

Abstract
Inflammation driven by immune cells and pro-inflammatory cytokines is implicated in pancreatic β-cell injury, leading to the development of diabetes mellitus. IL-27, a cytokine consisting of IL-27p28 and Epstein-Barr virus–induced gene 3 (EBI3), binds a membrane-bound heterodimeric receptor consisting of the IL-27 receptor α chain (WSX-1) and gp130. IL-27 has anti-inflammatory properties that regulate T-cell polarization and cytokine production. We evaluated blood glucose and islet proinsulin concentrations, inflammatory cell infiltration in islets, and expression of IL-1β mRNA in pancreas in wild-type (WT), EBI3−/−, and WSX-1−/− mice treated with streptozotocin (STZ). Hyperglycemia was augmented in EBI3−/− and WSX-1−/− mice compared with WT mice. Islet proinsulin levels after STZ treatment were lower in EBI3−/− and WSX-1−/− mice than in WT mice. The infiltration of islets by F4/80+CD11c−7/4− macrophages, CD4+ T cells, and CD8+ T cells was increased in EBI3−/− and WSX-1−/− mice compared with WT mice. The administration of recombinant IL-27, compared with control, decreased the blood glucose level, immune cell infiltration into islets, and IL-1β mRNA expression in the pancreas and increased islet proinsulin levels in WT and EBI3−/− mice. Thus, IL-27 inhibits STZ-induced hyperglycemia and pancreatic islet inflammation in mice and represents a potential novel therapeutic approach for β-cell protection in diabetes.
Insulin insufficiency, resulting from injury to the insulin-producing β cells in pancreatic islets, plays a pivotal role in the development of diabetes mellitus. Accumulating evidence suggests that inflammatory processes driven by T cells and macrophages are involved in β-cell injury.1 Autoantigens are thought to trigger imbalanced T-cell differentiation, resulting in the autoimmune-mediated β-cell injury involved in the pathogenesis of type 1 diabetes. T cells differentiate into effector T cells by engaging with β-cell autoantigens that are presented by local antigen-presenting cells, including macrophages.2, 3 CD4+ and CD8+ T cells are required to initiate islet inflammation in type 1 diabetes, and CD8+ T cells participate in the process of β-cell destruction. On the other hand, metabolic stress, including hyperglycemia in type 2 diabetes, stimulates β-cell production of inflammatory mediators, such as cytokines and chemokines that promote immune cell infiltration into islets, resulting in islet inflammation and resultant β-cell injury.4 Pro-inflammatory cytokines, such as IL-1β, reportedly promote β-cell injury by stimulating immune cell– and cytokine-mediated injury and increasing reactive oxygen species production in the pancreas.5
Streptozotocin (STZ) is a naturally occurring chemical that is selectively toxic to mammalian β cells by inducing alkylation-mediated DNA damage and has been used to induce β-cell injury in vivo, resulting in an experimental model of insulin insufficiency and diabetes.6 The administration of relatively low-dose STZ for 5 consecutive days induces diabetes in vivo with hyperglycemia and reduces insulin production associated with inflammatory changes of pancreatic islets involving T cells and macrophages.7
IL-27 is a heterodimeric cytokine consisting of IL-27p28 and Epstein-Barr virus–induced gene 3 (EBI3) and belongs to the IL-6 subfamily of type 1 cytokines. IL-27 binds to a membrane-bound heterodimeric receptor consisting of the IL-27 receptor α chain (WSX-1) and gp130 and activates cell signaling predominantly through multiple Jak and STAT proteins.8, 9 IL-27 receptor complex is expressed by T cells and innate immune cells, such as macrophages and dendritic cells, and by endothelial cells, indicating that multiple cell types are able to respond to IL-27.10 Infection of WSX-1–deficient mice that lack IL-27 receptors with pathogens resulted in augmented T-cell responses and enhanced pro-inflammatory cytokine production.11, 12 The anti-inflammatory effects of IL-27 signaling, which regulates polarization of T-cell subsets, such as types 1 and 17 helper T cells (Th1 and Th17 cells, respectively), and cytokine production have been demonstrated in animal models of experimental autoimmune encephalitis, allergic asthma, and delayed-type hypersensitivity of skin.13, 14, 15, 16
In the present study, we examined the involvement of IL-27 signaling in hyperglycemia and inflammation-mediated β-cell injury induced by STZ by comparing EBI3−/− and WSX-1−/− mice that lack IL-27 and IL-27 receptor, respectively, with wild-type (WT) mice.11, 17 EBI3−/− and WSX-1−/− mice exhibit augmented hyperglycemia and inflammatory β-cell injury after STZ administration compared with WT control mice. Also, the administration of recombinant IL-27 inhibits hyperglycemia and inflammatory β-cell injury induced by STZ in WT and EBI3−/− mice, suggesting that IL-27 represents a novel therapeutic approach for β-cell protection in diabetes.
Materials and Methods
Animal Model
EBI3−/− and WSX-1−/− mice,11, 17 backcrossed at least 10 times to C57BL/6J and littermate WT mice, were used in this study. Eight-week-old male C57BL6 WT, EBI3−/−, and WSX-1−/− mice were i.p. injected with STZ (Sigma-Aldrich, St. Louis, MO) dissolved in 0.1 mol/L citrate buffer (pH 4.0) at a dose of 40 mg/kg per day for 5 consecutive days, as previously described.7 Recombinant IL-27 was injected daily at a dose of 400 ng/day for 10 days from the beginning of STZ treatment. Nonfasting blood glucose, taken from tail veins, was measured using a glucometer (G-Checker; Gunze, Kyoto, Japan). Animal care and all experiments were conducted in accordance with the institutional guidelines of Saga University Faculty of Medicine, Saga, Japan.
Oral Glucose Tolerance Test
After fasting overnight for 16 hours, mice (n = 6 per group) were orally loaded with 10% glucose (2 g/kg). Blood glucose was measured before the loading and 30, 60, 90, and 120 minutes after the loading from the tail vein with a glucometer (G-Checker).
Recombinant IL-27
Mouse EBI3 and IL-27p28 cDNAs were isolated by RT-PCR using total RNA prepared from concanavalin A–activated splenocytes. The fragment encoding EBI3-(VPGVG)2 linker-IL-27p28 was generated by PCR and cloned into the p3xFLAG-CMV-14 vector (Sigma-Aldrich). 293F cells were then transiently transfected with the expression vector using 293fectin (Invitrogen, Carlsbad, CA). After 3 days, culture supernatant was collected and 3xFLAG-tagged recombinant single-chain IL-27 was purified by affinity chromatography using anti-FLAG (M2) affinity gel (Sigma-Aldrich). The protein concentration was determined by titration of immunoblots with anti-FLAG (M2) antibody.
