

Abstract
Anti-inflammatory properties of protein C (PC) concentrate are poorly studied compared to activated protein C, although PC is suggested to be safer in clinical use. We investigated how PC interferes with the leukocyte recruitment cascade during acute inflammation and its efficacy during murine endotoxemia. We found that similar to activated protein infusion, intravenous PC application reduced leukocyte recruitment in inflamed tissues in a dose- and time-dependent manner. During both tumor necrosis factor-α induced and trauma-induced inflammation of the cremaster muscle, intravital microscopy revealed that leukocyte adhesion and transmigration, but not rolling, were profoundly inhibited by 100 U/kg PC. Moreover, PC blocked leukocyte emigration into the bronchoalveolar space during lipopolysaccharide (LPS) induced acute lung injury. PC was efficiently activated in a murine endotoxemia model, which reduced leukocyte infiltration of organs and strongly improved survival (75% versus 25% of control mice). Dependent on the inflammatory model, PC provoked a significant inhibition of leukocyte recruitment as early as 1 hour after administration. PC-induced inhibition of leukocyte recruitment during acute inflammation critically involves thrombomodulin-mediated PC activation, subsequent endothelial PC receptor and protease-activated receptor-1-dependent signaling, and down-regulation of intercellular adhesion molecule 1 leading to reduced endothelial inflammatory response. We conclude that during acute inflammation and sepsis, PC is a fast acting and effective therapeutic approach to block leukocyte recruitment and improve survival.
Protein C (PC) is a vitamin K-dependent anticoagulant protein mainly synthesized by the liver, but also by endothelial cells, various leukocytes, and keratinocytes. PC is activated by thrombin bound to thrombomodulin (TM).1, 2 The conversion to activated protein C (APC) is augmented by endothelial protein C receptor (EPCR), which is besides other cell types also expressed on endothelial cells,3, 4, 5 pointing toward an important role of blood vessels to independently synthesize and activate PC to mediate APC triggered functions.1, 6, 7
During the past decade, tremendous research was performed on APC and its antithrombotic, pro-fibrinolytic, anti-apoptotic effects, and recently its cytoprotective properties.1, 4, 8 These APC-induced effects are partially independent of APCs antithrombotic function.9 In vitro studies demonstrated that APC controls leukocyte trafficking,5, 10, 11 and there is increasing evidence that APC reduced leukocyte infiltration during inflammation in vivo.1, 4, 8, 12, 13, 14 This anti-inflammatory effect may relate in part to a direct binding of APC to β1, β2, and β3-integrins.11, 15 The majority of APC-induced cytoprotective effects, however, is mediated by receptor-dependent mechanisms, including an interaction with EPCR and activation of the protease-activated receptor-1 (PAR-1).1, 16 A subsequent inhibition of NF-κB translocation reduces the production of proinflammatory cytokines and expression of cell adhesion molecules, such as intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin,17 leading to impaired leukocyte recruitment.
During inflammation, the cascade of leukocyte recruitment is initiated by the capture of free-flowing leukocytes to the vessel wall, followed by leukocyte rolling along and adhesion to the inflamed endothelial layer.18, 19 During rolling, leukocytes get into intimate contact with the endothelial surface, which allows endothelial bound chemokines to interact with their specific receptors on the leukocyte surface. This triggers the activation of β2-integrins, which leads to firm leukocyte arrest on the endothelium and finally to leukocyte transmigration.18, 19
Given its anti-inflammatory properties, APC was evaluated in clinical studies, revealing a reduced mortality in severely septic patients receiving APC.20, 21, 22, 23, 24, 25 Uncertainties, however, regarding the efficacy and safety of APC during septic shock still persist, stemming from a reportedly increased risk for serious bleeding in APC-treated patients.21, 23 Notably, there is first evidence that protein C concentrate exerts anti-inflammatory properties similar to APC, but in contrast to APC with reduced side effects.5, 7, 10, 26, 27, 28, 29, 30 During a clinical study, de Kleijn et al10 showed that protein C concentrate safely and effectively improves survival in septic patients with meningococcemia. Clinically used, PC doses varied from 50 U/kg to 600 U/kg per day depending on the underlying disease (ie, congenital or acquired PC deficiencies and severe sepsis).10, 28, 29, 30, 31, 32 Notably, a systematic in vivo investigation of a dose-dependent anti-inflammatory effect of PC concentrate is lacking so far. Moreover, none of the published protein C studies compared PC and APC therapy, and systematically investigated leukocyte recruitment in vivo, including rolling, firm adhesion, and transmigration of leukocytes taking PC activation and signaling into account.
This prompted us to elucidate how PC interferes with the cascade of leukocyte recruitment under in vivo conditions using intravital microscopy in different acute models of inflammation. Furthermore, we investigated its optimal timing, dosing, and possible mechanisms of action. Our findings reveal that PC effectively blocks leukocyte adhesion during inflammation and therefore may be considered as an alternative therapeutic approach in treating patients with inflammatory diseases.
Materials and Methods
Animals
C57BL/6 mice and lymphocyte function–associated antigen 1 (Lfa-1−/−) mice were purchased from Charles River and maintained at the Central Animal Facility of the University of Heidelberg, Germany. For all experiments, mice were at least 8 weeks of age. Icam-1−/− mice were generated as previously described, and were backcrossed for at least 15 generations into the C57BL/6 background.33 Thrombomodulin mutant mice (TMPro/Pro) were kindly provided by Hartmut Weiler (Blood Research Institute, Milwaukee, WI). They were generated using a targeting construct with a point mutation of the TM gene (Q404P) resulting in a strongly diminished TM-dependent PC activating capacity to approximately 5%, as previously described in detail.2 All animal experiments were approved by the Animal Care and Use Committee of the Regierungspraesidium Karlsruhe, Germany (AZ 35–9185.81/G-142/07 and AZ 35–9185.81/G-8/08).
Protein C, Cytokines, and Special Reagents
Protein C concentrate (Ceprotin) was kindly provided from Baxter (Unterschleissheim, Germany), dissolved as indicated in the drug data sheet to an isotonic working solution of 100 U/mL protein C (1 U = 4 μg PC), containing 8 mg/mL human serum albumin for stabilizing reasons. Isotonic human serum albumin (Sigma-Aldrich, Munich, Germany) at 8 mg/mL served as the standard control solution. PC solution and control solution were further dissolved in normal saline to 200 μL and were intravenously administered at indicated doses and time points during inflammation. For dose finding experiments, isotonic solution of human serum albumin at 64 mg/mL (equivalent to 800 U/kg PC) served as control solution of high-dose PC administration (800 U/kg PC). In all other experiments, PC was administered at 100 U/kg referring to 400 μg/kg.
Activated Protein C (Xigris; Eli Lilly, Bad Homburg, Germany) was diluted in normal saline to a working solution of 3 μg/mL and was systemically injected at 24 μg/kg/hour at 3 hours before microscopic observation or as indicated.
In certain experiments, the EPCR antibody RCR252 (30 μg/mouse; Abcam, Cambridge, UK) directed to the PC binding domain of EPCR or the selective PAR-1 Inhibitor SCH79797 (44 μg/mouse, Tocris Bioscience, Bristol, UK34) was injected into the tail vein 5 minutes before PC application.
In designated in vivo experiments, (recombinant murine) tumor necrosis factor-α (TNF-α) (R&D, Minneapolis, MN) was applied intrascrotally at 500 ng per mouse, recombinant murine keratinocyte-derived chemokine (Peprotech, London, UK) was systemically injected with a dose of 600 ng/mouse.
Coagulation Assays
To investigate the coagulation parameters during PC therapy, whole blood of protein C treated and control mice was obtained by cardiac puncture 3 hours after application of variable doses of PC in TNF-α-induced inflammation. Using citrated plasma samples, prothrombin time, partial thromboplastin time, fibrinogen, and protein C levels were measured by the laboratory core facility of the Department of Clinical Chemistry, University of Heidelberg, Germany. For prothrombin time, partial thromboplastin time and fibrinogen standard assays were performed. Levels of zymogen protein C were measured photometrically using a chromogenic substrate (PCa; American Diagnostica, Greenwich, CT) crossreacting with human and murine protein C.
Activity of human protein C was analyzed as previously described,35, 36 with some modifications. In detail, animals were injected with 100 U/kg of human PC (Ceprotin, Baxter) into the tail vein. As positive controls, in some experiments 50 milliunits of human α-thrombin (Hemochrom Diagnostica, Essen, Germany) were injected 10 minutes before blood sampling. In another set of experiments, animals were continuously injected with activated Protein C (Xigris; Eli Lilly, Bad Homburg, Germany) at 24 μg/kg/hour. At different time points after PC or APC administration, blood was taken as a final blood sample from the vena cava into 0.38% sodium citrate and 50 mmol/L benzamidine HCl. Human activated protein C was captured from plasma samples using an antibody specific for human activated protein C 1555 (from a kind gift of C.T. Esmon, Oklahoma City, OK), and the activity of the captured human PC was determined using a chromogenic substrate (PCa; American Diagnostica).4
Intravital Microscopy
As recently reported, we used the cremaster muscle models of trauma induced and TNF-α-induced inflammation.37 Briefly, for the preparation of intravital microscopy after i.p. injection of ketamine (125 mg/kg body weight; Pfizer, Karlsruhe, Germany) and xylazine (12.5 mg/kg body weight; Alverta, Neumuenster, Germany) mice were placed on a heating pad to maintain body temperature. Intravital microscopy was conducted on an upright microscope (Leica; Wetzlar, Germany) with a saline immersion objective (SW40/0.75 numerical aperture; Zeiss, Jena, Germany). Mice were intubated and the left carotid artery was cannulated for blood sampling and systemic antibody administration.
Cremaster Muscle Preparation
The surgical preparation of the cremaster muscle was conducted as previously described (trauma-induced inflammation).37 In certain experiments, recombinant murine CXCL-1 chemokine keratinocyte-derived chemokine was injected systemically at a dose of 600 ng/mouse. In another set of experiments, mice were injected with 500 ng TNF-α via intrascrotal 3 hours before intravital microscopy (TNF-α-induced inflammation). Microscopic observation of cremaster muscle venules of 20- to 40-μm diameter were recorded via CCD camera (CF8/1; Kappa, Gleichen, Germany) on a Panasonic S-VHS recorder (Panasonic, Hamburg, Germany). The number of adherent leukocytes (firm adhesion for >30) was assessed as adherent cells per mm2 vessel surface area. Rolling leukocyte flux fraction was defined as the percentage of rolling leukocytes to all leukocytes passing the same vessel in 1 minute.37 Individual leukocyte rolling velocities were measured from video recordings by analyzing 5 to 15 leukocytes/venule and measuring frame-to-frame displacement of rolling leukocytes.38
In a separate set of experiments, cremaster muscle whole mounts were obtained as previously described,37 and analyzed for intravascular and extravascular leukocytes using a Leica DMRB upright microscope and a 25/0.75NA oil immersion objective (both Leica).
Leukocyte Recruitment during LPS-Induced Acute Lung Injury
For induction of an acute pulmonary inflammation in mice, we adapted the described model of LPS-induced acute lung injury (ALI).39 Briefly, LPS from E. coli 0111:B4 (10 μg LPS/50 μL PBS; Sigma-Aldrich, Munich, Germany) was intratracheally instilled during anesthetic inhalation of isoflurane (Baxter) to trigger neutrophil infiltration into the lung. PBS served as a negative control. For PC timing experiments 100 U PC/kg or control solution were given intravenously 5.5 hours, 2 hours, 1 hours, or directly before bronchoalveolar lavage, as indicated. In a second approach, 100 U PC/kg was administered 0.5 hours after LPS instillation in WT mice, LFA-1, or ICAM-1 deficient mice to dissect the role of LFA-1 and ICAM-1. Six hours after LPS application mice were anesthetized by i.p. injection as already mentioned, the trachea was cannulated and a bronchoalveolar lavage of the left lung was performed using a rinse solution containing PBS and protease inhibitor solution (Protease Inhibitor Cocktail; Sigma-Aldrich, Munich, Germany) to harvest infiltrated cells. For leukocyte differentiation, stained cytospin preparations were analyzed on a Leica DMRB upright microscope (Leica, Wetzlar, Germany) and × 25/0.75 NA oil immersion objective (Leica).
