

Abstract
Mitochondria contain their own genetic material and evolved from prokaryotic ancestors some two billion years ago. They are the main source of the cell's energy supply and are involved in such important processes as apoptosis, mitochondrial diseases and aging. Mitochondria display a complex dynamical behavior involving cycles of fusion and fission, the function of which is as yet unknown. We recently proposed a concise theory that explains: (1) why fusion and fission have evolved, (2) how these processes relate to the accumulation of mitochondrial mutants during aging and (3) why mtDNA is located close to the respiration complexes where most radicals are generated. We also believe that this ‘organelle control’ theory may explain why mutations in mitochondrial tRNA genes are the most prevalent kind of defect associated with inherited human mitochondrial diseases, despite the fact that mt-tRNA genes account for only 5% of the mtDNA coding sequence.
Key words: aging, mitochondria, fusion, fission, evolution, organelle control theory, mutation, mitochondrial disease
All higher eukaryotes contain mitochondria, endosymbiotic organelles1 with their own small genomes (mtDNA), which produce the majority of ATP for the cell. During the course of evolution large parts of the original bacterial genome were transferred to the host cell nucleus, probably to minimize host-endosymbiont conflict,2 leaving only a small fraction of the genes required for mitochondrial functions encoded within mtDNA. Nevertheless, mitochondria undergo their own cycles of replication and removal within the cell, providing opportunities for selection at the level of the organelle.
MtDNA mutations are involved in many diseases3 and also in the aging process.4–8 Mitochondria are the major site of production of damaging Reactive Oxygen Species (ROS), which are formed as by-products of the respiratory chain reactions and are assumed to result in high levels of mtDNA mutation.
Another important finding of recent years is that mitochondria regularly fuse into a dynamic network within which they exchange proteins, mtDNA and lipids.9,10 Why mitochondria fuse and divide has long been a puzzle.11 In a recent publication12 we proposed that (1) mitochondrial fusion is necessary to control mitochondria in an optimal way after most genes have been migrated to the nucleus, (2) fusion also allows the accumulation of mitochondrial mutants during aging, (3) mitochondrial fission and selective turnover is then necessary to remove accumulated mutants and (4) this requires a physical connection between nucleoids and the inner mitochondrial membrane.
Although mitochondria perform in principle the same functions, their requirements for the various proteins quickly diverge because mitochondria at different cellular locations have different metabolic activities. This makes it impossible to supply different mitochondria with an optimal amount of proteins, if only a single set of mitochondrial genes exists in the nucleus. Mitochondrial fusion offers a solution by equilibrating the concentrations of all nuclear-encoded mitochondrial proteins over all organelles, effectively forming a single mitochondrial compartment in the cell. Transcriptional control of a single set of mitochondrial genes is then sufficient to respond in an optimal way to a single set of protein concentrations in the mitochondrial matrix.
Fusion, however, introduces new difficulties because it lessens the capacity for selection to deal with the frequent mtDNA mutations that arise because of the vulnerability of mtDNA to attack by ROS. Single cell studies have revealed that old cells tend to contain only a single type of mtDNA mutation, different for each cell, that has displaced the wild type (wt) mitochondria to a large degree.13,14 This suggests that either the mutations have a selection advantage over the wt, leading to a clonal expansion of the single mutant, or that clonal expansion happens slowly over time through random drift. Previous debate has placed these ideas in opposition. On the one hand, the reduced size of mtDNA deletion mutants might allow them to replicate faster than wt.15–17 On the other hand, the time required for the replication of the mtDNA is much shorter than the half-life of mitochondria, suggesting mtDNA replication is not a rate-limiting step—the so-called “relaxed replication” principle.18,19 In fact, once fusion becomes a regular feature of mitochondrial dynamics, the situation is different. Within the fused complex, the competing entities are no longer mitochondria, but individual mtDNA molecules. For the outcome of this competition only the mtDNA replication times are important. Indeed, using computer simulations it could be shown that this “survival of the tiny” idea can very well reproduce experimental findings regarding the distribution of deletion sizes in rat muscle fibers.20 Thus we propose that mitochondrial fusion is the underlying mechanism that opens the door for the clonal expansion of mitochondrial deletion mutants.