Histological and Immunofluorescence Findings
H&E staining was performed using paraffin-embedded pancreas specimens fixed in 4% paraformaldehyde. For immunofluorescence, sections were rinsed with PBS and blocked with PBS containing 1% bovine serum albumin. For the detection of neutrophils, sections were incubated with anti-neutrophil rat monoclonal antibody 7/4 conjugated with phycoerythrin (clone 7/4; Abcam, Cambridge, UK). Sections were triple stained with Alexa Fluor 488–conjugated anti-F4/80 antibody (clone BM8; eBioscience, San Diego, CA), Alexa Fluor 647–conjugated anti-CD11c antibody (clone N418; eBioscience), and phycoerythrin-conjugated 7/4 antibody for the detection of F4/80+CD11c−7/4− macrophages. To detect CD4 and CD8 antigen, sections were double stained with fluorescein isothiocyanate (FITC)–conjugated anti-CD4 antibody (clone GK1.5; eBioscience) and phycoerythrin-conjugated anti-CD8 antibody (clone 53–6.7; eBioscience). For the detection of proinsulin, sections were incubated with guinea pig anti-mouse insulin C chain polyclonal antibody (Takara Bio Inc., Otsu, Japan) that was raised against the amino-terminal part of mouse insulin C chain (71–84; SPGDLQTLALEVAR) as antigen and reacts with proinsulin, including insulin C chain, followed by incubation with Alexa Fluor 488–conjugated goat anti-guinea pig IgG antibody (Invitrogen). After rinsing, sections were mounted. Images were captured using confocal microscopy (LSM5 Pascal; Carl Zeiss, Jena, Germany). The cellular proinsulin levels were evaluated by measuring the fluorescence intensity of immunofluorescence microscopy images using the ImageJ software program (NIH, Bethesda, MD).
Quantification of Cell Number in Islets
The standard islet size was determined to undertake statistical analysis. Fifteen nonconsecutive islets were chosen from each group, and the islet area was measured using Axiovision software (Carl Zeiss). In the present study, the standard islet size was calculated as 7878 ± 1090 μm2. No significant differences in standard islet size were detected between WT, EBI3−/−, and WSX-1−/− mice. Ten nonconsecutive standard islets were randomly selected from 10 sections per mouse, and the numbers of cells were counted in samples from the seven mice in each group. Data shown are the mean ± SE of cell number per 10 islets.
Pancreatic Islet Isolation
A cold collagenase solution was injected from the common bile duct and then the pancreas was removed, followed by incubation with a collagenase solution at 37°C for 17 minutes. After digestion, the pancreas was poured through sterile mesh and mixed with a buffer containing HBSS, HEPES (10 mmol/L), and 5% fetal calf serum. Cells were collected by centrifugation at 250 × g for 1 minute. After washing three times, cells were suspended with 25% Ficoll solution. A 23% Ficoll solution was layered on top of the suspended pellets, and the remaining densities of Ficoll solutions were mounted on top of each density in 20% and 11% in order, followed by centrifugation at 800 × g for 10 minutes. The islets were collected from the second layer and washed three times. Collected islets were suspended with RPMI 1640 medium (Sigma-Aldrich) containing 10% fetal calf serum and incubated at 37°C in a 5% CO2 incubator.
RT-PCR Data
RNA was extracted from the thymus and isolated pancreatic islets of WT mice with Isogen, following the manufacturer's protocol (Nippon Gene, Tokyo, Japan). cDNA synthesis was performed using 2 μg of total RNA and a RETRO script kit, following the manufacturer's protocol (Ambion, Austin, TX). RT-PCR was performed as reported. The specific primer sets for the target genes were as follows: WSX-1, 5′-AAATGAAGCCAGACACACCC-3′ and 5′-CACACAAGGTCTTGGGTCCT-3′ (GenBank accession number NM_016671); and gp130, 5′-TCCGTGCAAGTGTTCTCAAG-3′ and 5′-CCCAGGTGTGACTTTGTCCT-3′ (GenBank accession number NM_010560). These primers were synthesized by JBioS (Saitama, Japan). β-Actin was amplified using QuantumRNA β-actin Internal Standards from Ambion. cDNA was amplified by PCR (WSX-1, 39 cycles of 40 seconds at 94°C, 20 seconds at 59°C, and 40 seconds at 72°C; and gp130, 38 cycles of 40 seconds at 94°C, 20 seconds at 60°C, and 40 seconds at 72°C). The RT-PCR products were analyzed by agarose gel electrophoresis (2.0%), followed by ethidium bromide staining.
Real-Time PCR
Total RNA was isolated from pancreatic tissues with Isogen, following the manufacturer's protocol. cDNA synthesis was performed using 2 μg of total RNA and the RETROscript Kit (Ambion). Real-time PCR was conducted using the SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) and was performed with an ABI 7300 Sequence Detection System (Applied Biosystems) using a standard temperature profile (2 minutes at 50°C, 10 minutes at 95°C, and 40 to 45 cycles of 15 seconds at 95°C and 1 minute at 60°C). Cycle threshold values were determined by automated threshold analysis with ABI Prism version 1.3.1 software (Applied Biosystems). The specific primer sets for mouse F4/80 antigen, mouse CD4, mouse CD8, and mouse IL-1β were obtained from Takara Bio Inc. β-Actin and glyceraldehyde-3-phosphate dehydrogenase, which served as internal controls, were commercially available from Takara Bio Inc. mRNA expression was normalized with respect to β-actin or glyceraldehyde-3-phosphate dehydrogenase expression and expressed relative to control levels.
Statistical Analysis
Data are expressed as mean ± SE. The significance in each group was evaluated with an unpaired Student's t-test, a one-way analysis of variance, and the Tukey's test. P < 0.05 was considered statistically significant.
Results
Nonfasting blood glucose levels were measured before and on days 7, 14, and 28 after the last dose of STZ treatment (Figure 1A). In this multiple low-dose STZ model, nonfasting blood glucose levels did not become significantly elevated after STZ treatment in control WT mice. In contrast, the nonfasting blood glucose levels were significantly higher on days 14 and 28 and tended to be higher on day 7 in EBI3−/− and WSX-1−/− mice compared with WT mice. No significant differences in nonfasting blood glucose levels were detected between WT, EBI3−/−, and WSX-1−/− mice treated with control vehicle at any point (data not shown). EBI3−/− and WSX-1−/− mice displayed significantly impaired glycemic control in a glucose tolerance test compared with WT mice on day 14 after the last dose of STZ treatment (Figure 1B). These data suggest that deficiency of EBI3 and WSX-1 exacerbates the effects of STZ with the induction of hyperglycemia and impaired glucose tolerance in these mice.
Figure 1.