Immunohistochemistry
To investigate the effect of protein C on TNF-α-stimulated cremaster muscle venules concerning the endothelial expression of E-selectin, P-selectin, VCAM-1, ICAM-1, TM, and EPCR, we performed immunohistochemical analysis of whole mount cremaster muscles as described.40, 41 Briefly, primary antibodies against murine E-selectin (9A9, monoclonal rat IgG1 anti-mouse and 30 μg/mouse; generous gift from B. Wolitzky, MitoKor, San Diego, CA), P-selectin (RB40.34, monoclonal rat IgG1 anti-mouse and 30 μg/mouse; generous gift from D. Vestweber, Max-Planck-Institute, Münster, Germany), VCAM-1 (MVCAMA 429 and 30 μg/mouse; Abd Serotec, Oxford, UK), or ICAM-1 (YN-1, monoclonal rat anti-mouse and 30 μg/mouse; ATCC, Wesel, Germany) were systemically injected and incubated for 10 minutes. Because of the intravascular antibody application after exteriorization of the cremaster muscle, binding of antibodies is mostly restricted to surface expressed antigens within the vasculature. Primary antibodies against TM and EPCR were administered after permeabilization. Analysis of stained slides was conducted semi-quantitatively in a blinded manner (0 = no, 1 = weak, 2 = medium, 3 = strong signal) on a Leica DMRB upright microscope (Leica) and a × 25/0.75 NA oil immersion objective (Leica). Photographs of the samples were taken using a color CCD camera (KAPPA, Gleichen, Germany).
Flow Cytometry
For flow cytometric analysis of ICAM-1 expression on endothelial cells, we used cultured murine aortic endothelial cells (MAECs). The MAECs were isolated and cultured as previously described.42 In brief, 3-mm long freshly harvested and cleaned aortic rings were seeded into Matrigel-coated culture dishes (BD, San Jose, CA) and incubated at 37°C, 5% CO2 in Dulbecco's Modified Eagle Medium (supplemented with 15% fetal bovine serum, 1% Pen/Strep, 90 μg/mL heparin, 60 μg/mL endothelial cell growth supplement, and 1 μg/mL amphothericin B; Fungizone, Invitrogen, Karlsruhe, Germany). After sufficient growing endothelial cells were passaged with Dispase (BD) and characterized by immunocytochemistry as described.43 For TNF-α-stimulation cells were grown in Costar 6-well plates (Corning Inc., Amsterdam, The Netherlands) and standard medium to near confluence and incubated with TNF-α at 25 ng/106 cells for 4 hours at 37°C. PC pretreatment with 5 U/106 cells was initiated 2 hours before TNF-α stimulation. Next, cells were harvested and incubated in the dark for 45 minutes on ice with fluorescein isothiocyanate-conjugated anti-human CD54 monoclonal antibody (mAb) or the respective isotype control antibody (both at 1 μg/105 cells, mouse IgG1; eBioscience, Frankfurt, Germany) to detect anti-ICAM-1 signal on 10.000 cells using the 4-decade FACSScan LSRII with DIVA software package (BD).
The expression of the β2 integrins LFA-1 and macrophage 1 antigen (Mac-1) on neutrophils of PC treated mice was assessed by flow cytometry as previously described.37 Briefly, we used the model of TNF-α-induced inflammation and systemically injected anti-Mac-1 mAb M1/70 (100 μg/mouse, rat IgG2b; eBioscience, San Diego, CA), LFA-1 mAb M17/4 (100 μg/mouse, rat IgG2b; eBioscience, San Diego, CA), or isotype control antibody (100 μg/mouse, rat IgG2b; PharMingen, San Diego, CA) into the carotid artery of control mice and mice treated with 100 U/kg PC for a duration of 3 hours. This procedure of “in vivo labeling” allowed us to exclude a significant up-regulation of integrins by isolation procedures. Twenty minutes after injection, whole blood was obtained, and red blood cell lysis was performed as previously mentioned. Cells were incubated in the dark, thereafter, with secondary phycoerythrin-conjugated goat anti-rat polyclonal antibodies IgG2b (1 μg/105 cells; Abcam) for 45 minutes on ice to assess the expression of the β2 integrins Mac-1 or LFA-1 on 10,000 cells/mouse within the neutrophil cluster defined by forward–side scatter analysis using the FACS-Scan LSRII (BD).
To detect any PC-induced alteration of CXCR2 (CD182) expression on neutrophils, we treated bone marrow neutrophils with PC in vitro. Briefly after red blood cell lysis (1 U PC) was added to 106 leukocytes, it was then incubated for 3 hours at room temperature. Cells were incubated in the dark, thereafter, for 45 minutes on ice with a phycoerythrin-conjugated anti-human CD182 mAb or the respective isotype control antibody (both at 2.5 μg/105 cells, mouse IgG1; eBioscience, Frankfurt, Germany) to detect anti-CXCR2 signal on 10,000 cells/sample within the neutrophil cluster in the previously described manner.
Model of Lethal Endotoxemia
Lethal endotoxemia was induced by a single i.p. injection of 40 mg/kg LPS (Escherichia coli, serotype O55:B5; Sigma-Aldrich, Munich, Germany), which was reconstituted in 100 μL of sterile PBS, as previously reported.9, 44 Control solution or 100 U/kg PC was administered intravenously at 30 minutes, 8 hours, and 24 hours after LPS challenge. In the first group, survival was observed for 14 days. In the second group, APC plasma concentrations were measured at approximately 24 hours after LPS injection as previously described. Mice were perfused with saline, thereafter, and the lungs were harvested. After fixation in 1% paraformaldehyde, they were prepared for paraffin-embedded sections. Sections were performed at 3-μm thickness and stained with H&E for microscopic evaluation.
Statistics
We used the log rank test of Kaplan-Meier survival analysis of SPSS (IBM, Munich, Germany) to compare the mortality of PC-treated and control mice during lethal endotoxemia. All other statistical analyses were performed using Sigma Stat 3.5 (Systat Software, Erkrath, Germany). Statistical significance between groups and treatments were compared with one-way analysis of variance followed by a multiple pairwise comparison test (Dunn's test) or by the Wilcoxon rank-sum test, as appropriate. Semi-quantitative results of the immunhistochemistry signals between PC-treated and control mice were compared using the Students t-test. Statistical significance was set at P < 0.05.
Results
Dose-Dependent Impact of PC on Leukocyte Recruitment in TNF-α-Stimulated Cremaster Muscle Venules
For the observation of leukocyte recruitment by intravital microscopy, mice were injected with PC in variable doses, different treatment intervals, and diverse inflammation models. In all treatment groups, mice appeared healthy throughout the in vivo experiments. There were no significant differences in hemodynamic parameters between the treatment groups (see Supplemental Table S1 at http://ajp.amjpathol.org).
First, we observed leukocyte recruitment in 3-hour TNF-α-stimulated cremaster muscle venules of mice treated with different doses of PC or control solution at the time of TNF-α administration. In this model, PC treatment resulted in reduced leukocyte adhesion in a dose dependent manner. PC doses of 50 U/kg, and more, significantly diminished the number of adherent leukocytes in cremaster muscle venules compared to respective control mice, reaching a plateau of approximately 60% reduced leukocyte adhesion by a PC dose of 400 U/kg (Figure 1A). Next, we investigated leukocyte rolling in TNF-α-stimulated cremaster muscle venules. In line with previous studies, performed in mice with defective leukocyte adhesion,45, 46 we found no significant change of the rolling flux fraction with increasing doses of PC (Figure 1B). PC doses of 400 and 800 U/kg resulted in an acceleration of rolling leukocytes, whereas 200 U/kg PC or less did not alter leukocyte rolling velocities compared to control mice (Figure 1C), suggesting that impaired leukocyte rolling is not causative for the reduced leukocyte adhesion in this setting.
Figure 1.
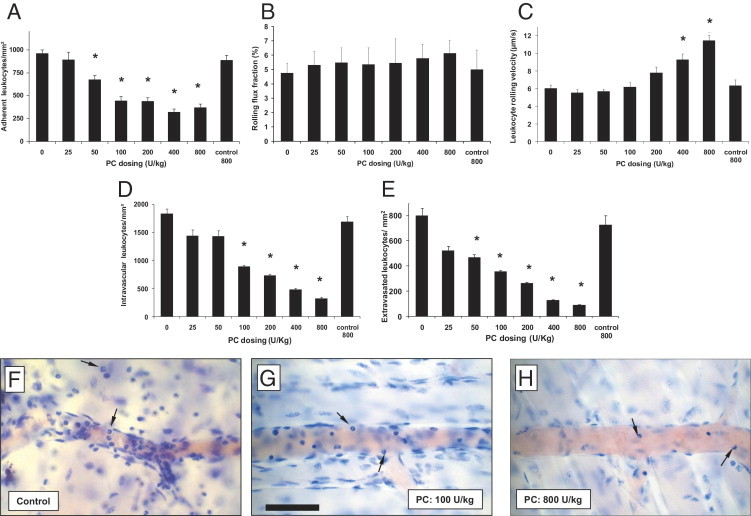
Dose-dependent effect of protein C (PC) on leukocyte recruitment in tumor necrosis factor-α (TNF-α)-stimulated cremaster muscle venules. Leukocyte adhesion (per mm2 of surface area), rolling flux fraction (RFF) (percentage), leukocyte rolling velocity (μm/s) in cremaster muscle venules, and intravascular and extravascular leukocytes (per mm2 surface area) in cremaster muscles were compared 3 hours after TNF-α stimulation and after intravenous administration of indicated doses of PC or isotonic control solution for 800 U/kg PC (control 800; contains as much human serum albumin as 800 U/kg PC). Adherent leukocytes (A), RFF (B), and rolling velocities (C) in cremaster muscle venules are presented for the different treatment groups. Intravascular (D) and perivascular (E) number of leukocytes were observed in Giemsa-stained cremaster muscle whole mounts obtained after the respective intravital microscopic experiments. The distribution of intravascular and perivascular leukocytes in TNF-α-treated cremaster muscles tissue is additionally illustrated by representative micrographs of cremaster muscle whole mounts of control mice (F), after 100 U/kg (G) and 800 U/kg PC (H). As indicated by arrows in (F–H) the majority of intravascular and extravascular leukocytes are neutrophils. Scale bar for (F–H) is shown in G and represents 50 μm. All values are presented as mean ± SEM from at least three mice per group. Significant differences (*P < 0.05) to control are indicated.
To elucidate the impact of PC on leukocyte transmigration, we performed Giemsa staining of whole mounts of TNF-α-treated cremaster muscles. Differentiation of intraluminal and transmigrated leukocytes showed a predominant role of neutrophils in this model (88% and 94%, respectively) (Figure 1, F–H). The number of intravascular leukocytes decreased with higher doses of PC (Figure 1D), affecting mostly neutrophils. Similarly, extravasation of leukocytes into cremaster muscle tissue was strongly inhibited by PC treatment in a dose-dependent manner (Figure 1E). Leukocyte transmigration was significantly reduced by treatment with 50 U/kg PC, which was stronger at 100 U/kg PC and almost completely blocked by 800 U/kg PC compared to respective control mice, as also shown in representative micrographs of cremaster muscle whole mount preparations in Figure 1 F–H, respectively.