In order to defend cells against the continual threat of being taken over by mtDNA deletions a mechanism to combat this accumulation is needed. Fission of the mitochondrial syncytium into smaller fragments—individual organelles—offers this defense since it has been found that mitochondrial fragments often display unequal membrane potential after fission and that fragments with lowered potential are more likely to undergo removal by mitophagy.21 What is needed to enable such a mechanism to counter the accumulation of mtDNA mutants is a link between genotype and phenotype. It is known that the mtDNA is attached to the inner mitochondrial membrane and we propose that it is actually connected to respiration complexes that are encoded by this specific molecule of mtDNA.12 In prokaryotes, a connection between translation and membrane insertion exists and is known as transertion.22
The essence of the organelle control theory is summarized in Figure 1. Fusion is needed to deliver optimal control of mitochondrial metabolism, but this leaves the cell vulnerable to mtDNA mutations. Fission provides the basis to check the accumulation of mutants through selective elimination, provided there exists some attachment of nucleoids to their own gene products (respiration complexes) in the inner mitochondrial membrane. Although it will be technically challenging to test this idea experimentally, there exists circumstantial evidence supporting this idea. Twig et al.10 demonstrated that mitochondrial fragments often show an unequal membrane potential after fission and proposed that this is a consequence of random sorting. However, if intact and damaged OXPHOS complexes could freely diffuse and mix in the membrane, such sorting would be significantly less likely than if the complexes are arranged in large clusters containing either intact or damaged proteins. Furthermore, the necessary link between genotype and phenotype can only be established between the mtDNA and membrane proteins, and not with diffusible matrix proteins. In agreement with this requirement, animal mitochondrial genomes code only for membrane proteins.
Figure 1.
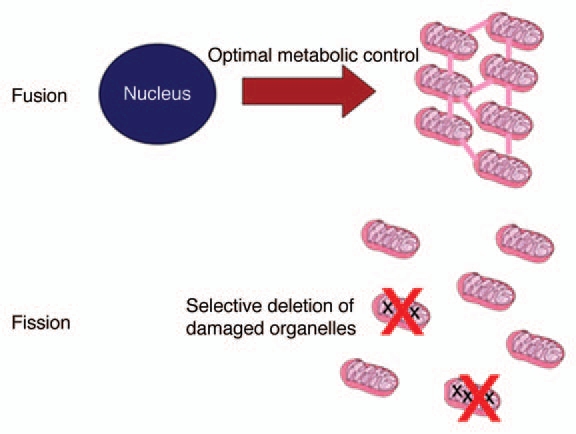
Key ingredients of the organelle control theory. Most mitochondrial proteins are encoded in the nucleus and an optimal supply of the different mitochondria can only be achieved if fusion equilibrates all protein concentrations. But since this also allows the accumulation of mtDNA mutants, fission and selective degradation of defective fragments is necessary. See text for details.
Intriguingly, the only significant set of mtDNA-encoded genes not specifying membrane proteins are those coding for mitochondrial tRNAs. During the transfer of mitochondria through germ-line, which happens only in females, there is an important bottleneck in mitochondrial copy number that occurs transiently during oocyte development. This bottleneck is thought to facilitate some form of quality control to ensure that the next generation starts life as free as possible from acquired mtDNA mutations. If the quality control mechanism is based on the integrity of the mitochondrial membrane potential, as seems to be the case for selective degradation of defective mitochondria in somatic cells, it would work well against mtDNA mutations in protein-coding genes but not against mtDNA mutations affecting tRNAs. It may therefore be significant that mt-tRNA mutations are the most prevalent genetic defect in cases of familial mitochondrial disease, even though the 22 mt-DNA genes account for only about 5% of the human mtDNA coding sequence.23
Acknowledgments
T.B.L.K. is supported by the BBSRC Centre for Integrated Systems Biology of Aging and Nutrition and by the UK NIHR Biomedical Research Centre for Aging and Age-related disease award to the Newcastle upon Tyne Foundation Hospitals NHS Trust. A.K. was supported by a grant of the BMBF (‘GerontoMitoSys’). Furthermore, this work was partly funded by a Glenn Foundation award to T.B.L.K.