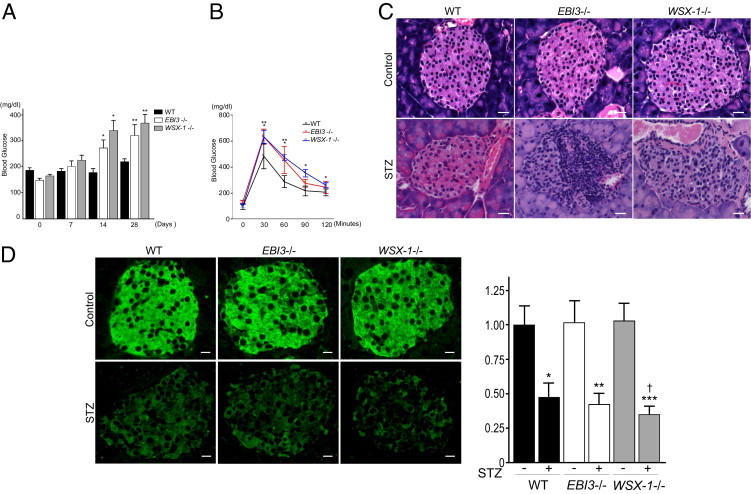
Exacerbation of STZ-induced hyperglycemia, impaired glucose tolerance, and β-cell injury by the deficiency of EBI3 and WSX-1. A: STZ-induced hyperglycemia in WT, EBI3−/−, and WSX-1−/− mice. Mice aged 8 weeks were injected i.p. with STZ (40 mg/kg per day) or control vehicle for 5 consecutive days (n = 10 to 12 per group). Nonfasting blood glucose levels were determined using blood from the tail vein before STZ treatment (day 0) and on days 7, 14, and 28 after STZ treatment in WT mice (black bar), EBI3−/− mice (white bar), and WSX-1−/− mice (gray bar). Data shown are the mean ± SE. *P < 0.05 versus WT mice on day 14; **P < 0.05 versus WT mice on day 28. B: Glucose tolerance test after an overnight fast in WT (black line), EBI3−/− (red line), and WSX-1−/− mice (blue line) on day 14 after STZ treatment (n = 6 per group). Data shown are the mean ± SE. *P < 0.05 for EBI3−/− mice versus WT mice at each point; **P < 0.05 for WSX-1−/− mice versus WT mice at each point. C: Pancreatic tissue was resected on day 28 and histologically examined. H&E staining revealed mononuclear cell infiltration within islets of STZ-treated WT, EBI3−/−, and WSX-1−/− mice, which was not evident in vehicle-treated WT, EBI3−/−, and WSX-1−/− mice. The mononuclear cell infiltrate was more marked in STZ-treated EBI3−/− and WSX-1−/− mice than in STZ-treated WT mice. Scale bar = 20 μm. D: Sections of pancreatic tissue were stained with anti-mouse insulin C-chain antibody that reacts with proinsulin and examined by immunofluorescence. No obvious changes in proinsulin staining of islets were observed in vehicle-treated WT, EBI3−/−, and WSX-1−/− mice, whereas STZ treatment decreased islet proinsulin staining compared with vehicle controls in each group. Proinsulin staining was weaker in STZ-treated EBI3−/− and WSX-1−/− mice than in STZ-treated WT mice (left panel). Scale bar = 20 μm. Fluorescence intensities, measured as described in Materials and Methods, were expressed as relative to that of WT mice treated with control vehicle (right panel). *P < 0.05 versus control vehicle treatment in WT mice; **P < 0.05 versus control vehicle treatment in EBI3−/− mice; ***P < 0.05 versus control vehicle treatment in WSX-1−/− mice; †P < 0.05 versus WT mice treated with STZ.
The histological analysis of pancreatic tissues was then undertaken to investigate the molecular mechanism underlying the promotion of STZ-induced hyperglycemia in EBI3−/− and WSX-1−/− mice. The administration of STZ resulted in both intraislet and peri-islet mononuclear cell infiltration in WT mice, with increased intraislet and peri-islet mononuclear cell infiltration evident in EBI3−/− and WSX-1−/− mice compared with WT mice (Figure 1C). Intraislet and peri-islet mononuclear cell infiltration was barely detectable in either group of mice injected with control vehicle (Figure 1C). These data indicate that deficiency of EBI3 and WSX-1 promotes STZ-induced pancreatic islet inflammation in mice.
Cellular proinsulin levels of pancreatic islets were evaluated by immunostaining with antibody against the insulin C chain that composes proinsulin. Immunofluorescence microscopy revealed decreased cellular proinsulin levels in pancreatic islets on day 28 after STZ treatment in each type of mice compared with control vehicle treatment (Figure 1D). However, cellular proinsulin levels were significantly lower in WSX-1−/− mice and tended to be lower in EBI3−/− mice compared with WT mice after STZ treatment (Figure 1D). No significant changes in intraislet proinsulin levels were observed in any mice treated with control vehicle (Figure 1D). These data indicate that insulin production in pancreatic islets was reduced to a greater extent in EBI3−/− and WSX-1−/− mice compared with WT mice after STZ treatment.
To evaluate the infiltration of polymorphonuclear leukocytes into pancreatic islets, the numbers of polymorphonuclear leukocytes in pancreatic islets were counted in H&E-stained pancreatic tissues (Figure 2A). The infiltration of polymorphonuclear leukocytes into pancreatic islets was hardly detectable in WT, EBI3−/−, and WSX-1−/− mice treated with control vehicle. The number of polymorphonuclear leukocytes was slightly, but not significantly, higher in EBI3−/− and WSX-1−/− mice compared with WT mice on day 7 after STZ treatment. The infiltration of neutrophils into pancreatic islets was examined by staining pancreatic tissues with anti-neutrophil antibody 7/4 (Figure 2A). The number of 7/4+ neutrophils in islets showed no significant differences between WT and EBI3−/− or WSX-1−/− mice on day 7 after STZ treatment. To assess macrophage infiltration into islets, we identified and counted intraislet and peri-islet F4/80+CD11c−7/4− cells as a macrophage subset by the observation of triple-stained pancreatic tissues using confocal microscopy that results in the exclusion of CD11c+ dendritic cells and 7/4+ neutrophils from F4/80+ cells (Figure 2B). The number of F4/80+CD11c−7/4− cells was significantly higher in EBI3−/− and WSX-1−/− mice compared with WT mice on day 28 after STZ treatment. The infiltration of F4/80+CD11c−7/4− cells was hardly detectable in WT, EBI3−/−, and WSX-1−/− mice treated with control vehicle. mRNA expression of F4/80 antigen in pancreatic tissues from STZ-treated mice, quantified by real-time PCR, revealed that F4/80 antigen expression is up-regulated in EBI3−/− and WSX-1−/− mice compared with WT mice (Figure 2B). Accumulating evidence has shown that T cells are important mediators and effectors in the pathogenesis of islet inflammation. Accordingly, the number of CD4+ and CD8+ T cells infiltrated into islets was assessed by double staining of pancreatic tissues with FITC-conjugated anti-CD4 antibody and phycoerythrin-conjugated anti-CD8 antibody (Figure 2C). The number of CD4+ cells was significantly higher in WSX-1−/− mice compared with WT mice on day 28 after STZ treatment. The infiltration of CD4+ cells was hardly detectable in WT, EBI3−/−, and WSX-1−/− mice treated with control vehicle. mRNA expression of CD4 in pancreatic tissues from STZ-treated mice quantified by real-time PCR tended to be up-regulated in EBI3−/− and WSX-1−/− mice compared with WT mice (Figure 2C). The number of CD8+ cells in islets was significantly higher in EBI3−/− and WSX-1−/− mice compared with WT mice on day 28 after STZ treatment (Figure 2C). The infiltration of CD8+ cells was hardly detectable in WT, EBI3−/−, and WSX-1−/− mice treated with control vehicle. mRNA expression of CD8 in pancreatic tissues from STZ-treated mice, quantified by real-time PCR, was significantly up-regulated in WSX-1−/− mice and tended to increase in EBI3−/− mice compared with WT mice (Figure 2C). These data suggest that STZ-induced islet inflammation, as represented by infiltration with macrophages and CD4+ and CD8+ T cells, was augmented by deficiency of EBI3 and WSX-1 compared with WT mice.