To confirm that the injected PC is circulating in the investigated mice, we analyzed PC plasma levels showing a PC-dose dependent and significant increase of PC plasma concentration (percentage of the level in pooled plasma from normal donors) (see Supplemental Table S2 at http://ajp.amjpathol.org). Administration of 100 U/kg PC significantly increased the PC level to 66% and 400 U/kg or more, which resulted in a PC plasma saturation expressed as 100% compared to human normal values. The basic plasmatic coagulation parameter prothrombin time, partial thromboplastin time, and fibrinogen did not change after PC treatment (see Supplemental Table S2 at http://ajp.amjpathol.org). Based on these results, we used a dose of 100 U/kg for PC therapy throughout all further experiments.
Timing of PC and APC Treatment for Leukocyte Recruitment in TNF-α-Stimulated Cremaster Muscle Venules
To further elucidate the anti-inflammatory potential of PC during the 3 hours TNF-α-induced inflammation, we injected 100 U/kg PC at different time points before observation of leukocyte adhesion in cremaster muscle venules by intravital microscopy. Microvascular parameters are listed in Supplemental Table S1 (available at http://ajp.amjpathol.org). As shown in Figure 2A, the earlier PC was applied, the stronger the PC-induced inhibition of leukocyte adhesion. When given 3 hours after TNF-α (ie, shortly before microscopic observation), the PC did not significantly alter leukocyte adhesion compared to control mice. In contrast, PC exerted a significant inhibition of leukocyte adhesion when applied for 1 hour, and even stronger when given 2 or 3 hours before observation.
Figure 2.
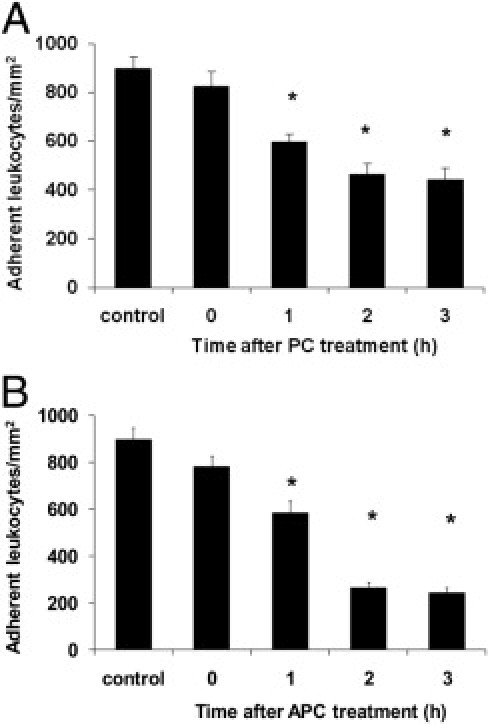
Time-dependent impact of protein C (PC) and activated protein C (APC) treatment for leukocyte adhesion in tumor necrosis factor-α (TNF-α)-stimulated cremaster muscle venules. Leukocyte adhesion (per mm2 of vessel surface area) of cremaster muscle venules were measured after 3 hours of TNF-α-induced inflammation and at different time points after injecting a single dose of 100 U/kg PC (A) or after starting continuous administration of 24 μg/kg/hour APC (B). Control mice received isotonic human serum albumin (8 mg/mL) 3 hours before observation. All values are presented as mean ± SEM from at least three mice per group. Significant differences (*P < 0.05) to the control are indicated.
In another set of experiments, we injected 24 μg/kg/hour APC for different time intervals, and we observed leukocyte adhesion in TNF-α-stimulated cremaster muscle venules by intravital microscopy. Similar to the PC-induced inhibition of leukocyte adhesion, APC treatment was effective when started at least 1 hour before microscopic observation (Figure 2B). A direct comparison of the APC and PC-induced inhibition of leukocyte adhesion after 3 hours revealed a significantly stronger effect in APC treated mice (not depicted).
Leukocyte Recruitment during Trauma-Induced Inflammation of Cremaster Muscle Venules and PC Treatment
Next, we observed the effect of PC on leukocyte recruitment in trauma-stimulated cremaster muscle venules, in which the leukocyte adhesion is induced by the surgical preparation of the cremaster muscle.47 Microvascular and hemodynamic parameters did not vary significantly between the treatment groups (see Supplemental Table S3 at http://ajp.amjpathol.org). Depending on the time of PC application, we found that treatment with 100 U/kg PC resulted in a marked reduction of the number of adherent cells when compared to control mice. Leukocyte adhesion was only marginally changed in mice when treated with PC shortly before, 1 hour before, or 2 hours before trauma-induced inflammation. When administered 3 hours before inflammatory stimulation, however, PC significantly reduced leukocyte adhesion (Figure 3A). We observed a moderate decrease in the number of rolling leukocytes during trauma-induced inflammation in the PC treatment group, although this reduction did not reach the level of significance (P = 0.49) (Figure 3B). Next, we used the chemokine (C-X-C motif) ligand 1 (CXCL1), also known as keratinocyte-derived chemokine, to trigger additional leukocyte adhesion in exteriorized cremaster muscle venules,48 and we analyzed CXCL1-induced leukocyte adhesion in relation to PC treatment. As previously described,48 the number of adherent cells strongly increased 3 minutes after injection of CXCL1 under control conditions (Figure 3C) (see Supplemental Video S1 at http://ajp.amjpathol.org). Similarly, we observed a considerable increase of leukocyte adhesion after CXCL1 injection in mice treated with PC immediately before or 1 hour before inflammatory stimulation. Longer PC pretreatment for 2 or 3 hours, however, strongly reduced or even abrogated CXCL1-induced leukocyte adhesion compared to control mice (Figure 3C) (see Supplemental Video S2 at http://ajp.amjpathol.org), suggesting that chemokine-mediated firm leukocyte arrest can be regulated by PC in a time-dependent manner. The PC-induced inhibition of CXCL1, furthermore, triggered leukocyte adhesion translated into significantly reduced transmigration of leukocytes into cremaster muscle tissue (Figure 3D).
Figure 3.
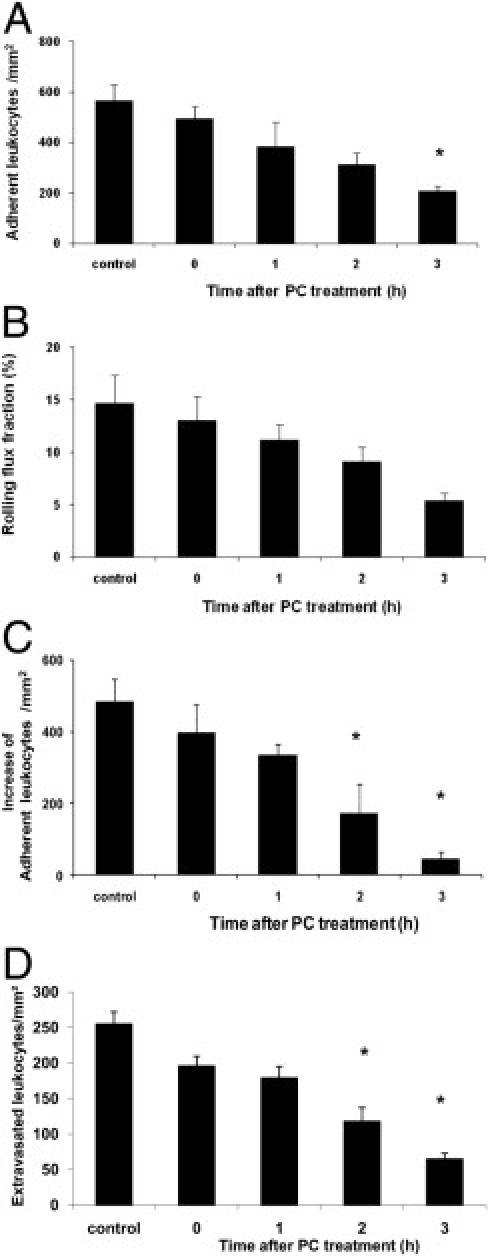
Leukocyte recruitment in trauma-induced inflammation after protein C (PC) treatment. Leukocyte adhesion (per mm2 of surface area) and rolling flux fraction (RFF) (percentage) after trauma-induced inflammation were observed in mice treated with 100 U/kg PC at different time points before microscopic observation and in control mice, which received isotonic human serum albumin (8 mg/mL) for 3 hours. First, the number of adherent leukocytes (A) RFF (B) in surgically prepared cremaster muscle venules were analyzed under baseline conditions. Next, the additional increase of leukocyte adhesion over baseline (per mm2 of surface area) is shown 3 minutes after systemic administration of 600 ng of chemokine (C-X-C motif) ligand 1 for the respective PC treatment intervals and control condition (C). Leukocyte transmigration (D) was observed in Giemsa-stained cremaster muscle whole mounts obtained after the respective intravital microscopic experiments. All values are presented as mean ± SEM from three or more mice per group. Significant differences (*P < 0.05) to control are indicated.
Leukocyte Recruitment in LPS-Induced ALI and PC Treatment
To address the question of whether PC interferes with leukocyte recruitment in a disease relevant model of acute inflammation, we investigated leukocyte emigration during LPS induced ALI.39 LPS induced a strong neutrophil transmigration into the bronchoalveolar space compared to PBS-stimulated control mice (Figure 4A). Recruitment of neutrophils was profoundly blocked by treatment with PC at 100 U/kg in a time-dependent manner (Figure 4A). During the 6-hour model of LPS-induced ALI PC reduced neutrophil emigration when it was given 1 hour prior to bronchoalveolar lavage (i.e. 5 hours after LPS instillation), but most efficiently when administered shortly after the LPS application (which was 5.5 hours before lavage).
Figure 4.
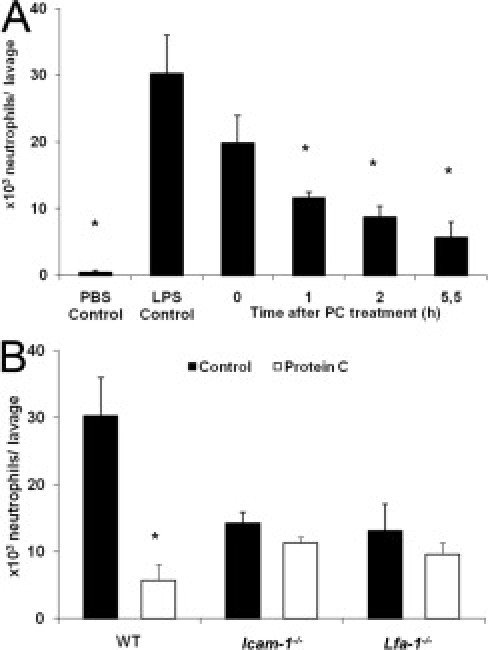
Impact of protein C (PC) on leukocyte emigration into the bronchoalveolar space in lipopolysaccharide (LPS) induced acute lung injury (ALI). In a 6-hour model of LPS-induced ALI, the number of intra-alveolar neutrophils, obtained by bronchoalveolar lavage were analyzed in wild-type (WT) mice treated with 100 U/kg PC at different time points and compared to WT control mice (A). Time between PC treatment and bronchoalveolar lavage is referred to as “Time after PC treatment”. PBS control mice were injected with PBS instead of LPS. In parallel, intercellular adhesion molecule 1 (Icam-1)−/− and lymphocyte function assiciated antigen-1 (Lfa-1)−/− mice were treated with 100 U/kg PC 30 minutes after LPS application and were compared to untreated control mice and respective WT mice (B). All values are presented as mean ± SEM from at least three mice per group. Significant differences (*P < 0.05) to LPS control mice are indicated.
