

Abstract
Oligodendrocyte precursor cells (OPCs), a major glial cell type giving rise to myelinating oligodendrocytes in the CNS, express calcium-permeable (CP-) AMPARs. Although CP-AMPARs are important in OPC proliferation and neuron-glia signalling, they render OPCs susceptible to ischemic damage in early development. Here we identify factors controlling dynamic regulation of AMPAR subtypes in OPCs from rat optic nerve and mouse cerebellar cortex. We find that activation of group 1 mGluRs drives an increase in the proportion of CP-AMPARs, reflected in increased single-channel conductance and inward rectification. This plasticity requires elevation of intracellular calcium, utilizes PI3 kinase, PICK-1 and the JNK pathway. In white matter, neurons and astrocytes release both ATP and glutamate. Surprisingly, activation of purinergic receptors in OPCs decreases CP-AMPAR expression, suggesting a capacity for homeostatic regulation. Finally, we show that stargazin-related transmembrane AMPAR regulatory proteins, which are key for AMPAR surface expression in neurons, regulate CP-AMPAR plasticity in OPCs.
Introduction
During central nervous system development, oligodendrocyte precursor cells (OPCs) give rise to oligodendrocytes that are responsible for axonal myelination. A population of ‘adult’ OPCs also persists in the mature brain, and these cells are capable of differentiating into oligodendrocytes if myelin is damaged. OPCs express AMPA-type glutamate receptors (AMPARs), activation of which is thought critical in a variety of important physiological and developmental processes, including OPC proliferation, migration and differentiation 1,2, neuron-glia signaling 3 and pathological changes that occur following ischemia.
AMPARs can assemble either as homo- or heterotetramers, with functional properties that are dictated by their subunit composition and by the presence of auxiliary transmembrane AMPAR regulatory proteins (TARPs) 4. The GluA2 subunit is a key determinant of AMPAR calcium permeability 5. Premyelinating OPCs (in vivo and in vitro) are known to contain all four subtypes of AMPAR subunit (GluA1-4) 6,7 and express a mixture of GluA2-lacking calcium-permeable (CP-) and GluA2-containing calcium-impermeable (CI-) AMPAR subtypes, the relative proportions of which vary during development and with brain region 7,8.
The presence of CP-AMPARs renders OPCs particularly vulnerable to hypoxic-ischemic excitotoxic injury in early development 9,10. Specifically, calcium influx through these receptors, when combined with disrupted calcium homeostasis, is a trigger that initiates OPC damage 11. The resultant hypomyelination is thought to be a major factor in white matter injury in premature infants 12. In keeping with this view, myelin damage and impaired axonal conduction can be suppressed by strategies that reduce calcium influx through these receptors. Despite the clear importance of CP-AMPARs in OPCs, their functional properties, and many of the factors involved in regulation of AMPAR subtype expression in these cells remain relatively unexplored. In particular, the molecular mechanisms that regulate CP AMPARs in OPCs, and how these relate to their neuronal counterparts, have not been defined.
Ultrastructural and functional studies have demonstrated that, in many brain regions, CP-AMPARs in OPCs are activated during transmission at discrete ‘neuron-glia synapses’ 3. In many CNS white matter tracts, devoid of nerve cell bodies or nerve terminals, synapses can also form directly between axons and OPC cell processes 8,13 These various neuron-glia synapses share features with conventional neuronal synapses, including the presence of action potential-evoked transmitter release that is calcium-dependent and quantal in nature 3,13. The synaptic currents at these sites are mediated in part by CP-AMPARs 3, as described for certain neuronal synapses and other neuron-glia synapses 14,15. Moreover, high frequency presynaptic activity at these neuron-glia synapses is reported to generate a form of long-term potentiation that is associated with a rapid rise in the proportion of CP-AMPARs 16, underscoring the dynamic nature of AMPAR expression in OPCs.
In addition to CP-AMPARs, OPCs express other receptor types capable of triggering an increase in intracellular calcium, and which are likely to be activated by released transmitter. Thus in white matter, ATP can activate P2Y and P2X7 receptors in these cells 17, and there is evidence that ATP release is involved in white matter damage during ischemia 18. OPCs in slices and in vitro also express group 1 mGluRs (predominantly mGluR5), the activation of which similarly results in elevation of intracellular calcium 11,19.
We have previously described mGluR-mediated regulatory mechanisms that govern the relative expression of CP-AMPARs in cerebellar stellate cells 20. Here we have examined the functional properties of AMPARs in OPCs, and identified distinct mGluR- and ATP-mediated changes in glial CP-AMPAR expression. In particular, we found that activation of group I mGluRs increases the surface expression and enhances the current generated by CP-AMPARs, whilst activation of purinergic P2Y receptors decreases the fraction of current mediated by CP-AMPARs. The delivery of CP-AMPARs depends on a rise in intracellular calcium, and involves PI3 kinase, PICK-1 and the JNK pathway. In addition, the stargazin family of auxiliary AMPAR subunits plays a critical role in this process. Our experiments thus establish the existence of, and mechanisms underlying, unexpected bidirectional AMPAR plasticity in OPCs.