Figure 2.
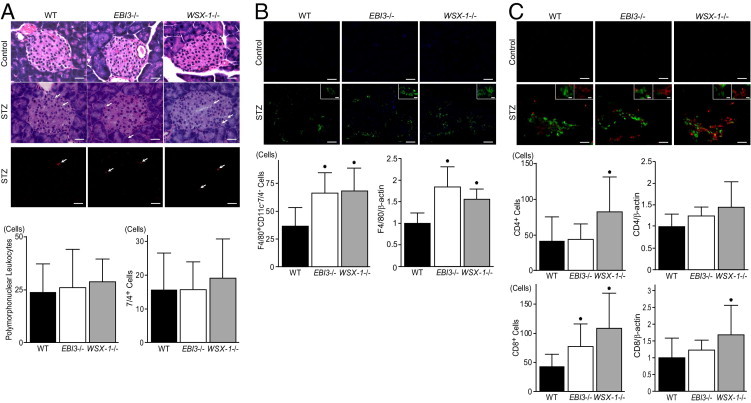
Promotion of STZ-induced immune cell infiltration into islets by the deficiency of EBI3 and WSX-1. A: Representative photomicrographs of H&E- stained islets from WT, EBI3−/−, and WSX-1−/− mice on day 7 after treatment with control vehicle and STZ (top panel). Arrows indicate polymorphonuclear cells. Scale bar = 20 μm. Representative photomicrographs of phycoerythrin-conjugated anti-neutrophil antibody 7/4–stained islets from WT, EBI3−/−, and WSX-1−/− mice on day 7 after STZ treatment (middle panel). Arrows indicate 7/4+ neutrophils. Scale bar = 20 μm. The number of polymorphonuclear cells (left bottom panel) and 7/4+ neutrophils (right bottom panel) in islets of mice treated with STZ is shown. No significant difference in the number of polymorphonuclear cells and 7/4+ neutrophils in islets of mice treated with STZ was detected between three groups. B: Representative photomicrographs of islets triple stained with Alexa Fluor 488–conjugated anti-F4/80, Alexa Fluor 647–conjugated anti-CD11c and phycoerythrin-conjugated 7/4 antibodies from WT, EBI3−/−, and WSX-1−/− mice on day 28 after treatment with control vehicle and STZ (top panel). Scale bar = 20 μm. Insets: Representative F4/80+CD11c−7/4− cells are shown. Scale bar = 5 μm. The number of F4/80+CD11c−7/4− cells in islets of mice treated with STZ is shown (left bottom panel). *P < 0.05 versus WT mice. mRNA expression levels of F4/80 antigen in pancreatic tissues from STZ-treated mice that were examined by real-time PCR, normalized with respect to β-actin expression, and expressed relative to that of WT mice, are shown (right bottom panel). *P < 0.05 versus WT mice. C: Representative photomicrographs of islets double stained with FITC-conjugated anti-CD4 antibody and phycoerythrin-conjugated anti-CD8 antibody from WT, EBI3−/−, and WSX-1−/− mice on day 28 after treatment with control vehicle and STZ (top panel). Scale bar = 20 μm. Insets: Representative CD4+ T cells (green) and CD8+ T cells (red) are shown. Scale bar = 5 μm. The number of CD4+ T cells (left middle panel) and CD8+ T cells (left bottom panel) in islets of mice treated with STZ is shown. *P < 0.05 versus WT mice. mRNA expression levels of CD4 and CD8 in pancreatic tissues from STZ-treated mice that were examined by real-time PCR, normalized with respect to β-actin expression, and expressed relative to that of WT mice are shown (middle right and bottom right panels, respectively). *P < 0.05 versus WT mice. For quantification, 10 nonconsecutive standard islets were randomly selected from 10 sections per mouse, and the numbers of cells were counted in samples from seven mice in each group. Data shown are the mean ± SE of cell number.
Gene expression of WSX-1 and gp130, which compose IL-27 receptors in pancreatic islets, was investigated by RT-PCR (Figure 3A). The expression of WSX-1 and gp130 was observed in pancreatic islets isolated from WT mice at relatively lower levels compared with thymus, suggesting a possibility that IL-27 acts on pancreatic islet cells, including β cells. We then explored the possibility of whether exogenous IL-27 might inhibit STZ-induced islet inflammation and, thus, ameliorate the reduced production of insulin by pancreatic islets and resultant hyperglycemia. Recombinant mouse IL-27 was injected daily for 10 days from the initiation of STZ treatment. As shown in Figure 3B, nonfasting blood glucose levels on day 14 after STZ treatment were significantly lower in WT mice treated with recombinant IL-27 compared with WT mice receiving vehicle. Treatment of EBI3−/− mice with recombinant IL-27 significantly decreased nonfasting blood glucose levels compared with the vehicle control group on days 7 and 14. Nonfasting blood glucose levels of WT and EBI3−/− mice treated with recombinant IL-27 showed no significant differences on days 7 and 14. In WSX-1−/− mice that lack the IL-27 receptor, recombinant IL-27 treatment showed no obvious effects on blood glucose levels. These data demonstrate that administration of recombinant IL-27 inhibits hyperglycemia induced by multiple low-dose STZ treatments in mice, indicating a beneficial effect of IL-27 on STZ-induced hyperglycemia. A histological examination by H&E-stained pancreatic tissue on day 14 revealed that recombinant IL-27 treatment decreased intraislet and peri-islet mononuclear cell infiltration induced by STZ compared with vehicle treatment in WT and EBI3−/− mice (Figure 3C). In WSX-1−/− mice, recombinant IL-27 treatment showed no obvious effects on mononuclear cell infiltration induced by STZ. Furthermore, the proinsulin levels of islets observed by immunofluorescence were increased by recombinant IL-27 treatment compared with vehicle treatment in WT and EBI3−/− mice on day 14 (Figure 3D). In WSX-1−/− mice, recombinant IL-27 treatment showed no obvious effects on proinsulin levels of islets compared with vehicle treatment.
Figure 3.