Because ICAM-1 and LFA-1 are known to mediate leukocyte recruitment into the lung during ALI,49 we investigated their role for PC-induced inhibition of leukocyte emigration into the bronchoalveolar space in our model of LPS-induced ALI. In accordance with a previous report,49 the number of transmigrated neutrophils was reduced by approximately 50% in the absence of ICAM-1 or LFA-1 compared to WT control mice (Figure 4B), suggesting that ICAM-1 and LFA-1 are relevant adhesion molecules in this setting. In contrast, PC treatment (30 minutes after LPS instillation) of Icam-1−/− mice and Lfa-1−/− mice did not further affect neutrophil emigration into acutely inflamed lungs, suggesting that both ICAM-1 and LFA-1 are involved in mediating PC-induced inhibition of leukocyte recruitment in this model.
Capture of Human-Activated Protein C in Murine Circulation after PC and APC Treatment
After having established that leukocyte recruitment is profoundly blocked by PC which is comparable to APC therapy, we explored whether the injected PC became activated in our experimental setting. We, therefore, quantified plasma concentration of human activated protein C in PC (100 U/kg) or APC (24 μg/kg/hour) treated mice, control mice, and thrombin-stimulated PC-treated mice from 10 to 180 minutes after treatment (Figure 5A). Co-injection of PC with thrombin maximally stimulated PC activation, and thus served as positive control. APC concentration significantly increased 30 minutes after PC therapy, being comparable to levels of APC-treated mice (Figure 5, A and B). APC concentration, however, decreased to baseline levels within 3 hours after PC treatment (Figure 5C). Altogether, our data demonstrate that human zymogen PC is efficiently activated in vivo shortly after administration.
Figure 5.
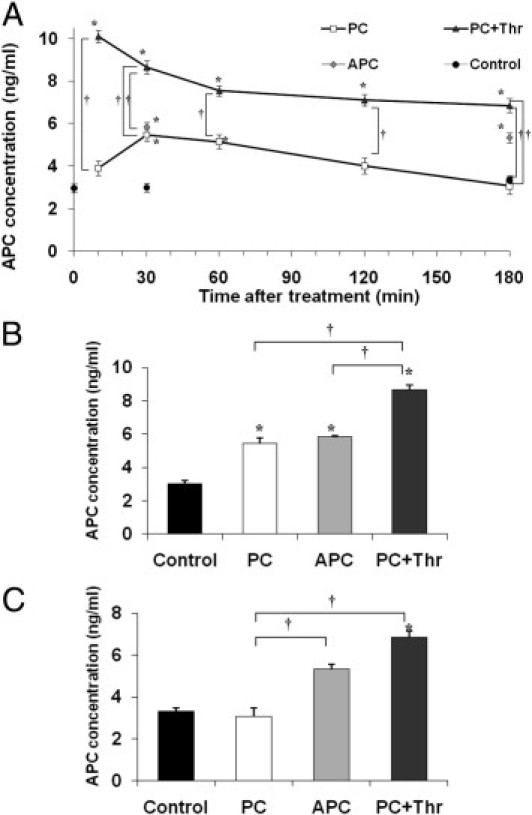
Plasma concentration of human activated protein C (PC) after PC or activated protein C (APC) treatment. Plasma concentration of human activated PC (ng/mL) was analyzed in tumor necrosis factor-α-stimulated mice injected with 100 U/kg PC or 24 μg/kg/hour APC for different treatment intervals (10 to 180 minutes) and compared to control mice. In some experiments, additionally administered human α-thrombin augmented PC activation and served as positive controls (PC+Thr). Human APC was captured from plasma samples using an antibody specific for human APC (A). In addition, separate bar graphs illustrate APC concentration (ng/mL) for respective treatment groups after 30 minutes (B) and 180 minutes (C). The values of at least three separate experiments per group are presented as mean ± SEM. Significant differences (*P < 0.05 to control and †P<0.05 to any other group) are indicated.
Expression of TM, EPCR, and Adhesion Molecules during TNF-α- Induced Inflammation and PC Pretreatment
Because of the fact that efficient activation of PC requires sufficient expression of TM and EPCR, despite acute inflammation,8, 20, 50, 51 we performed immunohistochemistry of TM and EPCR in TNF-α-stimulated cremaster muscles venules. TM and EPCR were clearly expressed on the endothelium of cremaster muscles venules during TNF-α-induced inflammation. Endothelial expression of EPCR was not affected by PC, although we observed a slightly reduced immunostaining for TM in PC-treated mice compared to control mice, as also shown in representative micrographs of cremaster muscle venules (Figure 6, F and G, respectively).
Figure 6.
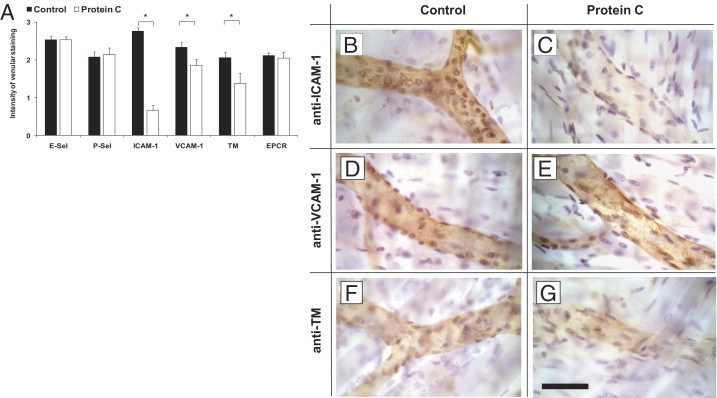
Expression of P- and E-selectin, intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), thrombomodulin (TM), and endothelial protein C receptor (EPCR) in tumor necrosis factor-α (TNF-α)-stimulated cremaster muscles. Immunohistochemistry was performed to assess endothelial expression of P-selectin, E-selectin, ICAM-1, VCAM-1, TM, and EPCR in TNF-α-stimulated cremaster muscle venules for protein C (PC) (100 U/kg) treated or control mice (at least three mice per group). For P-selectin (P-Sel), E-selectin (E-Sel), ICAM-1, and VCAM, primary antibodies were injected systemically and incubated for 10 minutes. Cremaster muscle whole mounts were then fixed and permeabilized. Antibodies against TM and EPCR were administered after permeabilization. Biotinylated secondary antibody, peroxidase-conjugated streptavidin, and diaminobenzidine (DAB) were used to detect endothelial expression as brown signal. Counterstaining was performed by H&E. The intensity of venular immunostaining was analyzed semi-quantitatively and presented as the mean ± SEM for all antibodies in PC-treated mice and compared to respective control mice (A) 0 = no, 1 = weak, 2 = medium, 3 = strong signal. Significant differences (*P < 0.05) to the control are indicated. Representative micrographs are depicted to illustrate the endothelial expression of ICAM-1 (B), VCAM-1, (D) and TM, (F) in control and PC-treated mice (C, E, G, respectively). Scale bar for (B–G) is shown in G and represents 50 μm.
To further study underlying mechanisms of PC-induced inhibition of leukocyte recruitment, we assessed endothelial expression of adhesion molecules in PC-treated and control mice (Figure 6A). Expression of E- and P-selectin in TNF-α- treated cremaster muscle venules were not affected by PC when compared to immmunostained control venules. In contrast, endothelial expression of ICAM-1 was almost completely absent after PC pretreatment (Figure 6C) compared to the strong signal in control mice (Figure 6B). To a minor degree, PC pretreatment also reduced endothelial expression of VCAM-1 in TNF-α-treated cremaster muscle venules (Figure 6, D and E, respectively). The results of semi-quantitative analysis of these immunostainings are also summarized in Figure 6A.
To further support the hypothesis that PC down-regulates endothelial ICAM-1 expression we investigated TNF-α-induced ICAM-1 expression in cultured MAECs by flow cytometry. In PC pretreated TNF-α-stimulated MAECs, ICAM-1 expression was strongly reduced compared to MAECs solely stimulated with TNF-α (Figure 7A). The PC-induced down-regulation of ICAM-1 expression, therefore, was similar to baseline expression of ICAM-1 in unstimulated MAECs.
Figure 7.
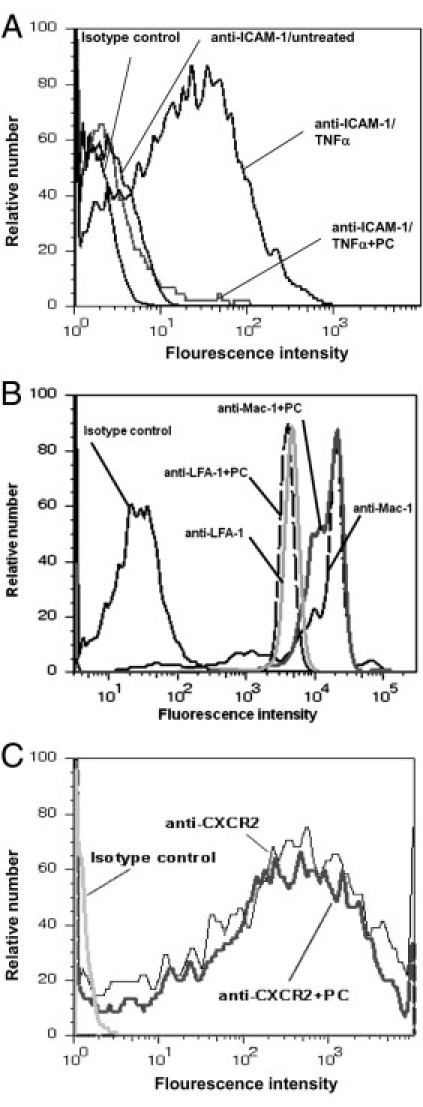
Tumor necrosis factor-α (TNF-α)-stimulated intercellular adhesion molecule 1 (ICAM-1) expression on endothelial cells, β2-integrin, and CXCR2 expression on neutrophils after PC treatment. ICAM-1 expression of cultured endothelial cells was measured after 4 hours of TNF-α stimulation (25 ng/mL) with or without prior PC pre-incubation (5 U/106 cells) for 2 hours (A). Macrophage 1 antigen (Mac-1) and lyphocyte function-associated antigen 1 (LFA-1) blocking monoclonal antibody (mAb) were systemically injected into mice treated with 100 U/kg PC for 3 hours and a secondary antibody was used to detect Mac-1 and LFA-1 expression on peripheral blood neutrophils compared to control mice (B). Surface expression of CXCR2 on bone marrow neutrophils after pre-incubation with protein C (PC) (1 U/106 cells for 3 hours) was compared to control (C). Representative histograms are shown from three separate experiments.
Next, we explored whether PC regulates expression of adhesion molecules on neutrophils and performed flow cytometric analyses of the β2-integrins Mac-1 and LFA-1 and the chemokine receptor CXCR2 on neutrophils. PC treatment neither changed the expression of Mac-1 and LFA-1 (Figure 7B) nor CXCR2 (Figure 7C) on neutrophils compared to controls, arguing against a PC-induced down-regulation of these molecules. The results indicate that direct binding of PC to functionally relevant sites of these molecules does not play a key role in this setting, as we used functionally blocking antibodies.
Role of Thrombomodulin and EPCR-Dependent Signaling for PC-Induced Inhibition of Leukocyte Recruitment in TNF-α-Induced Inflammation
Next, we addressed the question regarding the in vivo relevance of TM- and EPCR- dependent activation and signaling for PC induced inhibition of leukocyte recruitment by using thrombomodulin mutant mice (TMPro/Pro), the EPCR blocking mAb RC252 or PAR-1 inhibitor SCH79797 during TNF-α-induced inflammation.
TMPro/Pro mice are shown to have a strongly reduced PC activating capacity (approximately 5% of control levels) and a proinflammatory phenotype.2, 35 Notably, in TNF-α-stimulated cremaster muscles of TMPro/Pro mice leukocyte adhesion was comparable to WT mice in the absence of exogenous PC. Following therapeutic administration of PC (100 U/kg), however, an inhibition of leukocyte adhesion was only observed in WT mice, but not in TMPro/Pro mice. This demonstrates that TM-dependent PC activation is required for the therapeutic efficacy of PC (Figure 8A).