References
- 1.Schwartz RM, Dayhoff MO. Origins of prokaryotes, eukaryotes, mitochondria and chloroplasts. Science. 1978;199:395–403. doi: 10.1126/science.202030. [DOI] [PubMed] [Google Scholar]
- 2.Blackstone NW, Kirkwood TBL. Mitochondria and programmed cell death “Slave Revolt” or community homeostasis? In: Hammerstein P, editor. Genetic and Cultural Evolution of Cooperation. Cambridge: MIT Press; 2003. [Google Scholar]
- 3.Wallace DC. Mitochondrial disease in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 4.Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20:145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 5.Harman D. Free radical theory of aging: consequences of mitochondrial aging. Age. 1983;6:86–94. [Google Scholar]
- 6.Miquel J. An integrated theory of aging as the result of mitochondrial-DNA mutation in differentiated cells. Arch Gerontol Geriat. 1991;12:99–117. doi: 10.1016/0167-4943(91)90022-i. [DOI] [PubMed] [Google Scholar]
- 7.Linnane AW, Marzuki S, Ozawa T, Tanaka M. Mitochondrial DNA mutations as an important contributor to ageing and degenerative diseases. Lancet. 1989;333:642–645. doi: 10.1016/s0140-6736(89)92145-4. [DOI] [PubMed] [Google Scholar]
- 8.Miquel J, Economos AC, Fleming J, Johnson JE. Mitochondrial role in cell ageing. Exp Gerontol. 1980;15:575–591. doi: 10.1016/0531-5565(80)90010-8. [DOI] [PubMed] [Google Scholar]
- 9.Duvezin-Caubet S, Jagasia R, Wagener J, Hofmann S, Trifunovic A, Hansson A, et al. Proteolytic processing of OPA1 links mitochondrial dysfunction to alterations in mitochondrial morphology. J Biol Chem. 2006;281:37972–37979. doi: 10.1074/jbc.M606059200. [DOI] [PubMed] [Google Scholar]
- 10.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27:433–446. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skulachev VP. Power transmission along biological membranes. J Membr Biol. 1990;114:97–112. doi: 10.1007/BF01869092. [DOI] [PubMed] [Google Scholar]
- 12.Kowald A, Kirkwood TB. Evolution of the mitochondrial fusion-fission cycle and its role in aging. Proc Natl Acad Sci USA. 2011;108:10237–10242. doi: 10.1073/pnas.1101604108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Khrapko K, Bodyak N, Thilly WG, van Orsouw NJ, Zhang X, Coller HA, et al. Cell by cell scanning of whole mitochondrial genomes in aged human heart reveals a significant fraction of myocytes with clonally expanded deletions. Nucl Acids Res. 1999;27:2434–2441. doi: 10.1093/nar/27.11.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cao Z, Wanagat J, McKiernan SH, Aiken JM. Mitochondrial DNA deletion mutations are concomitant with ragged red regions of individual, aged muscle fibers: Analysis by laser-capture microdissection. Nucl Acids Res. 2001;29:4502–4508. doi: 10.1093/nar/29.21.4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayashi J, Ohta S, Kikuchi A, Takemitsu M, Goto Y, Nonaka I. Introduction of disease-related mitochondrial DNA deletions into HeLa cells lacking mitochondrial DNA results in mitochondrial dysfunction. Proc Natl Acad Sci USA. 1991;88:10614–10618. doi: 10.1073/pnas.88.23.10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wallace DC. Mitochondrial genetics: a paradigm for aging and degenerative diseases? Science. 1992;256:628–632. doi: 10.1126/science.1533953. [DOI] [PubMed] [Google Scholar]
- 17.Takai D, Isobe K, Hayashi JI. Transcomplementation between different types of respiration deficient mitochondria with different pathogenic mutant mitochondrial DNAs. J Biol Chem. 1999;274:11199–11202. doi: 10.1074/jbc.274.16.11199. [DOI] [PubMed] [Google Scholar]
- 18.Elson JL, Samuels DC, Turnbull DM, Chinnery PF. Random intracellular drift explains the clonal expansion of mitochondrial DNA mutations with age. Am J Hum Genet. 2001;68:802–806. doi: 10.1086/318801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Grey ADNJ. The Mitochondrial Free Radical Theory of Aging. Austin: RG Landes Company; 1999. [Google Scholar]
- 20.Kowald A, Jendrach M, Pohl S, Bereiter-Hahn J, Hammerstein P. On the relevance of mitochondrial fusions for the accumulation of mitochondrial deletion mutants: a modelling study. Aging Cell. 2005;4:273–283. doi: 10.1111/j.1474-9726.2005.00169.x. [DOI] [PubMed] [Google Scholar]
- 21.Twig G, Hyde B, Shirihai OS. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim Biophys Acta. 2008;1777:1092–1097. doi: 10.1016/j.bbabio.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woldringh CL. The role of co-transcriptional translation and protein translocation (transertion) in bacterial chromosome segregation. Mol Microbiol. 2002;45:17–29. doi: 10.1046/j.1365-2958.2002.02993.x. [DOI] [PubMed] [Google Scholar]
- 23.Elson JL, Swalwell H, Blakely EL, McFarland R, Taylor RW, Turnbull DM. Pathogenic mitochondrial tRNA mutations—which mutations are inherited and why? Hum Mutat. 2009;30:984–992. doi: 10.1002/humu.21113. [DOI] [PubMed] [Google Scholar]