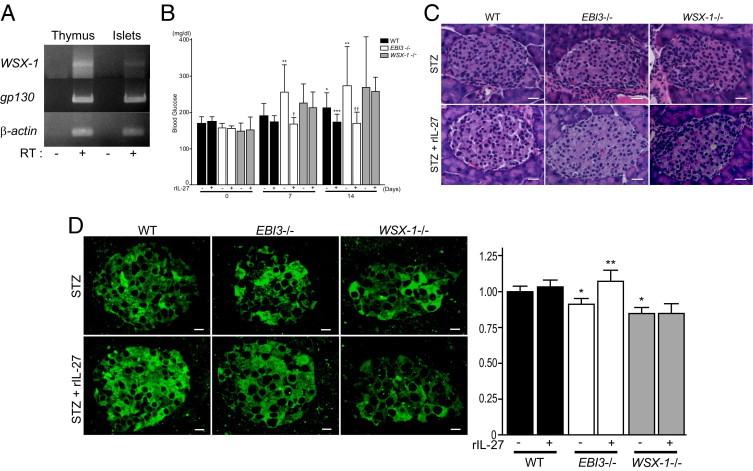
Effects of recombinant IL-27 (rIL-27) on hyperglycemia and β-cell injury induced by STZ. A: Gene expression of WSX-1, gp130, and β-actin in thymus and pancreatic islets. Total RNA extracted from thymus and pancreatic islets of WT mice was examined by RT-PCR in the absence or presence of reverse transcriptase (RT). A representative gel image is shown. B: Effects of rIL-27 on STZ-induced hyperglycemia. Nonfasting blood glucose levels were measured in WT mice (black bar), EBI3−/− mice (white bar), and WSX-1−/− mice (gray bar) treated with STZ in combination with rIL-27 or control vehicle, before (day 0) and on days 7 and 14 after STZ treatment (n = 10 to 12 per group). Data shown are the mean ± SE. *P < 0.05 versus WT mice treated with STZ alone on day 0; **P < 0.05 versus EBI3−/− mice treated with STZ alone on day 0; ***P < 0.05 versus WT mice treated with STZ alone on day 14; †P < 0.05 versus EBI3−/− mice treated with STZ alone on day 7; ††P < 0.05 versus EBI3−/− mice treated with STZ alone on day 14. C: Representative photomicrographs of H&E-stained islets from WT, EBI3−/−, and WSX-1−/− mice treated with STZ in combination with rIL-27 or control vehicle. Scale bar = 20 μm. D: Representative photomicrographs of islets from WT, EBI3−/−, and WSX-1−/− mice treated with STZ in combination with rIL-27 or control vehicle stained with anti-insulin C-chain antibody and examined by immunofluorescence (left panel). Scale bar = 20 μm. Fluorescence intensity was expressed as relative to that of WT mice treated with control vehicle (right panel). *P < 0.05 versus STZ treatment alone in WT mice; **P < 0.05 versus STZ treatment alone in EBI3−/− mice.
The effect of IL-27 treatment on the infiltrate of polymorphonuclear leukocytes, F4/80+CD11c−7/4− macrophages, and CD4+ and CD8+ T cells associated with STZ-induced islet inflammation was then examined. As shown in Figure 4A, intraislet infiltration of polymorphonuclear leukocytes observed by H&E staining was not significantly changed by recombinant IL-27 treatment compared with vehicle treatment in WT, EBI3−/−, and WSX-1−/− mice on day 7 after STZ treatment. The infiltration of 7/4+ neutrophils into islets examined by immunofluorescence showed no significant changes by recombinant IL-27 treatment compared with vehicle treatment in WT, EBI3−/−, and WSX-1−/− mice on day 7 after STZ treatment (Figure 4A). To assess macrophage infiltration of islets, F4/80+CD11c−7/4− cells detected by immunofluorescence of pancreatic tissues were quantified on day 14 after STZ treatment (Figure 4B). The number of F4/80+CD11c−7/4− cells in islets was significantly decreased by recombinant IL-27 treatment compared with vehicle treatment in both WT and EBI3−/− mice. mRNA expression of F4/80 antigen in pancreatic tissues, quantified by real-time PCR, significantly decreased in response to recombinant IL-27 treatment in WT and EBI3−/− mice but not in WSX-1−/− mice (Figure 4B). The infiltration of CD4+ and CD8+ T cells into islets was quantified by counting cell numbers in pancreatic tissue sections that were double stained with FITC-conjugated anti-CD4 antibody and phycoerythrin-conjugated anti-CD8 antibody (Figure 4C). The number of CD4+ T cells in islets was significantly decreased by recombinant IL-27 treatment compared with vehicle treatment in EBI3−/− mice and tended to decrease in WT mice on day 14 after STZ treatment. mRNA expression of CD4 in pancreatic tissues, quantified by real-time PCR, significantly decreased in WT and EBI3−/− mice treated with recombinant IL-27 compared with vehicle treatment (Figure 4C). Also, recombinant IL-27 treatment significantly decreased the infiltration of CD8+ T cells compared with vehicle treatment in EBI3−/− mice on day 14 after STZ treatment. The infiltration of CD8+ T cells was not significantly changed by recombinant IL-27 treatment in WT and WSX-1−/− mice. mRNA expression of CD8 in pancreatic tissues, quantified by real-time PCR, was significantly down-regulated in WT and EBI3−/− mice treated with recombinant IL-27 compared with vehicle treatment (Figure 4C). Thus, recombinant IL-27 treatment seems to decrease STZ-induced infiltration of macrophages and CD4+ and CD8+ T cells into islets compared with vehicle treatment in WT and EBI3−/− mice. In WSX-1−/− mice that lack the IL-27 receptor, recombinant IL-27 treatment showed no obvious effects on the infiltration of F4/80+CD11c−7/4− cells and CD4+ and CD8+ T cells compared with vehicle treatment. These data suggest that recombinant IL-27 administration inhibits islet inflammation induced by STZ in mice.
Figure 4.
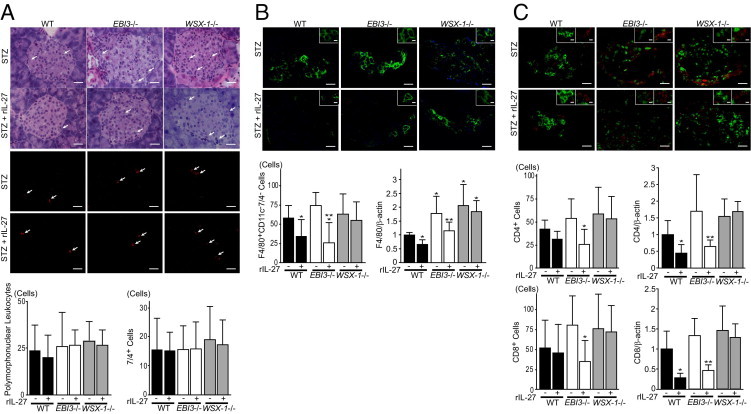
Effects of recombinant IL-27 (rIL-27) on immune cell infiltration into islets induced by STZ. A: Effects of rIL-27 administration on the infiltration of polymorphonuclear leukocytes and neutrophils into islets. Representative photomicrographs of pancreatic tissues visualized by H&E- and phycoerythrin-conjugated anti-neutrophil antibody 7/4 staining (top and middle panels, respectively) from WT, EBI3−/−, and WSX-1−/− mice treated with STZ in combination with rIL-27 or control vehicle on day 7 after STZ treatment are shown. Arrows indicate polymorphonuclear leukocytes and 7/4+ neutrophils. Scale bar = 20 μm. The number of polymorphonuclear leukocytes (left bottom panel) and 7/4+ neutrophils (right bottom panel) in islets is shown. B: Effects of rIL-27 administration on macrophage infiltration into islets. Representative photomicrographs of pancreatic tissues triple stained with Alexa Fluor 488–conjugated anti-F4/80, Alexa Fluor 647–conjugated anti-CD11c and phycoerythrin-conjugated 7/4 antibodies from WT, EBI3−/−, and WSX-1−/− mice treated with STZ in combination with rIL-27 or control vehicle on day 14 are shown (top panel). Scale bar = 20 μm. Insets: Representative F4/80+CD11c−7/4− cells are shown. Scale bar = 5 μm. The number of F4/80+CD11c−7/4− macrophages is shown (left bottom panel). *P < 0.05 versus WT mice treated with STZ alone; **P < 0.05 versus EBI3−/− mice treated with STZ alone. mRNA expression levels of F4/80 antigen in pancreatic tissues normalized with respect to β-actin expression and expressed relative to that of WT mice treated with STZ alone (right bottom panel). *P < 0.05 versus WT mice treated with STZ alone; **P < 0.05 versus EBI3−/− mice treated with STZ alone. C: Effects of rIL-27 administration on the infiltration of CD4+ T-cells and CD8+ T-cells into islets. Representative photomicrographs of pancreatic tissues double stained with FITC-conjugated anti-CD4 antibody and phycoerythrin-conjugated anti-CD8 antibody from WT, EBI3−/−, and WSX-1−/− mice treated with STZ in combination with rIL-27 or control vehicle on day 14 are shown (top panel). Scale bar = 20 μm. Insets: Representative CD4+ T cells (green) and CD8+ T cells (red) are demonstrated. Scale bar = 5 μm. The number of CD4+ T cells (middle left panel) and CD8+ T cells (bottom left panel) in islets of mice is shown. *P < 0.05 versus EBI3−/− mice treated with STZ alone. mRNA expression levels of CD4 and CD8 in pancreatic tissues that were examined by real-time PCR, normalized with respect to β-actin expression, and expressed relative to that of WT mice treated with STZ alone are shown (middle right and bottom right panels, respectively). *P < 0.05 versus WT mice treated with STZ alone; **P < 0.05 versus EBI3−/− mice treated with STZ alone. For quantification, 10 nonconsecutive standard islets were randomly selected from 10 sections per mouse and the numbers of cells were counted in samples from seven mice in each group. Data shown are the mean ± SE of cell number.
