle; sPAP, systolic pulmonary arterial pressure; sRVP, systolic right ventricular pressure.
P < 0.05 versus controls.
Development of Pulmonary Vascular Remodeling
Rats that received monocrotaline administration and an aortocaval shunt (M+F) showed progressive pulmonary vascular remodeling during PAH development (Table 1, Figure 1). Wall thickness and muscularization of the intra-acinar vessels occurred already 1 week after administration of monocrotaline and progressively increased over time. At M+F 14 (early-stage PAH), most intra-acinar vessels showed muscularization and sporadic neointimal lesions were observed. At M+F 30 (end-stage PAH), complex vascular lesions were seen in most intra-acinar vessels. These lesions caused vessel obliteration through both endothelial and smooth muscle cell proliferation (Figure 1, C and D). Immunofluorescence staining showed clear distinction between smooth muscle cells and endothelial cells within these lesions (Figure 1D). Vascular occlusive lesions were not seen in the preacinar vessels.
Figure 1.

Development of pulmonary vascular remodeling and neointimal lesions. A: Occlusion percentage of the intra-acinar vessels per experimental group (Materials and Methods). B: Muscularization per experimental group: percentage of intra-acinar vessels per degree of muscularization (including neointimal lesions). C: Typical examples (original magnification, ×400) of intra-acinar vessels per experimental group stained with Verhoeff. Note at M+F 30 the neointimal lesion with destruction of inner elastic membrane and disorganized cell proliferation. D: Typical examples of immunofluorescence double staining (original magnification, ×400) of intra-acinar vessels per experimental group: anti-SMA (red; smooth muscle cells), anti–von Willebrand factor (green; endothelial cells), and Dako (blue; nuclei). Note at M+F 30 luminal obstruction due to endothelial proliferation. Data are presented as mean ± SEM. *P < 0.05 versus controls. CON, pooled sham groups. Scale bar = 50 μm.
Increased Egr-1 Expression after Increased Blood Flow
One day before flow addition (M 7) Egr-1 levels were not increased (Figure 2A). The induction of increased pulmonary blood flow was followed by a nearly threefold increase of pulmonary Egr-1 mRNA expression (P < 0.05 versus control; Figure 2A). At M+F 14 (early-stage PAH), Egr-1 mRNA levels were decreased back to the control level. At M+F 30 (end-stage PAH), Egr-1 mRNA expression increased again, although not reaching significance using a Bonferroni post hoc analysis.
Figure 2.
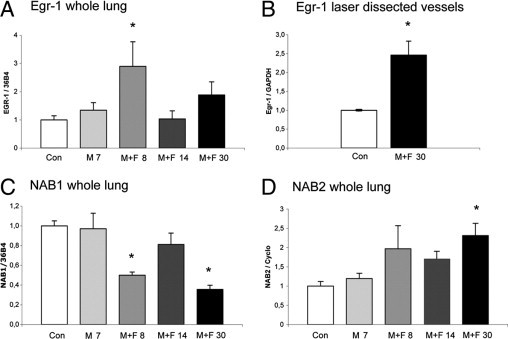
mRNA expression during pulmonary vascular remodeling in flow-associated PAH. A: Egr-1 expression relative to 36B4 per experimental group in whole lung. Bars indicate mRNA increase compared with controls. Expression levels in the control group are set to 1. Bars indicate mRNA increase compared with controls. B: Egr-1 expression relative to GAPDH in laser-dissected intra-acinar vessels at M+F 30 compared with laser-dissected intra-acinar vessels of controls. NAB1 expression relative to 36B4 (C) and NAB2 relative to cyclophilin (D) per experimental group in whole lung. Data are presented as mean ± SEM. *P < 0.05 versus controls. CON, pooled sham groups.
To investigate specifically the vascular expression of Egr-1 at end-stage PAH, we investigated mRNA expression in laser-dissected intra-acinar vessels. These vessels showed a 2.5-fold increase in Egr-1 at M+F 30 (end-stage PAH) compared with the control group (P < 0.05) (Figure 2B).
Two co-repressors of Egr-1, NAB1 (constitutive) and NAB2 (inducible), block Egr-1 activation through direct interaction. NAB1 mRNA expression showed a biphasic decrease, parallel with Egr-1 mRNA increase, namely at M+F 8 (early activation) and at M+F 30 (end-stage PAH; Figure 2C). At the same time points, NAB2 mRNA expression showed a similar up-regulation compared with Egr-1, however only reaching significance (P < 0.05 versus control) at M+F 30 (end-stage PAH; Figure 2D).
Progressive Increase of Egr-1 Protein Expression and Shift in Localization
Immunohistochemistry revealed only sporadic Egr-1 protein staining in pulmonary vessels of control rats and 1 day before flow addition (M 7). In contrast, strong Egr-1 staining was observed in intra-acinar vessels at M+F 8 (Figure 3). During disease development Egr-1 protein expression increased progressively with more than 90% of vessels staining positive for Egr-1 in end-stage disease (P < 0.05 versus control).
Figure 3.