Figure 8.
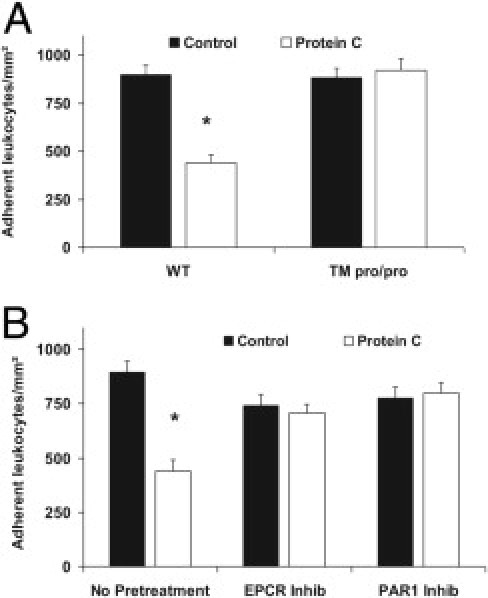
Role of thrombomodulin (TM), endothelial protein C receptor (EPCR), and protease-activated receptor-1 (PAR1) for protein C (PC)-induced inhibition of leukocyte adhesion during tumor necrosis factor-α (TNF-α)-induced inflammation. Leukocyte adhesion in 3 hours of TNF-α-stimulated cremaster muscle venules of TM mutant (TM pro/pro) mice with and without administration of 100 U/kg PC for 3 hours before observation were compared to wild-type (WT) control mice (A). Furthermore, PC-treated (100 U/kg, 3 hours) mice were pretreated with the EPCR-blocking antibody RCR252 or PAR-1 inhibitor SCH79797 to observe leukocyte adhesion in TNF-α-stimulated cremaster muscle venules and compared with respective controls (B). All values were generated from at least three mice per group and are presented in adherent leukocytes per mm2 as mean ± SEM. Significant differences (*P < 0.05) to control are indicated.
To explore the role of EPCR- and PAR-1-dependent signaling for PC induced anti-inflammatory effects during TNF-α-induced inflammation, we pretreated PC treated and control mice with EPCR blocking mAb RC252 or PAR-1 inhibitor SCH79797. In contrast to mice without pretreatment, PC therapy did not reduce leukocyte adhesion in mice pretreated with anti-EPCR mAb RC252 or PAR-1 inhibitor SCH79797, demonstrating that EPCR and PAR-1 are crucially involved in mediating PC-induced inhibition of leukocyte recruitment (Figure 8B).
Using immunohistochemistry, we then investigated the role of TM, EPCR, and PAR-1 for the observed PC-induced down-regulation of endothelial expression of ICAM-1 and VCAM-1 during TNF-α-induced inflammation. Semi-quantitative results of anti-ICAM-1-staining intensity (see Supplemental Figure S1A at http://ajp.amjpathol.org) are additionally illustrated by representative micrographs (see Supplemental Figure S1, B–G at http://ajp.amjpathol.org). In contrast to PC-treated WT mice, PC failed to reduce ICAM-1 expression in cremaster muscle venules of TM Pro/Pro or WT mice pretreated with the EPCR blocking mAb RC252 or PAR-1 inhibitor SCH79797. Endothelial ICAM-1 expression in respective control mice without PC (ie, TM Pro/Pro or WT mice pretreated with the EPCR blocking mAb RC252 or PAR-1 inhibitor SCH79797) did not differ from WT control mice (not depicted). VCAM-1 expression was not significantly different between the investigated groups (see Supplemental Figure S2 at http://ajp.amjpathol.org). Based on these results we conclude that PC primarily reduces ICAM-1 expression through a mechanism requiring TM, EPCR, and PAR-1.
Role of ICAM-1 for PC-Induced Inhibition of Leukocyte Recruitment in TNF-α and Trauma-Induced Inflammation
Using the cremaster muscle models, we observed a significant reduction of adherent leukocytes in Icam-1 −/− mice, following TNF-α treatment or during trauma induced inflammation (with and without concomitant CXCL1 stimulation). The PC-mediated attenuation of leukocyte adhesion was markedly reduced in the TNF-α model (Figure 9A) and completely abolished in the trauma model (Figure 9, B and C), suggesting that the inhibitory effect of PC on leukocytes adhesion depends largely on ICAM-1.
Figure 9.
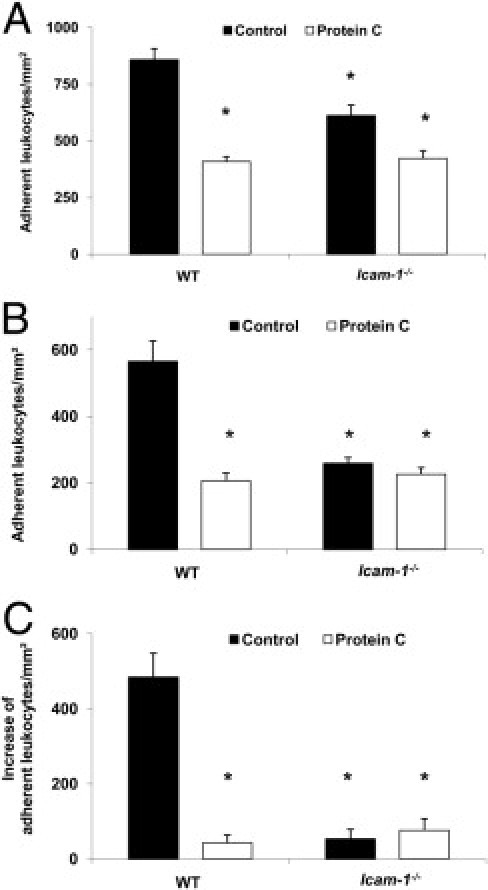
Role of intercellular adhesion molecule 1 (ICAM-1) for protein C (PC)-induced inhibition of leukocyte adhesion during tumor necrosis factor-α (TNF-α)- and trauma-induced inflammation. Leukocyte adhesion in cremaster muscle venules (per mm2 of surface area) of intercellular adhesion molecule 1 (Icam-1)−/− mice and wild-type (WT) mice with and without PC treatment (100 U/kg) 3 hours before observation is shown for TNF-α-stimulation (A) and trauma induced inflammation (B). Next, the additional increase of leukocyte adhesion over baseline trauma-induced adhesion is shown 3 minutes after systemic administration of 600 ng of CXCL1 (C). All values are presented as mean ± SEM from three or more mice per group. Significant differences (*P < 0.05) to WT control mice are indicated.
PC Treatment during Lethal Endotoxemia
After having established that PC blocks leukocyte recruitment in various inflammatory models of single organs, we addressed the question of whether PC exerts anti-inflammatory potential during systemic inflammation and sepsis. We used an established murine model of lethal endotoxemia.9, 44 After the challenge with Escherichia coli LPS (40 mg/kg i.p.), the mice were treated with 3 × 100 U/kg PC or control solution (30 minutes, and 8 and 24 hours after LPS). PC therapy during lethal endotoxemia significantly improved survival for a 14-day period compared to control mice (75% versus 25%, respectively) (Figure 10A).
Figure 10.
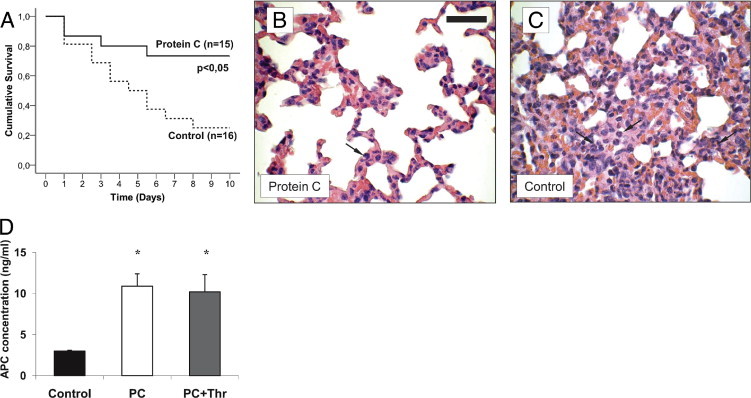
Protein C (PC) treatment during lethal endotoxemia. Lethal endotoxemia was induced by Escherichia coli LPS (40 mg/kg i.p.) and treated by i.v. of 100 U/kg PC or control solution at 30 minutes, and at 8 and 24 hours after LPS challenge. Kaplan-Meier survival analysis is shown for PC treated (n = 15) and control (n = 16) mice (A). Lungs of PC treated (B) and control mice (C) were harvested at the second day after LPS challenge and prepared as 3-μm paraffin-embedded sections for H&E staining. Micrographs are representative for at least three mice per group. Arrows indicate infiltrating neutrophils. Reference bar for (B) and (C) is shown in (B) and represents 50 μm. Plasma concentrations of human activated protein C (APC) (ng/mL) were measured at approximately 24 hours after LPS injection in PC-treated and control mice (D). Some PC-treated mice were additionally administered human α-thrombin-boostered PC activation and served as positive controls (PC+Thr). All values are presented as mean ± SEM from at least four mice per group. Significant differences (*P < 0.05) to control mice are indicated.
Next, we determined leukocyte infiltration into the lung. To this end, we obtained lungs on the day 2 of LPS-induced sepsis. Leukocyte infiltration was markedly reduced in PC-treated mice compared to control mice (Figure 10, B and C). Similar results were observed in liver and kidney sections (not depicted), suggesting that PC blocks inflammation of various organs during sepsis.
To ascertain that PC is efficiently activated in LPS-treated mice, we quantified APC plasma concentration in septic mice treated with 100 U/kg PC. Then, 24 hours after induction of sepsis, APC concentration in PC-treated mice significantly increased as much as 10 ng/mL compared to control mice (3 ng/mL) (Figure 10D). This increase of APC plasma levels is similar to that observed in PC and thrombin co-injected mice (positive control) (Figure 10D), pointing to sufficient PC activation during systemic inflammation.
Discussion
Anti-inflammatory properties of PC have increasingly been studied during the past decade,1, 8, 12, 17, 20, 26, 30, 52, 53, 54 although this is the first study investigating PC effects on the multi-step cascade of leukocyte recruitment during acute inflammation using intravital microscopy. We found that PC is capable to profoundly block leukocyte adhesion and consecutively, leukocyte transmigration in various in vivo models of acute inflammation without significant changes of hemodynamic or basic clotting parameter. These effects are shown to be dose-dependent. PC doses of 50 to 200 U/kg, which are commonly used in the treatment of congenital PC deficiency,31, 55, 56 strongly inhibited leukocyte adhesion and transmigration, which is consistent with previous studies.28, 29, 30 As with the results obtained from the cremaster muscle models of inflammation, 100 U/kg PC also significantly reduced leukocyte recruitment into the lung during LPS-induced ALI in a time-dependent manner that has been shown for APC only.24, 57 Interestingly, in our experimental setting, increasing PC doses to 400 to 800 U/kg did not lead to any further reduction in leukocyte recruitment. Notably, de Kleijn et al10 used as much as 600 U/kg PC for efficient treatment of patients with meningococcemia. Our results thus further support the hypothesis that PC exerts anti-inflammatory properties in a single dose of 100 U/kg, to be given every 6 to 12 hours, according to its half life.32, 58, 59
Because previous studies suggested an early use of APC to treat septic patients,20, 24 we explored optimal timing of PC and APC to interfere with leukocyte recruitment. Both PC and APC acted in a time-dependent manner and exerted the strongest blocking potential when given shortly (as much as 1 hour) after onset of inflammation, allowing an in vivo acting time of at least 2 hours, which is supported by a very recent study of PC treatment in a model of cecal ligation and puncture-induced sepsis.26 PC began to block leukocyte recruitment, nevertheless, as early as 1 hour after application, depending on the inflammatory model, pointing to additional rapid interaction or signaling through the PC pathway beside changes of transcriptional activity. To evaluate how fast PC enters the PC pathway in our experimental setting, we investigated the availability and activation of the zymogen PC, although systemic PC activity might underestimate the amount of activated protein C generated within the specific microenvironment at sites of inflammation.16 In accordance with very recent reports using PC in septic patients or severe inflammation,10, 26 we found that 30 minutes after PC administration systemic APC concentrations were comparable to continuous APC infusion, indicating that the injected PC underwent fast and sufficient on demand activation during acute inflammation. This observation might also explain the inhibitory potential of both PC and APC to block leukocyte recruitment in our inflammatory models.