We quantified pancreatic mRNA expression of cytokines and chemokines that have been implicated in islet inflammation. We found that mRNA expression of IL-1β, a cytokine that has promoted islet inflammation by recruiting leukocytes, tended to be higher in the pancreas on day 7 after STZ treatment in EBI3−/− and WSX-1−/− mice than in WT mice. Recombinant IL-27 treatment significantly decreased IL-1β mRNA expression in the pancreas compared with vehicle treatment in WT and EBI3−/− mice (Figure 5). In WSX-1−/− mice, recombinant IL-27 treatment showed no obvious effects on IL-1β mRNA expression in the pancreas. These data suggest that recombinant IL-27 treatment inhibits cytokine expression in pancreatic tissue that promotes islet inflammation.
Figure 5.
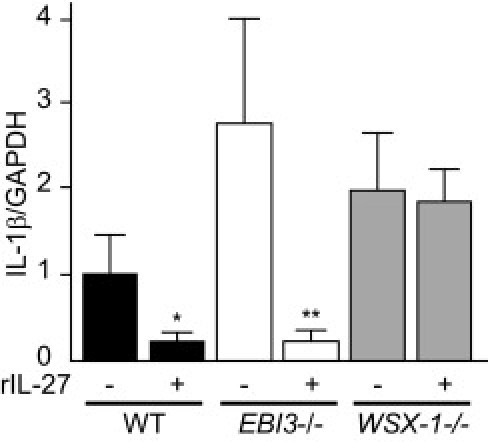
Quantification of IL-1β mRNA expression in the pancreas. IL-1β expression was examined by real-time PCR using RNA extracted from pancreatic tissue of WT, EBI3−/−, and WSX-1−/− mice treated with STZ in combination with recombinant IL-27 (rIL-27) or control vehicle on day 7. The mRNA expression levels were normalized with respect to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression and expressed relative to that of WT mice treated with STZ alone. *P < 0.05 versus WT mice treated with STZ alone; **P < 0.05 versus EBI3−/− mice treated with STZ alone.
Discussion
Injury to β cells and the consequent decreased insulin secretion play a crucial role in the development of diabetes. In type 1 diabetes, autoantigens that are incompletely identified trigger cytotoxic T-cell infiltration of islets with subsequent β-cell injury.18 Metabolic stress, such as hyperglycemia, that accompanies type 2 diabetes induces the expression of pro-inflammatory cytokines in islets that are implicated in the promotion of islet inflammation. Also, Fas up-regulation and NF-κB activation in β cells promote β-cell apoptosis.19 Thus, islet inflammation appears to be involved in the development of types 1 and 2 diabetes. Animal models of hyperglycemia accompanied with β-cell injury using STZ that induces selective DNA damage to β cells are well established.6 The infiltration of macrophages and T cells has been observed in islets after STZ treatment, and this represents a useful model to study mechanisms underlying islet inflammation.7
Accumulating evidence has demonstrated the roles of pro-inflammatory cytokines in islet inflammation and β-cell injury. Deficiency of the IL-1 type 1 receptor slows the rate of progression to overt diabetes in nonobese diabetic (NOD) mice.20 Also, the administration of IL-1 receptor antagonist to mice blocks STZ-induced hyperglycemia.21 These data suggest that IL-1β promotes diabetes in animal models. Several reports have also demonstrated a role for tumor necrosis factor (TNF)-α in experimental diabetes. Specific TNF-α expression in the β cells of NOD mice augments islet inflammation, with a decreased diabetes incidence, whereas administration of neutralizing anti-TNF-α antibody increases diabetic incidence in NOD mice.22, 23 Taken together, these data suggest that TNF-α promotes islet inflammation under metabolic stress but does not play a causative role in the development of diabetes. Also, other studies24 have shown that interferon (IFN)-γ administration has no obvious effects on diabetes incidence in NOD mice. However, β-cell–specific expression of IFN-γ induces hyperglycemia with islet inflammation.22 Deficiency of the receptor for IFN-γ has no obvious effects on the incidence of STZ-induced diabetes or islet inflammation.25 These data suggest an inconclusive role for IFN-γ in the development of diabetes and islet inflammation. In our present study, mRNA expression of TNF-α and IFN-γ in pancreatic tissue was hardly quantifiable by real-time PCR because of relatively lower levels of expression (data not shown). In contrast, IL-1β mRNA expression seems to be up-regulated in pancreatic tissue after STZ treatment, suggesting that inhibition of IL-1β expression by IL-27 could, at least in part, contribute to the anti-inflammatory properties of IL-27 on islet inflammation.