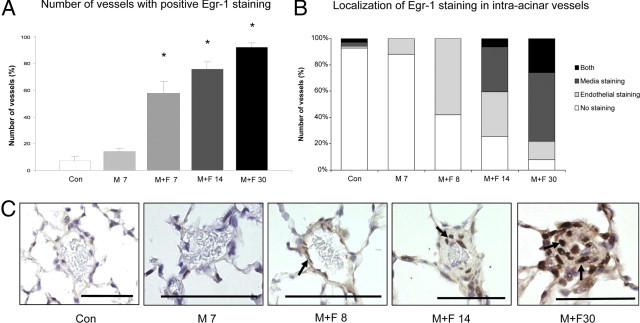
Egr-1 staining in intra-acinar vessels in flow-associated PAH. A: Percentage of intra-acinar vessels staining positive for Egr-1 per experimental group. B: Localization of Egr-1 staining in intra-acinar vessels per experimental group. C: Typical examples (original magnification, ×400) of Egr-1 staining (arrows) in intra-acinar vessels per experimental group. Note that Egr-1 staining is located in endothelial cells 1 day after flow addition (T8). During disease development, Egr-1 staining is seen in the intima and media layer with strong Egr-1 staining in neointimal lesions. Data are presented as mean ± SEM. *P < 0.05 versus controls. CON, pooled sham groups. Scale bar = 50 μm.
Qualitative analysis revealed that 1 day after increased pulmonary blood flow (M+F 8) Egr-1 was located mainly in endothelial cells (Figure 3, B and C). During disease development, Egr-1 showed a shift in localization. At M+F 14 (early-stage PAH) Egr-1 was mainly observed in the media layer of the vessel (P < 0.05 versus control). At M+F 30 (end-stage PAH) Egr-1 was seen throughout the intima and media layer (P < 0.001 versus control) with high production of Egr-1 in neointimal lesions. In the larger preacinar vessels, Egr-1 staining was only seen at M+F 30 (end-stage PAH; data not shown).
Only Sporadic Egr-1 Expression in Non-Neointimal Pulmonary Vascular Remodeling
To check whether Egr-1 expression is specifically up-regulated during flow-induced neointimal development, we investigated Egr-1 expression in a non-neointimal, non–flow-associated model of pulmonary hypertension, namely, monocrotaline only.
Four weeks after monocrotaline injection (M 30) rats developed pulmonary hypertension (mean systolic right ventricular pressure: 50 ± 14 mm Hg; mean Fulton index: 0.32 ± 0.03). Wall thickness and vessel occlusion increased over time (mean wall thickness: 3.0 ± 0.6 μm for the M 30 group versus 0.1 ± 0.1 μm for the control group; occlusion score: 18.2% ± 3.0% for the M 30 group versus 0.5% ± 0.2% for the control group, Figure 4E, Table 2) Neointimal lesions did not develop at M 30 (Table 2).
Figure 4.
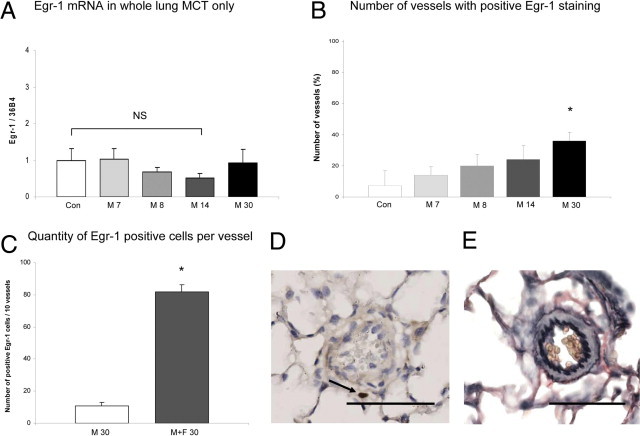
Egr-1 expression during pulmonary vascular remodeling in non-neointimal PH. To further emphasize the relationship among increased flow, Egr-1 up-regulation, and neointimal development, we investigated Egr-1 expression in a non-flow, non-neointmal PAH model (monocrotaline-only rats). See detailed description in Materials and Methods. A: Egr-1 expression relative to 36B4 per experimental group in whole lung. Bars indicate mRNA increase compared with controls. Expression levels in the control group are set to 1. Egr-1 mRNA expression is not up-regulated in whole lung in monocrotaline-induced vascular remodeling. B: Percentage of intra-acinar vessels staining positive for Egr-1 per experimental group. The number of vessels that show positive Egr-1 staining is increased in the M 30 group (*P < 0.05 versus controls). However, this number is far less compared with the number of vessels in the M+F 30 group as seen in Figure 3A. Also the number of positive Egr-1 cells per vessel in the end-stage groups differ (C). In addition, localization of Egr-1 staining differed in the M 30 group compared with M+F 30 group as seen in D. Typical example (original magnification, ×400) of Egr-1 staining in the M 30 group where Egr-1 staining was mainly found perivascularly. E: Typical example (original magnification, ×400) a Verhoeff staining of a peri-acinar vessel with media hypertrophy seen in the M 30 group. Administration of monocrotaline only never resulted in neointimal formation at M 30. Data are presented as mean ± SEM. *P < 0.05 versus controls. CON, pooled sham groups Scale bar = 50 μm.