We tested the hypothesis that sufficient activation of PC results in anti-inflammatory capacities not only during local inflammation of single organs, but also during systemic inflammation and sepsis. Using an established murine model of lethal endotoxemia,9, 44 we demonstrated that administration of the zymogen PC at 100 U/kg (30 minutes, and 8 and 24 hours after LPS challenge) strongly increased plasma APC concentration 24 hours after LPS application. At this time, inflammation had largely spread systemically15 and various organs were massively infiltrated by leukocytes in untreated control, but not in PC-treated mice. In agreement with the report of Messaris et al,26 we found that sepsis-induced mortality was significantly reduced in mice treated with zymogen PC at 100 U/kg for 30 minutes, and for 8 hours and 24 hours after LPS challenge. Notably, the survival improvement of PC-treated mice was comparable to APC treatment in the same experimental setting as described in the study of Cao et al.15 We provide, therefore, strong evidence for efficient activation of PC and its anti-inflammatory properties during systemic inflammation and sepsis.
The in vivo capacity of PC activation during acute inflammation10, 26 is a matter of debate, as the expression of TM and EPCR is expected to be reduced in this setting.8, 20, 50, 51 Indeed, based on our findings of TMPro/Pro mice TM-mediated PC activation is relevant for PC-induced inhibition of leukocyte adhesion during inflammation. Of note, we observed that TM and EPCR are strongly expressed in inflamed venular endothelium, indicating that the capacity to activate PC is maintained at least at the early stages of inflammation.
PC treatment failed to reduce leukocyte adhesion in TMPro/Pro mice, whereas no difference in leukocyte adhesion was apparent in animals without PC treatment. This emphasizes the important role of TM for PC activation. Given the established anti-inflammatory properties of the lectin-like domain of TM, which remains intact in TMPro/Pro mice, we cannot exclude an additional and PC-independent inhibitory effect of TM on leukocyte recruitment.
After in vivo activation of PC, its anti-inflammatory properties are further mediated by EPCR- and PAR-1-dependent signaling, as already described for the cytoprotective effects of APC.1, 3, 6, 16 Very recently, Guitton et al60 discovered that the protective APC signaling during inflammation crucially involves NFκB and ERK1/2 mitogen-activated protein kinases, which in turn might regulate endothelial inflammatory response via key leukocyte adhesion pathways.1, 6, 16 In the used inflammatory models, the majority of firm leukocyte adhesion is mediated by binding of the β2-intergins LFA-1 and Mac-1 to ICAM-1 and other, yet undefined ligands.19 In our study, expression of β2-integrins Mac-1 and LFA-1 on neutrophils was not altered in PC-treated mice, which, however, does not generally exclude a direct effect of PC on leukocytes such as integrin activation or function. This is interesting regarding two recent reports about direct effects and binding of APC to β1-, β2-, and β3-integrins.11, 15 In one of the studies, APC was found to directly bind to Mac-1 on macrophages, which was crucial for the anti-inflammatory effect of APC during LPS-induced sepsis in mice.15 Notably, the experimental design and the investigated leukocyte subsets in this study are different from our study.
On the endothelial site, TM, EPCR-, and PAR-1-dependent mechanisms trigger PC-induced down-regulation of the integrin ligand ICAM-1, an adhesion molecule that controls firm leukocyte arrest in our used inflammatory models. Of importance, PC failed to further block leukocyte adhesion in Icam-1−/− mice, suggesting that ICAM-1 is crucially involved in mediating PC-induced inhibition of leukocyte adhesion during acute inflammation, as described for APC-induced anti-inflammatory effects.8 Future studies, however, might further explore the mechanisms of PC-induced regulation of inflammatory response dissecting the role of integrins and their ligands.
Taken together, we demonstrated that protein C concentrate inhibits leukocyte recruitment in different in vivo models of inflammation in a dose- and time-dependent manner. Intravital microscopy of inflamed cremaster muscle venules revealed that 100 U/kg PC is sufficient to block leukocyte adhesion and transmigration, but not leukocyte rolling as early as 1 hour after administration. The similar potential of PC and APC to interfere with leukocyte recruitment is entailed to rapid and sufficient TM-mediated protein C activation and EPCR-dependent signaling, which in turn critically involves ICAM-1, a key adhesion molecule in our experimental models of inflammation. Sustained PC activating potential, even during lethal endotoxemia may explain why PC therapy improves survival of septic mice in our experimental setting. These results may contribute to discuss PC as an alternative anti-inflammatory treatment to APC and initiate further studies investigating protein C concentrate as a safe and effective therapeutic strategy in patients with inflammatory diseases.
Acknowledgments
We thank Melitta Weissinger for her excellent technical assistance in performing intravital microscopy, Simone Schmidt for her support in performing immunostaining experiments, Dr. Marcus Mall for his support in performing experiments of LPS induced ALI, Dr. Harmut Weiler (Blood Research Institute, Milwaukee, WI) for providing TMPro/Pro mice, Barry Wolitzky (MitoKor, San Diego, CA) and Dr. Dietmar Vestweber (Max-Planck-Institute, Münster, Germany) for providing the murine E-and P-selectin blocking antibodies, respectively, Dr. Charles T. Esmon (Oklahoma City, OK) for providing the human APC antibody, Drs. Bruno Eberspaecher and Bernhard Kaumanns (Baxter, Unterschleissheim, Germany, respectively) for providing the Ceprotin, and Britta Engelhardt, (Bern, Switzerland) for providing Icam1−/− mice.
Footnotes
Supported in part by grants from the Deutsche Forschungsgemeinschaft (SFB938 to A.B., and IS 67/2-4 and IS 67/5-1 to B.I., respectively).
D.F. designed the research, analyzed the data, and wrote the manuscript. J.T., A.D., M.St., J.Pf., and H.W. performed research and analyzed the data. L.K. edited the manuscript. K.B. and N.B. performed research, analyzed data, and edited the manuscript. A.B. provided Icam1−/− mice and MAECs, and edited the manuscript. M.Sp. and B.I. contributed analytical tools, consulted research design, and edited the manuscript. J.Po. consulted research design.
Supplemental material for this article can be found on http://ajp.amjpathol.org or at doi: 10.1016/j.ajpath.2011.07.023.
Supplementary data
Role of thrombomodulin (TM), endothelial protein C receptor (EPCR), and protease-activated receptor-1 (PAR-1) for protein C (PC)-induced inhibition of intercellular adhesion molecule 1 (ICAM-1) expression in tumor necrosis factor-α (TNF-α)-stimulated cremaster muscles. Endothelial ICAM-1 expression assessed by immunohistochemistry in 3 hours of TNF-α stimulated cremaster muscle venules of PC-treated (100 U/kg, 3 hours) TM deficient (TMPro/Pro) mice, wild-type (WT) mice, and WT mice pretreated with the EPCR-blocking antibody RCR252 or PAR-1 inhibitor SCH79797 were compared to untreated WT control mice. The primary antibody against ICAM-1 was injected systemically and incubated for 10 minutes. Cremaster muscle whole mounts were then fixed and permeabilized. Biotinylated secondary antibody, peroxidase-conjugated streptavidin, and diaminobenzidine were used to detect endothelial expression as brown signal. Counterstaining was performed by H&E. The intensity of venular immunostaining was analyzed semi-quantitatively (0 = no, 1 = weak, 2 = medium, 3 = strong signal) and was presented as mean ± SEM from at least three mice per group (A). The asterisk indicates significant differences (P < 0.05) to all other groups. In addition, representative micrographs are depicted to illustrate the endothelial expression of ICAM-1 (B–G). Reference bar for (B–G) is shown in (C) and represents 50 μm.
Role of thrombomodulin (TM), endothelial protein C receptor (EPCR), and protease-activated receptor-1 (PAR-1) for protein C (PC)-induced inhibition of vascular cell adhesion molecule 1 (VCAM-1) expression in tumor necrosis factor-α (TNF-α)-stimulated cremaster muscles. Endothelial VCAM-1 expression assessed by immunohistochemistry in 3 hours TNF-α-stimulated cremaster muscle venules of PC treated (100 U/kg, 3 hours) TM deficient (TMPro/Pro) mice, wild-type (WT) mice, and WT mice pretreated with the EPCR-blocking antibody RCR252 or PAR-1 inhibitor SCH79797 were compared to untreated WT control mice. The primary antibody against VCAM-1 was injected systemically and incubated for 10 minutes. Cremaster muscle whole mounts were then fixed and permeabilized. Biotinylated secondary antibody, peroxidase-conjugated streptavidin, and diaminobenzidine were used to detect endothelial expression as brown signal. Counterstaining was performed by H&E. The intensity of venular immunostaining was analyzed semi-quantitatively (0 = no, 1 = weak, 2 = medium, 3 = strong signal) and presented as mean ± SEM from at least three mice per group.
Chemokine (C-X-C motif) ligand 1 (CXCL1)-induced leukocyte adhesion in control mice. Intravital microscopy was used to visualize leukocyte arrest in an unstimulated cremaster muscle venule (vessel diameter = 27 μm) of a control mouse before and after systemic injection of 600 ng CXCL1. Superimposing CXCL1 in the top left corner indicates the time of injection. CXCL1 injection led to a rapid and effective arrest of leukocytes to the venular wall.
Chemokine (C-X-C motif) ligand 1 (CXCL1)-induced leukocyte adhesion in protein C (PC)-treated mice. This video exemplifies the impairment of CXCR2-mediated leukocyte arrest in cremaster muscle venules of mice treated with 100 U/kg PC for 3 hours. After systemic injection of 600 ng CXCL1 into a PC-treated mouse, leukocyte arrest is dramatically reduced in the observed cremaster muscle venule (vessel diameter = 31 μm).