The IL-27 increases pro-inflammatory cytokine production by splenocytes derived from diabetic NOD mice.26 These researchers also showed that IL-27–treated diabetic splenocytes promoted the development of diabetes, indicating that IL-27 accelerates T-cell–mediated autoimmune diabetes. Our study using mice lacking IL-27 and the IL-27 receptor, which were administered with recombinant IL-27, revealed inhibitory roles of IL-27 in STZ-induced islet inflammation and β-cell injury. A reduction of CD8+ T cells infiltrated into islets suggests that CD8+-mediated β-cell destruction induced by STZ is suppressed by IL-27. The effects of IL-27 on T-cell polarization and activation within pancreatic tissue and pancreatic lymph nodes that contribute to the progression of islet inflammation remain to be determined. The current study also shows that IL-27 prevents macrophage infiltration into inflamed islets, suggesting inhibitory effects of IL-27 on macrophage chemoattractants and macrophage activation in the development of islet inflammation. On the other hand, we demonstrated gene expression of IL-27 receptor components in isolated islets from WT mice, suggesting a possibility of IL-27 receptor expression in β cells. Mensah-Brown et al27 showed that recombinant IL-27 treatment accelerates mononuclear cell infiltration into islets with relatively preserved insulin production. These data raise the possibility that IL-27 has direct protective roles against β cells. The differential isolation of intact islet cells and immune cells from islets infiltrated with immune cells in mouse pancreas that are rich in digestive enzymes is technically difficult. Expression changes of IL-27 and IL-27 receptor in islet cells during diabetogenesis are to be studied using cell culture of islet cells. The current study indicates that IL-27 inhibits the islet infiltration of CD8+ T cells that mediate autoimmunity implicated in type 1 diabetes, whereas further studies using models of type 1 diabetes, such as NOD mice, are necessary to elucidate the involvement of immunoregulation by IL-27 in type 1 diabetes. The roles of IL-27 in islet inflammation and β-cell injury induced by a high-fat diet that are implicated in type 2 diabetes promoted by metabolic stress are under investigation in mice.
IL-27 is a heterodimeric cytokine and exhibits anti-inflammatory properties by regulating T-cell polarization and cytokine production. In the present study, IL-27 inhibited pancreatic gene expression of IL-1β that reportedly promotes β-cell apoptosis through the action of Fas, NF-κB, or mitogen-activated protein kinases.28, 29, 30 Although we observed gene expression of EBI3, IL27p28, WSX-1, and gp130 in islet cells, the direct effects of IL-27 on β-cell function, including insulin secretion and β-cell apoptosis, are yet to be clarified. IL-27 may exert an immunoregulatory role for T cells producing the immunosuppressive cytokine IL-10, and this may account for some of the anti-inflammatory properties of IL-27, although the role of IL-10 in islet inflammation is incompletely understood.31
An additional anti-inflammatory property of IL-27 is to inhibit differentiation of Th17 cells that promote inflammation by producing pro-inflammatory cytokines, such as IL-17, IL-6, TNF-α, and IFN-γ.32 IL-27 has therapeutic effects in models of autoimmune encephalomyelitis, allergic asthma, and delayed-type hypersensitivity of skin by preventing Th17 cell differentiation and pro-inflammatory cytokine production.13, 14, 15, 16 Limited information is available regarding the roles of Th17 cells and IL-17 in pancreatic islet inflammation. Mensah-Brown et al33 described that the cytokine IL-23 that promotes Th17 cell differentiation exacerbated both STZ-induced hyperglycemia and islet inflammation. However, further studies are necessary to elucidate the specific roles of Th17 cells and IL-17 in IL-27–mediated immunosuppression of islet inflammation.
In conclusion, we demonstrated in vivo that deficiency of IL-27 signaling exacerbates STZ-induced hyperglycemia and islet inflammation, whereas the administration of recombinant IL-27 is markedly inhibitory. These data provide novel evidence that immunosuppressive IL-27 is a potentially useful therapeutic agent to promote β-cell protection in diabetes.
Footnotes
Supported in part by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
H.Y. and K.N. are both considered senior authors.
References
- 1.Cnop M., Welsh N., Jonas J.C., Jörns A., Lenzen S., Eizirik D.L. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes. 2005;54(Suppl 2):S97–S107. doi: 10.2337/diabetes.54.suppl_2.s97. [DOI] [PubMed] [Google Scholar]
- 2.Pearl-Yafe M., Kaminitz A., Yolcu E.S., Yaniv I., Stein J., Askenasy N. Pancreatic islets under attack: cellular and molecular effectors. Curr Pharm Des. 2007;13:749–760. doi: 10.2174/138161207780249155. [DOI] [PubMed] [Google Scholar]
- 3.Santamaria P. The long and winding road to understanding and conquering type 1 diabetes. Immunity. 2010;32:437–445. doi: 10.1016/j.immuni.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 4.Ehses J.A., Perren A., Eppler E., Ribaux P., Pospisilik J.A., Maor-Cahn R., Gueripel X., Ellingsgaard H., Schneider M.K., Biollaz G., Fontana A., Reinecke M., Homo-Delarche F., Donath M.Y. Increased number of islet-associated macrophages in type 2 diabetes. Diabetes. 2007;56:2356–2370. doi: 10.2337/db06-1650. [DOI] [PubMed] [Google Scholar]
- 5.Mandrup-Poulsen T., Bendtzen K., Dinarello C.A., Nerup J. Human tumor necrosis factor potentiates human interleukin 1-mediated rat pancreatic beta-cell cytotoxicity. J Immunol. 1987;139:4077–4082. [PubMed] [Google Scholar]
- 6.Like A.A., Rossini A.A. Streptozotocin-induced pancreatic insulitis: new model of diabetes mellitus. Science. 1976;193:415–417. doi: 10.1126/science.180605. [DOI] [PubMed] [Google Scholar]
- 7.O'Brien B.A., Harmon B.V., Cameron D.P., Allan D.J. Beta-cell apoptosis is responsible for the development of IDDM in the multiple low-dose streptozotocin model. J Pathol. 1996;178:176–181. doi: 10.1002/(SICI)1096-9896(199602)178:2<176::AID-PATH433>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 8.Villarino A.V., Huang E., Hunter C.A. Understanding the pro- and anti-inflammatory properties of IL-27. J Immunol. 2004;173:715–720. doi: 10.4049/jimmunol.173.2.715. [DOI] [PubMed] [Google Scholar]
- 9.Yoshida H., Miyazaki Y. Regulation of immune responses by interleukin-27. Immunol Rev. 2008;226:234–247. doi: 10.1111/j.1600-065X.2008.00710.x. [DOI] [PubMed] [Google Scholar]