Table 2.
Animal Characteristics (Monocrotaline Only)
| Parameter | Controls | M 7 | M 8 | M 14 | M 30 |
|---|---|---|---|---|---|
| Pathology | |||||
| Body weight at day 1, g | 346 ± 20 | 382 ± 10 | 314 ± 7 | 310 ± 8 | 253 ± 16 |
| Body weight at sacrifice, g | 373 ± 24 | 379 ± 7 | 323 ± 6 | 318 ± 12 | 340 ± 18 |
| RV weight, g | 0.22 ± 0.03 | 0.21 ± 0.03 | 0.22 ± 0.04 | 0.18 ± 0.01 | 0.24 ± 0.03 |
| Fulton index [RV/(IVS/LV)] | 0.29 ± 0.04 | 0.26 ± 0.04 | 0.27 ± 0.04 | 0.27 ± 0.03 | 0.32 ± 0.03 |
| Liver wet/dry weight ratio | 2.8 ± 0.3 | 3.0 ± 0.2 | 3.2 ± 0.09 | 3.3 ± 0.1 | 3.2 ± 0.2 |
| Pulmonary vascular remodeling | |||||
| Intra-acinar vessels (<50 μm) | |||||
| Wall thickness, μm | 0.1 ± 0.1 | 0.5 ± 0.2 | 0.9 ± 0.3⁎ | 1.5 ± 0.2⁎ | 3.0 ± 0.6⁎ |
| Wall/lumen ratio | 0.003 ± 0.002 | 0.022 ± 0.010 | 0.035 ± 0.01⁎ | 0.05 ± 0.008⁎ | 0.13 ± 0.03⁎ |
| Muscularization | |||||
| Nonmuscularized, % of vessels | 92 ± 6 | 66 ± 6⁎ | 59 ± 17⁎ | 36 ± 6⁎ | 13 ± 6⁎ |
| Partially muscularized, % of vessels | 7 ± 5 | 24 ± 7⁎ | 34 ± 8⁎ | 42 ± 5⁎ | 28 ± 10⁎ |
| Totally muscularized, % of vessels | 0.8 ± 1 | 6.9 ± 3⁎ | 7 ± 2⁎ | 22 ± 3⁎ | 60 ± 9⁎ |
| Neointima, % of vessels | 0.0 ± 0 | 0.5 ± 1.1 | 0 ± 0.0 | 0 ± 0.0 | 0 ± 0.0 |
| Preacinar vessels (>50 μm) | |||||
| Wall thickness, μm | 7 ± 2 | 11 ± 2 | 5 ± 1 | 6 ± 2 | 9 ± 2 |
| Wall/lumen ratio | 0.07 ± 0.02 | 0.14 ± 0.03⁎ | 0.07 ± 0.04 | 0.09 ± 0.01 | 0.08 ± 0.02 |
Muscularization per experimental group is presented as the percentage of intra-acinar vessels per degree of muscularization. Data are presented as mean ± SD from 5 to 10 rats per group.
IVS, intraventricular septum; LV, left ventricle; RV, right ventricle.
P < 0.05 versus controls.
Egr-1 mRNA expression did not increase at the selected time points (M7, M 8, M 14, M 30 groups; Figure 4A). Also no changes were seen in NAB1 or NAB2 expression after monocrotaline injection only (see Supplemental Figure S1 at http://ajp.amjpathol.org).
Immunohistochemistry revealed a moderate increase in Egr-1 protein expression only at M 30 (Figure 4B). This increase in Egr-1–positive vessels (Figure 4B) was far less compared with the number of vessels in the M+F 30 group (Figure 3A). In addition, the number of positive Egr-1 cells per vessel in the M 30 group was much lower compared with the M+F group (Figure 4C). When Egr-1 expression occurred, these cells were mainly located perivascularly (Figure 4D).
Increased MCP-1 Expression and Macrophage Activation in End-Stage PAH
MCP-1 is a downstream target of Egr-1. MCP-1 mRNA expression increased during vascular remodeling (P < 0.05 for the M+F 30 group versus controls; Figure 5A). MCP-1 was also increased in the M 30 group (P < 0.05 for the M group versus controls; Figure 5A). One of the effects of MCP-1 is macrophage infiltration. Macrophage count was highly increased in the M+F 30 group compared with the controls (P < 0.05) and compared with the M 30 group (P < 0.05; Figure 5, C–D).
Figure 5.