References
- 1.Jackson C.J., Xue M. Activated protein C–an anticoagulant that does more than stop clots. Int J Biochem Cell Biol. 2008;40:2692–2697. doi: 10.1016/j.biocel.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 2.Weiler-Guettler H., Christie P.D., Beeler D.L., Healy A.M., Hancock W.W., Rayburn H., Edelberg J.M., Rosenberg R.D. A targeted point mutation in thrombomodulin generates viable mice with a prethrombotic state. J Clin Invest. 1998;101:1983–1991. doi: 10.1172/JCI2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zheng X., Li W., Song Y., Hu Y., Ferrell G.L., Esmon N.L., Esmon C.T. Nonhematopoietic EPCR regulates the coagulation and inflammatory responses during endotoxemia. J Thromb Haemost. 2007;5:1394–1400. doi: 10.1111/j.1538-7836.2007.02592.x. [DOI] [PubMed] [Google Scholar]
- 4.Isermann B., Vinnikov I.A., Madhusudhan T., Herzog S., Kashif M., Blautzik J., Corat M.A., Zeier M., Blessing E., Oh J., Gerlitz B., Berg D.T., Grinnell B.W., Chavakis T., Esmon C.T., Weiler H., Bierhaus A., Nawroth P.P. Activated protein C protects against diabetic nephropathy by inhibiting endothelial and podocyte apoptosis. Nat Med. 2007;13:1349–1358. doi: 10.1038/nm1667. [DOI] [PubMed] [Google Scholar]
- 5.Sturn D.H., Kaneider N.C., Feistritzer C., Djanani A., Fukudome K., Wiedermann C.J. Expression and function of the endothelial protein C receptor in human neutrophils. Blood. 2003;102:1499–1505. doi: 10.1182/blood-2002-12-3880. [DOI] [PubMed] [Google Scholar]
- 6.Riewald M., Petrovan R.J., Donner A., Mueller B.M., Ruf W. Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science. 2002;296:1880–1882. doi: 10.1126/science.1071699. [DOI] [PubMed] [Google Scholar]
- 7.Feistritzer C., Schuepbach R.A., Mosnier L.O., Bush L.A., Di Cera E., Griffin J.H., Riewald M. Protective signaling by activated protein C is mechanistically linked to protein C activation on endothelial cells. J Biol Chem. 2006;281:20077–20084. doi: 10.1074/jbc.M600506200. [DOI] [PubMed] [Google Scholar]
- 8.Scaldaferri F., Sans M., Vetrano S., Graziani C., De Cristofaro R., Gerlitz B., Repici A., Arena V., Malesci A., Panes J., Grinnell B.W., Danese S. Crucial role of the protein C pathway in governing microvascular inflammation in inflammatory bowel disease. J Clin Invest. 2007;117:1951–1960. doi: 10.1172/JCI31027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kerschen E.J., Fernandez J.A., Cooley B.C., Yang X.V., Sood R., Mosnier L.O., Castellino F.J., Mackman N., Griffin J.H., Weiler H. Endotoxemia and sepsis mortality reduction by non-anticoagulant activated protein C. J Exp Med. 2007;204:2439–2448. doi: 10.1084/jem.20070404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.de Kleijn E.D., de Groot R., Hack C.E., Mulder P.G., Engl W., Moritz B., Joosten K.F., Hazelzet J.A. Activation of protein C following infusion of protein C concentrate in children with severe meningococcal sepsis and purpura fulminans: a randomized, double-blinded, placebo-controlled, dose-finding study. Crit Care Med. 2003;31:1839–1847. doi: 10.1097/01.CCM.0000072121.61120.D8. [DOI] [PubMed] [Google Scholar]
- 11.Elphick G.F., Sarangi P.P., Hyun Y.M., Hollenbaugh J.A., Ayala A., Biffl W.L., Chung H.L., Rezaie A.R., McGrath J.L., Topham D.J., Reichner J.S., Kim M. Recombinant human activated protein C inhibits integrin-mediated neutrophil migration. Blood. 2009;113:4078–4085. doi: 10.1182/blood-2008-09-180968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nick J.A., Coldren C.D., Geraci M.W., Poch K.R., Fouty B.W., O'Brien J., Gruber M., Zarini S., Murphy R.C., Kuhn K., Richter D., Kast K.R., Abraham E. Recombinant human activated protein C reduces human endotoxin-induced pulmonary inflammation via inhibition of neutrophil chemotaxis. Blood. 2004;104:3878–3885. doi: 10.1182/blood-2004-06-2140. [DOI] [PubMed] [Google Scholar]
- 13.Yuda H., Adachi Y., Taguchi O., Gabazza E.C., Hataji O., Fujimoto H., Tamaki S., Nishikubo K., Fukudome K., D'Alessandro-Gabazza C.N., Maruyama J., Izumizaki M., Iwase M., Homma I., Inoue R., Kamada H., Hayashi T., Kasper M., Lambrecht B.N., Barnes P.J., Suzuki K. Activated protein C inhibits bronchial hyperresponsiveness and Th2 cytokine expression in mice. Blood. 2004;103:2196–2204. doi: 10.1182/blood-2003-06-1980. [DOI] [PubMed] [Google Scholar]
- 14.Nagai M., Terao S., Yilmaz G., Yilmaz C.E., Esmon C.T., Watanabe E., Granger D.N. Roles of inflammation and the activated protein C pathway in the brain edema associated with cerebral venous sinus thrombosis. Stroke. 2010;41:147–152. doi: 10.1161/STROKEAHA.109.562983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao C., Gao Y., Li Y., Antalis T.M., Castellino F.J., Zhang L. The efficacy of activated protein C in murine endotoxemia is dependent on integrin CD11b. J Clin Invest. 2010;120:1971–1980. doi: 10.1172/JCI40380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Weiler H. Regulation of inflammation by the protein C system. Crit Care Med. 2010;38:S18–S25. doi: 10.1097/CCM.0b013e3181c9cbb5. [DOI] [PubMed] [Google Scholar]
- 17.Joyce D.E., Gelbert L., Ciaccia A., DeHoff B., Grinnell B.W. Gene expression profile of antithrombotic protein c defines new mechanisms modulating inflammation and apoptosis. J Biol Chem. 2001;276:11199–11203. doi: 10.1074/jbc.C100017200. [DOI] [PubMed] [Google Scholar]
- 18.Springer T.A. Traffic signals on endothelium for lymphocyte recirculation and leukocyte emigration. Annu Rev Physiol. 1995;57:827–872. doi: 10.1146/annurev.ph.57.030195.004143. [DOI] [PubMed] [Google Scholar]
- 19.Ley K., Laudanna C., Cybulsky M.I., Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 20.Faust S.N., Levin M., Harrison O.B., Goldin R.D., Lockhart M.S., Kondaveeti S., Laszik Z., Esmon C.T., Heyderman R.S. Dysfunction of endothelial protein C activation in severe meningococcal sepsis. N Engl J Med. 2001;345:408–416. doi: 10.1056/NEJM200108093450603. [DOI] [PubMed] [Google Scholar]
- 21.Bernard G.R., Vincent J.L., Laterre P.F., LaRosa S.P., Dhainaut J.F., Lopez-Rodriguez A., Steingrub J.S., Garber G.E., Helterbrand J.D., Ely E.W., Fisher C.J., Jr. Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med. 2001;344:699–709. doi: 10.1056/NEJM200103083441001. [DOI] [PubMed] [Google Scholar]
- 22.Vincent J.L., Angus D.C., Artigas A., Kalil A., Basson B.R., Jamal H.H., Johnson G., III, Bernard G.R. Effects of drotrecogin alfa (activated) on organ dysfunction in the PROWESS trial. Crit Care Med. 2003;31:834–840. doi: 10.1097/01.CCM.0000051515.56179.E1. [DOI] [PubMed] [Google Scholar]
- 23.Bernard G.R. Drotrecogin alfa (activated) (recombinant human activated protein C) for the treatment of severe sepsis. Crit Care Med. 2003;31:S85–S93. doi: 10.1097/00003246-200301001-00012. [DOI] [PubMed] [Google Scholar]
- 24.Abraham E., Laterre P.F., Garg R., Levy H., Talwar D., Trzaskoma B.L., Francois B., Guy J.S., Bruckmann M., Rea-Neto A., Rossaint R., Perrotin D., Sablotzki A., Arkins N., Utterback B.G., Macias W.L. Drotrecogin alfa (activated) for adults with severe sepsis and a low risk of death. N Engl J Med. 2005;353:1332–1341. doi: 10.1056/NEJMoa050935. [DOI] [PubMed] [Google Scholar]
- 25.Frommhold D., Birle A., Linderkamp O., Zilow E., Poschl J. Drotrecogin alpha (activated) in neonatal septic shock. Scand J Infect Dis. 2005;37:306–308. doi: 10.1080/00365540510031412. [DOI] [PubMed] [Google Scholar]
- 26.Messaris E., Betrosian A.P., Memos N., Chatzigianni E., Boutsikou M., Economou V., Dontas I., Theodossiades G., Konstadoulakis M.M., Douzinas E.E. Administration of human protein C improves survival in an experimental model of sepsis. Crit Care Med. 2010;38:209–216. doi: 10.1097/CCM.0b013e3181b4a972. [DOI] [PubMed] [Google Scholar]
- 27.Grinnell B.W., Hermann R.B., Yan S.B. Human protein C inhibits selectin-mediated cell adhesion: role of unique fucosylated oligosaccharide. Glycobiology. 1994;4:221–225. doi: 10.1093/glycob/4.2.221. [DOI] [PubMed] [Google Scholar]
- 28.Rintala E., Kauppila M., Seppala O.P., Voipio-Pulkki L.M., Pettila V., Rasi V., Kotilainen P. Protein C substitution in sepsis-associated purpura fulminans. Crit Care Med. 2000;28:2373–2378. doi: 10.1097/00003246-200007000-00032. [DOI] [PubMed] [Google Scholar]
- 29.Schellongowski P., Bauer E., Holzinger U., Staudinger T., Frass M., Laczika K., Locker G.J., Quehenberger P., Rabitsch W., Schenk P., Knobl P. Treatment of adult patients with sepsis-induced coagulopathy and purpura fulminans using a plasma-derived protein C concentrate (Ceprotin) Vox Sang. 2006;90:294–301. doi: 10.1111/j.1423-0410.2006.00760.x. [DOI] [PubMed] [Google Scholar]
- 30.Crivellari M., Della Valle P., Landoni G., Pappalardo F., Gerli C., Bignami E., Marino G., Zangrillo A., D'Angelo A. Human protein C zymogen concentrate in patients with severe sepsis and multiple organ failure after adult cardiac surgery. Intensive Care Med. 2009;35:1959–1963. doi: 10.1007/s00134-009-1584-3. [DOI] [PubMed] [Google Scholar]
- 31.Knoebl P.N. Human protein C concentrates for replacement therapy in congenital and acquired protein C deficiency. Drugs Today (Barc) 2008;44:429–441. doi: 10.1358/dot.2008.44.6.1217993. [DOI] [PubMed] [Google Scholar]
- 32.Tcheng W.Y., Dovat S., Gurel Z., Donkin J., Wong W.Y. Severe congenital protein C deficiency: description of a new mutation and prophylactic protein C therapy and in vivo pharmacokinetics. J Pediatr Hematol Oncol. 2008;30:166–171. doi: 10.1097/MPH.0b013e31815d8943. [DOI] [PubMed] [Google Scholar]
- 33.Xu H., Gonzalo J.A., St Pierre Y., Williams I.R., Kupper T.S., Cotran R.S., Springer T.A., Gutierrez-Ramos J.C. Leukocytosis and resistance to septic shock in intercellular adhesion molecule 1-deficient mice. J Exp Med. 1994;180:95–109. doi: 10.1084/jem.180.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ahn H.S., Foster C., Boykow G., Stamford A., Manna M., Graziano M. Inhibition of cellular action of thrombin by N3-cyclopropyl-7-[4-(1-methylethyl)phenyl]methyl]-7H-pyrrolo[3, 2-f]quinazoline-1,3-diamine (SCH 79797), a nonpeptide thrombin receptor antagonist. Biochemical Pharmacology. 2000;60:1425–1434. doi: 10.1016/s0006-2952(00)00460-3. [DOI] [PubMed] [Google Scholar]
- 35.Weiler H., Lindner V., Kerlin B., Isermann B.H., Hendrickson S.B., Cooley B.C., Meh D.A., Mosesson M.W., Shworak N.W., Post M.J., Conway E.M., Ulfman L.H., von Andrian U.H., Weitz J.I. Characterization of a mouse model for thrombomodulin deficiency. Arterioscler Thromb Vasc Biol. 2001;21:1531–1537. doi: 10.1161/hq0901.094496. [DOI] [PubMed] [Google Scholar]