- 10.Stumhofer J.S., Hunter C.A. Advances in understanding the anti-inflammatory properties of IL-27. Immunol Lett. 2008;117:123–130. doi: 10.1016/j.imlet.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoshida H., Hamano S., Senaldi G., Covey T., Faggioni R., Mu S., Xia M., Wakeham A.C., Nishina H., Potter J., Saris C.J., Mak T.W. WSX-1 is required for the initiation of Th1 responses and resistance to L. major infection. Immunity. 2001;15:569–578. doi: 10.1016/s1074-7613(01)00206-0. [DOI] [PubMed] [Google Scholar]
- 12.Hamano S., Himeno K., Miyazaki Y., Ishii K., Yamanaka A., Takeda A., Zhang M., Hisaeda H., Mak T.W., Yoshimura A., Yoshida H. WSX-1 is required for resistance to Trypanosoma cruzi infection by regulation of proinflammatory cytokine production. Immunity. 2003;19:657–667. doi: 10.1016/s1074-7613(03)00298-x. [DOI] [PubMed] [Google Scholar]
- 13.Stumhofer J.S., Laurence A., Wilson E.H., Huang E., Tato C.M., Johnson L.M., Villarino A.V., Huang Q., Yoshimura A., Sehy D., Saris C.J., O'Shea J.J., Hennighausen L., Ernst M., Hunter C.A. Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol. 2006;7:937–945. doi: 10.1038/ni1376. [DOI] [PubMed] [Google Scholar]
- 14.Batten M., Li J., Yi S., Kljavin N.M., Danilenko D.M., Lucas S., Lee J., de Sauvage F.J., Ghilardi N. Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat Immunol. 2006;7:929–936. doi: 10.1038/ni1375. [DOI] [PubMed] [Google Scholar]
- 15.Miyazaki Y., Inoue H., Matsumura M., Matsumoto K., Nakano T., Tsuda M., Hamano S., Yoshimura A., Yoshida H. Exacerbation of experimental allergic asthma by augmented Th2 responses in WSX-1-deficient mice. J Immunol. 2005;175:2401–2407. doi: 10.4049/jimmunol.175.4.2401. [DOI] [PubMed] [Google Scholar]
- 16.Miyazaki Y., Shimanoe Y., Wang S., Yoshida H. Amelioration of delayed-type hypersensitivity responses by IL-27 administration. Biochem Biophys Res Commun. 2008;373:397–402. doi: 10.1016/j.bbrc.2008.06.038. [DOI] [PubMed] [Google Scholar]
- 17.Igawa T., Nakashima H., Sadanaga A., Masutani K., Miyake K., Shimizu S., Takeda A., Hamano S., Yoshida H. Deficiency in EBV-induced gene 3 (EBI3) in MRL/lpr mice results in pathological alteration of autoimmune glomerulonephritis and sialadenitis. Mod Rheumatol. 2009;19:33–41. doi: 10.1007/s10165-008-0117-1. [DOI] [PubMed] [Google Scholar]
- 18.Anderson M.S., Bluestone J.A. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol. 2005;23:447–485. doi: 10.1146/annurev.immunol.23.021704.115643. [DOI] [PubMed] [Google Scholar]
- 19.Donath M.Y., Ehses J.A., Maedler K., Schumann D.M., Ellingsgaard H., Eppler E., Reinecke M. Mechanisms of beta-cell death in type 2 diabetes. Diabetes. 2005;54(Suppl 2):S108–S113. doi: 10.2337/diabetes.54.suppl_2.s108. [DOI] [PubMed] [Google Scholar]
- 20.Thomas H.E., Irawaty W., Darwiche R., Brodnicki T.C., Santamaria P., Allison J., Kay T.W. IL-1 receptor deficiency slows progression to diabetes in the NOD mouse. Diabetes. 2004;53:113–121. doi: 10.2337/diabetes.53.1.113. [DOI] [PubMed] [Google Scholar]
- 21.Sandberg J.O., Andersson A., Eizirik D.L., Sandler S. Interleukin-1 receptor antagonist prevents low dose streptozotocin induced diabetes in mice. Biochem Biophys Res Commun. 1994;202:543–548. doi: 10.1006/bbrc.1994.1962. [DOI] [PubMed] [Google Scholar]
- 22.Eizirik D.L., Mandrup-Poulsen T. A choice of death: the signal-transduction of immune-mediated beta-cell apoptosis. Diabetologia. 2001;44:2115–2133. doi: 10.1007/s001250100021. [DOI] [PubMed] [Google Scholar]
- 23.Yang X.D., Tisch R., Singer S.M., Cao Z.A., Liblau R.S., Schreiber R.D., McDevitt H.O. Effect of tumor necrosis factor alpha on insulin-dependent diabetes mellitus in NOD mice, I: the early development of autoimmunity and the diabetogenic process. J Exp Med. 1994;180:995–1004. doi: 10.1084/jem.180.3.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mandrup-Poulsen T. The role of interleukin-1 in the pathogenesis of IDDM. Diabetologia. 1996;39:1005–1029. doi: 10.1007/BF00400649. [DOI] [PubMed] [Google Scholar]
- 25.Gysemans C.A., Pavlovic D., Bouillon R., Eizirik D.L., Mathieu C. Dual role of interferon-gamma signalling pathway in sensitivity of pancreatic beta cells to immune destruction. Diabetologia. 2001;44:567–574. doi: 10.1007/s001250051662. [DOI] [PubMed] [Google Scholar]
- 26.Wang R., Han G., Wang J., Chen G., Xu R., Wang L., Li X., Shen B., Li Y. The pathogenic role of interleukin-27 in autoimmune diabetes. Cell Mol Life Sci. 2008;65:3851–3860. doi: 10.1007/s00018-008-8540-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mensah-Brown E.P., Shahin A., Al-Shamsi M., Lukic M.L. New members of the interleukin-12 family of cytokines: IL-23 and IL-27 modulate autoimmune diabetes. Ann N Y Acad Sci. 2006;1079:157–160. doi: 10.1196/annals.1375.024. [DOI] [PubMed] [Google Scholar]
- 28.Maedler K., Størling J., Sturis J., Zuellig R.A., Spinas G.A., Arkhammar P.O., Mandrup-Poulsen T., Donath M.Y. Glucose- and interleukin-1beta-induced beta-cell apoptosis requires Ca2+ influx and extracellular signal-regulated kinase (ERK) 1/2 activation and is prevented by a sulfonylurea receptor 1/inwardly rectifying K+ channel 6.2 (SUR/Kir6.2) selective potassium channel opener in human islets. Diabetes. 2004;53:1706–1713. doi: 10.2337/diabetes.53.7.1706. [DOI] [PubMed] [Google Scholar]
- 29.Giannoukakis N., Rudert W.A., Trucco M., Robbins P.D. Protection of human islets from the effects of interleukin-1beta by adenoviral gene transfer of an Ikappa B repressor. J Biol Chem. 2000;275:36509–36513. doi: 10.1074/jbc.M005943200. [DOI] [PubMed] [Google Scholar]
- 30.Cardozo A.K., Heimberg H., Heremans Y., Leeman R., Kutlu B., Kruhøffer M., Ørntoft T., Eizirik D.L. A comprehensive analysis of cytokine-induced and nuclear factor-kappa B-dependent genes in primary rat pancreatic beta-cells. J Biol Chem. 2001;276:48879–48886. doi: 10.1074/jbc.M108658200. [DOI] [PubMed] [Google Scholar]
- 31.Jankovic D., Trinchieri G. IL-10 or not IL-10: that is the question. Nat Immunol. 2007;8:1281–1283. doi: 10.1038/ni1207-1281. [DOI] [PubMed] [Google Scholar]
- 32.Yoshida H., Nakaya M., Miyazaki Y. Interleukin 27: a double-edged sword for offense and defense. J Leukoc Biol. 2009;86:1295–1303. doi: 10.1189/jlb.0609445. [DOI] [PubMed] [Google Scholar]
- 33.Mensah-Brown E.P., Shahin A., Al-Shamisi M., Wei X., Lukic M.L. IL-23 leads to diabetes induction after subdiabetogenic treatment with multiple low doses of streptozotocin. Eur J Immunol. 2003;36:216–223. doi: 10.1002/eji.200535325. [DOI] [PubMed] [Google Scholar]