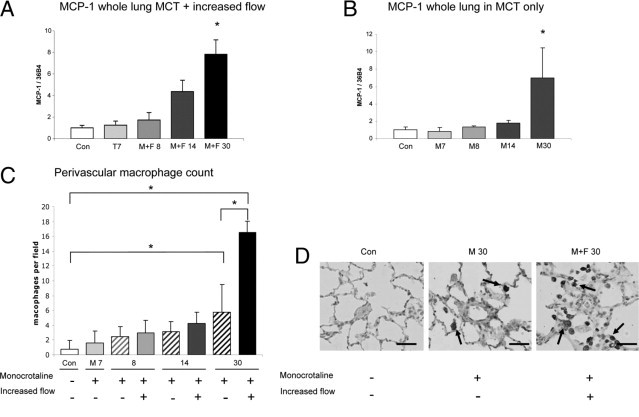
MCP-1 expression and macrophage infiltration during pulmonary vascular remodeling. MCP-1 expression relative to 36B4 per experimental group in whole lung of the M+F (A) and M (B) groups. Bars indicate mRNA increase compared with controls. Expression levels in the control group are set to 1. Bars indicate mRNA increase compared with controls. C: Perivascular macrophage count per field at ×400 magnification. D: Typical examples (original magnification, ×400) of CD68+ staining. Note the marked increase in macrophage infiltration around the neointimal lesion in the M+F 30 group compared with a normal vessel in the control group and a vessel with media hypertrophy in the M 30 group. Data are presented as mean ± SEM. *P < 0.05 versus controls. CON, pooled sham groups. Scale bar = 50 μm.
Increased Egr-1 Expression in Human PAH Is Associated with a Congenital Shunt
To study whether Egr-1 expression is also increased in human disease, we investigated Egr-1 expression in pulmonary vascular lesions of patients with end-stage PAH associated with congenital cardiac shunts. Egr-1 expression was nearly absent in pulmonary vessels from the controls (Figure 6, A and D). In contrast, strong Egr-1 staining was found in pulmonary vessels of end-stage flow-associated PAH patients (Figure 6, B, C, and E-H). In vessels with media hypertrophy (Figure 6, B and E) and plexiform lesions, Egr-1 was mainly found in the endothelial cells bordering the lumen (Figure 6, C, F, and G). In vessels with concentric intima fibrosis Egr-1 expression was found in cells throughout the vessel wall (Figure 6H).
Figure 6.
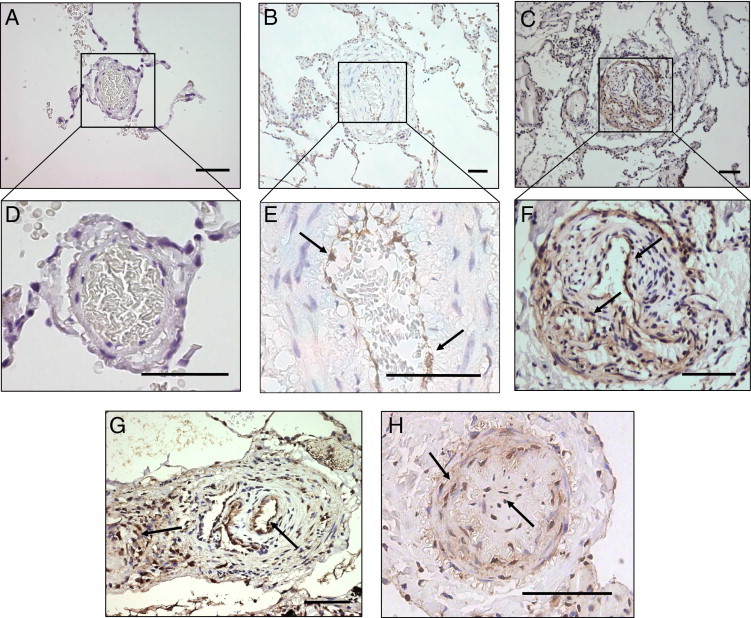
Egr-1 staining in human PAH lesions. A: Egr-1 staining of a normal lung vessel from a control subject free of lung disease (original magnification, ×200). B: Egr-1 staining of vessel with media hypertrophy in PAH patient associated with a congenital cardiac shunt (original magnification, ×200). C: Egr-1 staining of plexiform lesion in PAH patient associated with a congenital cardiac shunt (original magnification, ×200). D: Enlarged view of the same vessel in A, showing no Egr-1 staining. E: Enlarged view of the same vessel in B, showing marked Egr-1 staining in the endothelial layer. F: Enlarged view of the plexiform lesion in C, showing Egr-1 staining mainly in endothelial cells bordering the lumen (arrows). G: Egr-1 staining of a larger plexiform lesion (original magnification, ×100) showing as similar pattern of Egr-1 expression (arrows) compared with F. H: Egr-1 staining of a concentric intima lesion with diffuse staining throughout the vessel wall. Scale bar = 100 μm.
Discussion
In this study of experimental flow-associated PAH, we investigated the spatiotemporal expression of Egr-1 during the development of pulmonary neointimal lesions. In our PAH model, induction of increased pulmonary blood flow was followed by a biphasic response in Egr-1 mRNA expression, namely, a rapid response early after flow induction and a late response in end-stage PAH. During disease progression, Egr-1 protein expression in the intra-acinar vessels gradually increased. Moreover, with disease progression, Egr-1 protein expression migrated through the pulmonary vessel wall, from the endothelial cell layer to the media, with strong Egr-1 staining in neointimal lesions in end-stage PAH. Rats that were subjected to monocrotaline only did not show Egr-1 mRNA up-regulation, showed sporadic perivascular Egr-1 staining, and did not develop end-stage neointimal lesions. Increased Egr-1 expression during increased pulmonary blood flow was confirmed in end-stage lesions of PAH patients with a congenital cardiac shunt. These results suggest that Egr-1 plays an important role in the development of pulmonary vascular remodeling in flow-associated PAH, including the formation of neointimal lesions.