- 36.Liaw P.C., Ferrell G., Esmon C.T. A monoclonal antibody against activated protein C allows rapid detection of activated protein C in plasma and reveals a calcium ion dependent epitope involved in factor Va inactivation. J Thromb Haemost. 2003;1:662–670. doi: 10.1046/j.1538-7836.2003.00153.x. [DOI] [PubMed] [Google Scholar]
- 37.Frommhold D., Kamphues A., Hepper I., Pruenster M., Lukic I.K., Socher I., Zablotskaya V., Buschmann K., Lange-Sperandio B., Schymeinsky J., Ryschich E., Poeschl J., Kupatt C., Nawroth P.P., Moser M., Walzog B., Bierhaus A., Sperandio M. RAGE and ICAM-1 cooperate in mediating leukocyte recruitment during acute inflammation in vivo. Blood. 2010;116:841–849. doi: 10.1182/blood-2009-09-244293. [DOI] [PubMed] [Google Scholar]
- 38.Frommhold D., Mannigel I., Schymeinsky J., Mocsai A., Poeschl J., Walzog B., Sperandio M. Spleen tyrosine kinase Syk is critical for sustained leukocyte adhesion during inflammation in vivo. BMC Immunol. 2007;8:31. doi: 10.1186/1471-2172-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reutershan J., Basit A., Galkina E.V., Ley K. Sequential recruitment of neutrophils into lung and bronchoalveolar lavage fluid in LPS-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2005;289:L807–L815. doi: 10.1152/ajplung.00477.2004. [DOI] [PubMed] [Google Scholar]
- 40.Jung U., Ley K. Regulation of E-selectin: P-selectin and ICAM-1 expression in mouse cremaster muscle vasculature. Microcirculation. 1997;4:311–319. doi: 10.3109/10739689709146794. [DOI] [PubMed] [Google Scholar]
- 41.Isermann B., Hendrickson S.B., Zogg M., Wing M., Cummiskey M., Kisanuki Y.Y., Yanagisawa M., Weiler H. Endothelium-specific loss of murine thrombomodulin disrupts the protein C anticoagulant pathway and causes juvenile-onset thrombosis. J Clin Invest. 2001;108:537–546. doi: 10.1172/JCI13077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kobayashi M., Inoue K., Warabi E., Minami T., Kodama T. A simple method of isolating mouse aortic endothelial cells. J Atheroscler Thromb. 2005;12:138–142. doi: 10.5551/jat.12.138. [DOI] [PubMed] [Google Scholar]
- 43.Gerritsen M.E., Shen C.P., McHugh M.C., Atkinson W.J., Kiely J.M., Milstone D.S., Luscinskas F.W., Gimbrone M.A., Jr. Activation-dependent isolation and culture of murine pulmonary microvascular endothelium. Microcirculation. 1995;2:151–163. doi: 10.3109/10739689509146763. [DOI] [PubMed] [Google Scholar]
- 44.Kerlin B.A., Yan S.B., Isermann B.H., Brandt J.T., Sood R., Basson B.R., Joyce D.E., Weiler H., Dhainaut J.F. Survival advantage associated with heterozygous factor V Leiden mutation in patients with severe sepsis and in mouse endotoxemia. Blood. 2003;102:3085–3092. doi: 10.1182/blood-2003-06-1789. [DOI] [PubMed] [Google Scholar]
- 45.Dunne J.L., Ballantyne C.M., Ley K. Different roles of LFA-1 and Mac-1 in slow rolling and firm arrest during cytokine-induced inflammation. FASEB J. 2001;15:A332. [Google Scholar]
- 46.Dunne J.L., Ballantyne C.M., Beaudet A.L., Ley K. Control of leukocyte rolling velocity in TNF-alpha-induced inflammation by LFA-1 and Mac-1. Blood. 2002;99:336–341. doi: 10.1182/blood.v99.1.336. [DOI] [PubMed] [Google Scholar]
- 47.Frommhold D., Ludwig A., Bixel M.G., Zarbock A., Babushkina I., Weissinger M., Cauwenberghs S., Ellies L.G., Marth J.D., Beck-Sickinger A.G., Sixt M., Lange-Sperandio B., Zernecke A., Brandt E., Weber C., Vestweber D., Ley K., Sperandio M. Sialyltransferase ST3Gal-IV controls CXCR2-mediated firm leukocyte arrest during inflammation. J Exp Med. 2008;205:1435–1446. doi: 10.1084/jem.20070846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Smith M.L., Olson T.S., Ley K. CXCR2- and E-selectin-induced neutrophil arrest during inflammation in vivo. J Exp Med. 2004;200:935–939. doi: 10.1084/jem.20040424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Basit A., Reutershan J., Morris M.A., Solga M., Rose C.E., Jr., Ley K. ICAM-1 and LFA-1 play critical roles in LPS-induced neutrophil recruitment into the alveolar space. Am J Physiol Lung Cell Mol Physiol. 2006;291:L200–L207. doi: 10.1152/ajplung.00346.2005. [DOI] [PubMed] [Google Scholar]
- 50.Conway E.M., Nowakowski B., Steiner-Mosonyi M. Human neutrophils synthesize thrombomodulin that does not promote thrombin-dependent protein C activation. Blood. 1992;80:1254–1263. [PubMed] [Google Scholar]
- 51.Van der M.A., Vandamme M., Squiban C., Gaugler M.H., Mouthon M.A. Inflammatory reaction and changes in expression of coagulation proteins on lung endothelial cells after total-body irradiation in mice. Radiat Res. 2003;160:637–646. doi: 10.1667/rr3087. [DOI] [PubMed] [Google Scholar]
- 52.Murakami K., Okajima K., Uchiba M., Johno M., Nakagaki T., Okabe H., Takatsuki K. Activated protein C attenuates endotoxin-induced pulmonary vascular injury by inhibiting activated leukocytes in rats. Blood. 1996;87:642–647. [PubMed] [Google Scholar]
- 53.Mizutani A., Okajima K., Uchiba M., Noguchi T. Activated protein C reduces ischemia/reperfusion-induced renal injury in rats by inhibiting leukocyte activation. Blood. 2000;95:3781–3787. [PubMed] [Google Scholar]
- 54.Van de W.M., Collen D., Conway E.M. Thrombomodulin-protein C-EPCR system: integrated to regulate coagulation and inflammation. Arterioscler Thromb Vasc Biol. 2004;24:1374–1383. doi: 10.1161/01.ATV.0000134298.25489.92. [DOI] [PubMed] [Google Scholar]
- 55.Fischer D., Schloesser R.L., Nold-Petry C.A., Nold M.F., Veldman A. Protein C concentrate in preterm neonates with sepsis. Acta Paediatr. 2009;98:1526–1529. doi: 10.1111/j.1651-2227.2009.01404.x. [DOI] [PubMed] [Google Scholar]
- 56.Dreyfus M., Ladouzi A., Chambost H., Gruel Y., Tardy B., Ffrench P., Bridey F., Tellier Z. Treatment of inherited protein C deficiency by replacement therapy with the French purified plasma-derived protein C concentrate (PROTEXEL) Vox Sang. 2007;93:233–240. doi: 10.1111/j.1423-0410.2007.00953.x. [DOI] [PubMed] [Google Scholar]
- 57.Abraham E. Effects of recombinant human activated protein C in human models of endotoxin administration. Proc Am Thorac Soc. 2005;2:243–247. doi: 10.1513/pats.200501-004AC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dreyfus M., Magny J.F., Bridey F., Schwarz H.P., Planche C., Dehan M., Tchernia G. Treatment of homozygous protein C deficiency and neonatal purpura fulminans with a purified protein C concentrate. N Engl J Med. 1991;325:1565–1568. doi: 10.1056/NEJM199111283252207. [DOI] [PubMed] [Google Scholar]
- 59.Dreyfus M., Masterson M., David M., Rivard G.E., Muller F.M., Kreuz W., Beeg T., Minford A., Allgrove J., Cohen J.D., Christoph J., Bergmann F., Mitchell V.E., Haworth C., Nelson K., Schwarz H.P. Replacement therapy with a monoclonal antibody purified protein C concentrate in newborns with severe congenital protein C deficiency. Semin Thromb Hemost. 1995:371–381. doi: 10.1055/s-2007-1000658. [DOI] [PubMed] [Google Scholar]
- 60.Guitton C., Cottereau A., Gerard N., Quillard T., Chauveau A., Devalliere J., Tonnerre P., Charreau B. Protective cross talk between activated protein C and TNF signaling in vascular endothelial cells: implication of EPCR, noncanonical NF-kappaB, and ERK1/2 MAP kinases. Am J Physiol Cell Physiol. 2011;300:C833–C842. doi: 10.1152/ajpcell.00003.2010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Role of thrombomodulin (TM), endothelial protein C receptor (EPCR), and protease-activated receptor-1 (PAR-1) for protein C (PC)-induced inhibition of intercellular adhesion molecule 1 (ICAM-1) expression in tumor necrosis factor-α (TNF-α)-stimulated cremaster muscles. Endothelial ICAM-1 expression assessed by immunohistochemistry in 3 hours of TNF-α stimulated cremaster muscle venules of PC-treated (100 U/kg, 3 hours) TM deficient (TMPro/Pro) mice, wild-type (WT) mice, and WT mice pretreated with the EPCR-blocking antibody RCR252 or PAR-1 inhibitor SCH79797 were compared to untreated WT control mice. The primary antibody against ICAM-1 was injected systemically and incubated for 10 minutes. Cremaster muscle whole mounts were then fixed and permeabilized. Biotinylated secondary antibody, peroxidase-conjugated streptavidin, and diaminobenzidine were used to detect endothelial expression as brown signal. Counterstaining was performed by H&E. The intensity of venular immunostaining was analyzed semi-quantitatively (0 = no, 1 = weak, 2 = medium, 3 = strong signal) and was presented as mean ± SEM from at least three mice per group (A). The asterisk indicates significant differences (P < 0.05) to all other groups. In addition, representative micrographs are depicted to illustrate the endothelial expression of ICAM-1 (B–G). Reference bar for (B–G) is shown in (C) and represents 50 μm.
Role of thrombomodulin (TM), endothelial protein C receptor (EPCR), and protease-activated receptor-1 (PAR-1) for protein C (PC)-induced inhibition of vascular cell adhesion molecule 1 (VCAM-1) expression in tumor necrosis factor-α (TNF-α)-stimulated cremaster muscles. Endothelial VCAM-1 expression assessed by immunohistochemistry in 3 hours TNF-α-stimulated cremaster muscle venules of PC treated (100 U/kg, 3 hours) TM deficient (TMPro/Pro) mice, wild-type (WT) mice, and WT mice pretreated with the EPCR-blocking antibody RCR252 or PAR-1 inhibitor SCH79797 were compared to untreated WT control mice. The primary antibody against VCAM-1 was injected systemically and incubated for 10 minutes. Cremaster muscle whole mounts were then fixed and permeabilized. Biotinylated secondary antibody, peroxidase-conjugated streptavidin, and diaminobenzidine were used to detect endothelial expression as brown signal. Counterstaining was performed by H&E. The intensity of venular immunostaining was analyzed semi-quantitatively (0 = no, 1 = weak, 2 = medium, 3 = strong signal) and presented as mean ± SEM from at least three mice per group.
Chemokine (C-X-C motif) ligand 1 (CXCL1)-induced leukocyte adhesion in control mice. Intravital microscopy was used to visualize leukocyte arrest in an unstimulated cremaster muscle venule (vessel diameter = 27 μm) of a control mouse before and after systemic injection of 600 ng CXCL1. Superimposing CXCL1 in the top left corner indicates the time of injection. CXCL1 injection led to a rapid and effective arrest of leukocytes to the venular wall.
Chemokine (C-X-C motif) ligand 1 (CXCL1)-induced leukocyte adhesion in protein C (PC)-treated mice. This video exemplifies the impairment of CXCR2-mediated leukocyte arrest in cremaster muscle venules of mice treated with 100 U/kg PC for 3 hours. After systemic injection of 600 ng CXCL1 into a PC-treated mouse, leukocyte arrest is dramatically reduced in the observed cremaster muscle venule (vessel diameter = 31 μm).