Egr-1 Transcription during Pulmonary Vascular Remodeling
Using microarray analysis, we recently identified Egr-1 to be specifically up-regulated by increased pulmonary blood flow in end-stage PAH.8 However, the spatiotemporal expression of Egr-1 during the development of neointimal lesions in flow-associated PAH has not yet been described. We show that during pulmonary vascular remodeling in flow-associated PAH, Egr-1 transcription is biphasic. It is plausible that in pulmonary vascular remodeling in flow-associated PAH, early Egr-1 up-regulation (after 24 hours) in endothelial cells is due to the direct increase in shear stress in the pulmonary vascular bed after the induction of increased flow. This finding is supported by the fact that we did not see Egr-1 up-regulation in the M 8 group (Figure 4). Also, previous studies have shown Egr-1 up-regulation in endothelial cells of the systemic vasculature directly after vessel injury, including vessel injury due to shear stress.19, 20, 21 In flow-associated PAH, shear stress caused by increased pulmonary blood flow is a prerequisite for neointimal development.1, 7 Because Egr-1 expression is increased specifically after flow induction and because Egr-1 is seen to progress throughout the vessel wall during which neointimal lesions develop, we suggest that early Egr-1 activation may play an important role in turning on the process of neointimal vascular remodeling in flow-associated PAH.
Egr-1 may also participate in chronic phases of neointimal development. In end-stage PAH (both human and experimental) Egr-1 is strongly expressed in both rat and human neointimal lesions. However, the location of Egr-1–positive cells in humans and rats appears different, especially in human plexiform lesions, where Egr-1 expression is predominantly seen in endothelial cells. This difference could be attributed to the fact that, even though the neointimal PAH rat model shows great similarities to the human disease, it obviously is not identical, illustrated by the fact that the typical plexiform lesions have not been described in a flow-associated rat model. Another possible explanation between the differences in Egr-1 expression could be formed by the fact that the human lung samples used were from end-stage PAH patients who all had previously received specific PAH treatment. This might have affected the pattern of Egr-1 expression in humans.
Besides Egr-1 production in endothelial cells subjected to high flow in end-stage disease, Egr-1 transcription is also seen in cells throughout the vessel wall (human concentric intima fibrosis, rat neointimal lesions) that are not subjected to flow (ie, shear stress). This observation shows that Egr-1 could also be up-regulated by other stressors at this time point, for instance by specific Egr-1 downstream genes, such as platelet-derived growth factor, or by hypoxia. Both might contribute to vascular remodeling in end-stage disease in PAH patients.3, 4, 5 In hypoxia, Egr-1 has been shown to be up-regulated in lungs22, 23 and isolated fibroblasts24 subjected to chronic hypoxia. Focusing on the pulmonary vessels, Yan and colleagues22 have shown that in lungs of mice subjected to hypoxia, Egr-1 is specifically produced in pulmonary vessel smooth muscle cell. However, these studies failed to show typical neointimal lesions, which indicates that hypoxia-driven Egr-1 expression alone does not lead to neointimal development. Interestingly, in another model, transforming growth factor α–induced pulmonary vascular remodeling worsened in Egr-1−/− mice.25 Future interventional studies are justified to investigate the role of these specific triggers in chronic phases of neointimal development and the effect of Egr-1 up-regulation on pulmonary vascular remodeling in flow-associated PAH.
In our study, Egr-1 mRNA expression showed a different pattern compared with that of Egr-1 protein expression. Although transcriptional expression in whole lung decreased in early-stage PAH, Egr-1 protein expression in the intra-acinar vessels gradually progressed. A possible explanation for this difference is that Egr-1 mRNA expression was assessed in whole lung tissue, whereas protein expression was investigated specifically in the pulmonary vessels. This could have made protein expression more profound, as supported by the more profound Egr-1 expression in the laser dissected vessels. In addition, Egr-1 itself is known to bind to its own gene, allowing Egr-1 to attenuate its own transcription.26 Such negative feedback could be a possible explanation for the down-regulation of Egr-1 transcription seen at early-stage PAH.22
For activity, Egr-1 is also dependent on the expression of its repressors NAB1 and NAB2. NAB2 is a downstream target of Egr-1 and is able to directly repress Egr-1 activity.26, 27 The fact that NAB2 showed a similar expression pattern compared with Egr-1 illustrates that the Egr-1 product is active during its up-regulation. NAB2 may have its direct effect on preventing a permanent transcription of Egr-1, as seen at early-stage PAH (M+F 14). In normal conditions Egr-1 up-regulation is suppressed by the constitutive expression of NAB1.26, 27 Little is known about factors that regulate NAB1 expression. However, the fact that NAB1 mRNA levels in whole lung were seen to decrease at the same time as Egr-1 mRNA levels increased suggests that NAB1 expression could be down-regulated by the same stressors that induce Egr-1 expression.
We also investigated MCP-1 transcription during disease development. MCP-1 is a downstream target of Egr-1 and a proinflammatory cytokine known to play a role in PAH.28 Although we observed an increase in MCP-1 expression in end-stage PAH, this increase could not solely be attributed to Egr-1 up-regulation due to increased flow. Namely, MCP-1 was also increased by monocrotaline only. Previously, studies have also shown that MCP-1 plays a role in monocrotaline-induced media hypertrophy.29 Apparently, in the described flow-associated PAH model, MCP-1 is also activated by other factors than Egr-1. Interestingly though, perivascular macrophage infiltration, a known result of MCP-1 activation, was threefold higher after increased pulmonary blood flow (M+F 30 versus M 30) and seen to be located around neointimal lesions. This finding shows that inflammatory processes, in this case macrophage infiltration, may contribute to neointimal development as seen in end-stage vascular remodeling in human PAH.29, 30, 31
Egr-1 is also located upstream of numerous other genes, such as platelet-derived growth factor3, 5, 32 and tissue factor,9 that participate in PAH.9 Egr-1 could thus be an important early factor in PAH by regulating different genes involved in neointimal development. In systemic vessels, the importance of Egr-1 expression during vascular remodeling has been confirmed given that neointima formation is reduced when Egr-1 expression is blocked.33, 34, 35, 36 In addition, these studies demonstrate that Egr-1–induced neointima formation is mediated through both proliferative and inflammatory pathways.33, 34, 35, 36
Taken together, this is the first study to demonstrate the spatiotemporal correlation between increased Egr-1 expression and increased pulmonary blood flow, as an inductor of neointimal development. These data suggest that Egr-1 may be able to initiate and maintain the process of neointimal development in flow-associated PAH and therefore also justifies further exploration of the exact role of Egr-1 in pulmonary vascular remodeling in PAH.
The spatiotemporal expression of Egr-1 in flow-associated PAH, as described for the first time in this study, identifies Egr-1 as a candidate gene to master the flow-induced pulmonary vascular remodeling process associated with the development of neointimal lesions. Further studies are needed to study the specific role of Egr-1 in flow-associated PAH.
Footnotes
Supplemental material for this article can be found at http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.07.030.
Supplementary Data
Whole lung expression of NAB1 and NAB2 relative to 36B4 per experimental group in rats that received monocrotaline only. Data are presented as mean ± SEM. CON, pooled sham groups.
References
- 1.Berger R.M. Possibilities and impossibilities in the evaluation of pulmonary vascular disease in congenital heart defects. Eur Heart J. 2000;21:17–27. doi: 10.1053/euhj.1999.1698. [DOI] [PubMed] [Google Scholar]
- 2.Wagenvoort C.A., Mooi W.J. In: Munro Neville A., Walker F., Gottlieb L.S., editors. Chapman and Hall Medical; London: 1989. Biopsy pathology of the pulmonary vasculature; pp. 56–113. [Google Scholar]
- 3.Sakao S., Tatsumi K., Voelkel N.F. Reversible or irreversible remodeling in pulmonary arterial hypertension. Am J Respir Cell Mol Biol. 2010;43:629–634. doi: 10.1165/rcmb.2009-0389TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Humbert M., Morrell N.W., Archer S.L., Stenmark K.R., MacLean M.R., Lang I.M., Christman B.W., Weir E.K., Eickelberg O., Voelkel N.F., Rabinovitch M. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:13S–24S. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- 5.Chan S.Y., Loscalzo J. Pathogenic mechanisms of pulmonary arterial hypertension. J Mol Cell Cardiol. 2008;44:14–30. doi: 10.1016/j.yjmcc.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stenmark K.R., Meyrick B., Galie N., Mooi W.J., McMurtry I.F. Animal models of pulmonary arterial hypertension: the hope for etiological discovery and pharmacological cure. Am J Physiol Lung Cell Mol Physiol. 2009;297:L1013–L1032. doi: 10.1152/ajplung.00217.2009. [DOI] [PubMed] [Google Scholar]
- 7.van Albada M.E., Schoemaker R.G., Kemna M.S., Cromme-Dijkhuis A.H., van Veghel R., Berger R.M. The role of increased pulmonary blood flow in pulmonary arterial hypertension. Eur Respir J. 2005;26:487–493. doi: 10.1183/09031936.05.00015405. [DOI] [PubMed] [Google Scholar]
- 8.van Albada M.E., Bartelds B., Wijnberg H., Mohaupt S., Dickinson M.G., Schoemaker R.G., Kooi K., Gerbens F., Rolf M.F., Berger R.M.F. Gene expression profile in flow-associated pulmonary arterial hypertension with neointimal lesions. Am J Physiol Lung Cell Mol Physiol. 2010;298:L483–L491. doi: 10.1152/ajplung.00106.2009. [DOI] [PubMed] [Google Scholar]
- 9.White R.J., Meoli D.F., Swarthout R.F., Kallop D.Y., Galaria I.I., Harvey J.L., Miller C.M., Blaxall B.C., Hall C.M., Pierce R.A., Cool C.D., Taubman M.B. Plexiform-like lesions and increased tissue factor expression in a rat model of severe pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol. 2007;293:L583–L590. doi: 10.1152/ajplung.00321.2006. [DOI] [PubMed] [Google Scholar]
- 10.Botney M.D. Role of hemodynamics in pulmonary vascular remodeling: implications for primary pulmonary hypertension. Am J Respir Crit Care Med. 1999;159:361–364. doi: 10.1164/ajrccm.159.2.9805075. [DOI] [PubMed] [Google Scholar]
- 11.Khachigian L.M. Early growth response-1 in cardiovascular pathobiology. Circ Res. 2006;98:186–191. doi: 10.1161/01.RES.0000200177.53882.c3. [DOI] [PubMed] [Google Scholar]
- 12.McCaffrey T.A., Fu C., Du B., Eksinar S., Kent K.C., Bush H., Jr, Kreiger K., Rosengart T., Cybulsky M.I., Silverman E.S., Collins T. High-level expression of Egr-1 and Egr-1-inducible genes in mouse and human atherosclerosis. J Clin Invest. 2000;105:653–662. doi: 10.1172/JCI8592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fahmy R.G., Khachigian L.M. Locked nucleic acid-modified DNA enzymes targeting early growth response-1 inhibit vascular smooth muscle cell growth. Nucleic Acids Res. 2004;32:2281–2285. doi: 10.1093/nar/gkh543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohtani K., Egashira K., Usui M., Ishibashi M., Hiasa K.I., Zhao Q., Aoki M., Kaneda Y., Morishita R., Takeshita A. Inhibition of neointimal hyperplasia after balloon injury by cis-element ‘decoy’ of early growth response gene-1 in hypercholesterolemic rabbits. Gene Ther. 2004;11:126–132. doi: 10.1038/sj.gt.3302153. [DOI] [PubMed] [Google Scholar]
- 15.Khachigian L.M., Collins T. Inducible expression of Egr-1-dependent genes: a paradigm of transcriptional activation in vascular endothelium. Circ Res. 1997;81:457–461. doi: 10.1161/01.res.81.4.457. [DOI] [PubMed] [Google Scholar]
- 16.Silverman E.S., Khachigian L.M., Santiago F.S., Williams A.J., Lindner V., Collins T. Vascular smooth muscle cells express the transcriptional corepressor NAB2 in response to injury. Am J Pathol. 1999;155:1311–1317. doi: 10.1016/S0002-9440(10)65233-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Houston P., Dickson M.C., Ludbrook V., White B., Schwachtgen J.L., McVey J.H., Mackman N., Reese J.M., Gorman D.G., Campbell C., Braddock M. Fluid shear stress induction of the tissue factor promoter In vitro and in vivo is mediated by Egr-1. Arterioscler Thromb Vasc Biol. 1999;19:281–289. doi: 10.1161/01.atv.19.2.281. [DOI] [PubMed] [Google Scholar]
- 18.van Suylen R.J., Smits J.F., Daemen M.J. Pulmonary artery remodeling differs in hypoxia- and monocrotaline-induced pulmonary hypertension. Am J Respir Crit Care Med. 1998;157:1423–1428. doi: 10.1164/ajrccm.157.5.9709050. [DOI] [PubMed] [Google Scholar]
- 19.Khachigian L.M., Lindner V., Williams A.J., Collins T. Egr-1-induced endothelial gene expression: a common theme in vascular injury. Science. 1996;271:1427–1431. doi: 10.1126/science.271.5254.1427. [DOI] [PubMed] [Google Scholar]
- 20.Schwachtgen J.L., Houston P., Campbell C., Sukhatme V., Braddock M. Fluid shear stress activation of Egr-1 transcription in cultured human endothelial and epithelial cells is mediated via the extracellular signal-related kinase 1/2 Mitogen-activated protein kinase pathway. J Clin Invest. 1998;11:2540–2549. doi: 10.1172/JCI1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khachigian L.M., Anderson K.A., Halnon N.J., Resnick N., Gimbrone M.A., Jr., Collins T. Egr-1 is activated in endothelial cells exposed to fluid shear stress and interacts with a novel shear-stress response element in the PDGF A-chain promoter. Arterioscl Thromb Vasc Biol. 1997;17:2280–2286. doi: 10.1161/01.atv.17.10.2280. [DOI] [PubMed] [Google Scholar]
- 22.Yan S.F., Lu J., Xu L., Zou Y.S., Tongers J., Kisiel W., Mackman N., Pinsky D.J., Stern D.M. Pulmonary expression of early growth response-1: biphasic time course and effect of oxygen concentration. J Appl Physiol. 2000;88:2303–2309. doi: 10.1152/jappl.2000.88.6.2303. [DOI] [PubMed] [Google Scholar]
- 23.Nozik-Grayck E., Suliman H.B., Majka S., Albietz J., Van Rheen Z., Roush K., Stenmark K.R. Lung EC-SOD overexpression attenuates hypoxic induction of Egr-1 and chronic hypoxic pulmonary vascular remodeling. Am J Physiol Lung Cell Mol Physiol. 2008;295:L422–L430. doi: 10.1152/ajplung.90293.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banks M.F., Gerasimovskaya E.V., Tucker D.A., Frid M.G., Carpenter T.C., Stenmark K.R. Egr-1 antisense oligonucleotides inhibit hypoxia-induced proliferation of pulmonary artery adventitial fibroblasts. J Appl Physiol. 2005;98:732–738. doi: 10.1152/japplphysiol.00821.2004. [DOI] [PubMed] [Google Scholar]
- 25.Kramer E.L., Mushaben E.M., Pastura P.A., Acciani T.H., Deutsch G.H., Khurana Hershey G.K., Korfhagen T.R., Hardie W.D., Whitsett J.A., Le Cras T.D. Early growth response-1 suppresses epidermal growth factor receptor–mediated airway hyperresponsiveness and lung remodeling in mice. Am J Respir Cell Mol Biol. 2009;41:415–425. doi: 10.1165/rcmb.2008-0470OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thiel G., Cibelli G. Regulation of life and death by the zinc finger transcription factor Egr-1. J Cell Physiol. 2002;193:287–292. doi: 10.1002/jcp.10178. [DOI] [PubMed] [Google Scholar]
- 27.Russo M.W., Sevetson B.R., Milbrandt J. Identification of NAB1, a repressor of NGFI-A- and Krox20-mediated transcription. Proc Natl Acad Sci U S A. 1995;92:6873–6877. doi: 10.1073/pnas.92.15.6873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanchez O., Marcos E., Perros F., Fadel E., Tu L., Humbert M., Darteyelle P., Simonneau G., Adnot S., Eddahibi S. Role of endothelium-derived CC chemokine ligand 2 in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2007;176:1041–1047. doi: 10.1164/rccm.200610-1559OC. [DOI] [PubMed] [Google Scholar]
- 29.Ikeda Y., Yonemitsu Y., Kataoka C., Kitamoto S., Yamaoka T., Nishida K., Takeshita A., Egashira K., Sueishi K. Anti-monocyte chemoattractant protein-1 gene therapy attenuates pulmonary hypertension in rats. Am J Physiol Heart Circ Physiol. 2002;283:H2021–H2028. doi: 10.1152/ajpheart.00919.2001. [DOI] [PubMed] [Google Scholar]
- 30.Hassoun P.M., Mouthon L., Barbera J.A., Eddahibi S., Flores S.C., Grimminger F., Jones P.L., Maitland M.L., Michelakis E.D., Morrell N.W., Newnam J.H., Rabinovitch M., Schermuly R., Stenmark K.R., Voelkel N.F., Yuan J.X.J., Humbert M. Inflammation, growth factors, and pulmonary vascular remodeling. J Am Coll Cardiol. 2009;54:S10–S19. doi: 10.1016/j.jacc.2009.04.006. [DOI] [PubMed] [Google Scholar]
- 31.Hamada H., Terai M., Kimura H., Hirano K., Oana S., Niimi H. Increased expression of mast cell chymase in the lungs of patients with congenital heart disease associated with early pulmonary vascular disease. Am J Respir Crit Care Med. 1999;160:1303–1308. doi: 10.1164/ajrccm.160.4.9810058. [DOI] [PubMed] [Google Scholar]
- 32.Perros F., Montani D., Dorfmuller P., Durand-Gasselin I., Tcherakian C., Le P.J., Mazmanian M., Fadel E., Mussot S., Mercier O., Hervé P., Emilie D., Eddahibi S., Simonneau G., Souza R., Humbert M. Platelet-derived growth factor expression and function in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2008;178:81–88. doi: 10.1164/rccm.200707-1037OC. [DOI] [PubMed] [Google Scholar]
- 33.Santiago F.S., Lowe H.C., Kavurma M.M., Chesterman C.N., Baker A., Atkins D.G., Khachigian L.M. New DNA enzyme targeting Egr-1 mRNA inhibits vascular smooth muscle proliferation and regrowth after injury. Nat Med. 1999;5:1264–1269. doi: 10.1038/15215. [DOI] [PubMed] [Google Scholar]
- 34.Lowe H.C., Fahmy R.G., Kavurma M.M., Baker A., Chesterman C.N., Khachigian L.M. Catalytic oligodeoxynucleotides define a key regulatory role for early growth response factor-1 in the porcine model of coronary in-stent restenosis. Circ Res. 2001;89:670–677. doi: 10.1161/hh2001.097867. [DOI] [PubMed] [Google Scholar]
- 35.Fahmy R.G., Khachigian L.M. Suppression of growth factor expression and human vascular smooth muscle cell growth by small interfering RNA targeting EGR-1. J Cell Biochem. 2007;100:1526–1535. doi: 10.1002/jcb.21145. [DOI] [PubMed] [Google Scholar]
- 36.Santiago F.S., Atkins D.A., Khachigian L.M. Vascular smooth muscle cell proliferation and regrowth after injury in vitro is dependent upon NGFI-A/Egr-1. Am J Pathol. 1999;155:897–905. doi: 10.1016/S0002-9440(10)65189-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Whole lung expression of NAB1 and NAB2 relative to 36B4 per experimental group in rats that received monocrotaline only. Data are presented as mean ± SEM. CON, pooled sham groups.