Results
mGluR activation increases CP-AMPARs in CG4 OPCs
To test whether mGluR activation alters the proportion of GluA2-containing CP-AMPARs in oligodendrocyte lineage cells, we first measured glutamate-evoked whole-cell current-voltage (I-V) relationships in the CG4 OPC cell line (Fig. 1a). We assessed the presence of GluA2-lacking CP-AMPARs by examining the voltage-dependent block produced by intracellular spermine (100 μM). In these experiments, the agonist solution contained 100 μM glutamate plus 50 μM cyclothiazide (to reduce AMPAR desensitization). In separate experiments, we confirmed that the response to glutamate could be fully blocked by the AMPAR antagonist GYKI 52466 dihydrochloride (50 μM) (data not shown). In control cells, the I-V relationships (−100 to +60 mV) showed modest rectification, with a mean Rectification Index (RI, +60/−60 mV; see Methods) of 0.69 ± 0.05 (n = 10) (Fig. 1b). Following treatment with the group 1 mGluR (mGluR1/5) selective agonist DHPG (100 μM; 30 minutes at 37°C) the I-V relationships became more rectifying (Fig. 1c-e), with RI reduced to 0.33 ± 0.04 (n = 8; P = 0.0036). This increase in rectification (decrease in RI) is consistent with an increase in the proportion of CP-AMPARs following DHPG treatment.
Figure 1. DHPG increases rectification of AMPARs in CG4 OPCs.
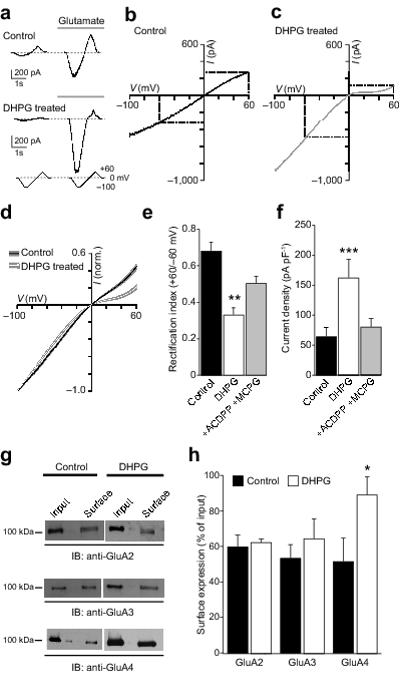
(a) Representative whole-cell current responses to voltage ramps (0, −100, +60, 0 mV) from a control CG4 OPC (upper panel) and one treated with 100 μM DHPG (lower panel). (b) I-V relationship (−100/+60 mV) for the control cell shown in a. The RI (+60/−60 mV) was 0.54. (c) Same as b for the DHPG-treated cell shown in a. Rectification was greater (RI = 0.38). (d) Averaged normalised whole-cell I-V relationships from untreated (n = 10) and DHPG-treated (n = 8) CG4 cells. Filled areas indicate S.E.M. (e) Pooled data showing the effect of DHPG treatment on RI and block of DHPG effect by the mGluR antagonists ACDPP (10 μM) and MCPG (1 mM) (error bars denote S.E.M). (f) Pooled data showing the effect of DHPG on current density (−100 mV) and block by ACDPP and MCPG. (g) Representative Western blots showing the effect of DHPG on cell surface expression of GluA2, GluA3 and GluA4. (h) Pooled data from three experiments of the type shown in g (error bars denote S.E.M).
At negative potentials (−100 mV), where CP-AMPARs are largely unaffected by polyamine block 21, DHPG increased the current density from 64 ± 15 to 162 ± 31 pA.pF−1 (P = 0.00016) (Fig. 1f). Such an increase could reflect a change in receptor number, but is also consistent with the higher single-channel conductance of CP-AMPARs compared with CI-AMPARs 22. The effects of DHPG, on both rectification and current density, could be prevented by co-treatment with the antagonists ACDPP (10 μM) and MCPG (1 mM) (Fig. 1e, f). In these conditions, RI (0.54 ± 0.05, n = 6) and current density (83.3 ± 16.2 pA.pF−1) were not significantly different from control values (P = 0.5235 and 0.09, respectively).
To determine if the increase in AMPAR current density and inward rectification that occurred following DHPG treatment was accompanied by an alteration in the cell membrane expression of AMPAR subunits, we performed cell-surface biotinylation experiments. The surface expression of GluA4 was significantly increased (from 51.7 ± 13.4 to 89.6 ± 10.8 % of input, n = 3; P = 0.022). The cell surface expression of GluA2 and GluA3 remained unaltered (60.1 ± 6.4 versus 62.3 ± 2.1 %, and 53.7 ± 7.6 versus 64.7 ± 11.0 %, respectively; both n = 3 and P = 0.70) (Fig. 1g, h). These results suggest that activation of group I mGluRs increased the number of surface CP-AMPARs and enhanced current density in CG4 OPCs by promoting the expression of GluA4-containing AMPARs.
Activation of mGluRs increases AMPAR channel conductance
If DHPG treatment did indeed increase the proportion of functional CP-AMPARs in CG4 OPCs, one would predict an increase in the mean single-channel conductance. To examine this, we recorded currents from outside-out membrane patches (−60 mV) in response to rapid application of 10 mM glutamate (100 ms), and used non-stationary fluctuation analysis (NSFA; see Methods) to determine the weighted mean single-channel conductance and the peak open probability (Po, peak) (Fig. 2a). DHPG treatment produced a significant increase in single-channel conductance (from 30.1 ± 2.2 pS to 45.4 ± 3.1 pS, n = 14 and 12, respectively; P = 0.0033) (Fig. 2b, c) that could be prevented by the mGluR antagonists MCPG and ACDPP (34.2 ± 4.1 pS, n = 5; P = 0.61 compared to control) (Fig. 2d). Po, peak and the time course of desensitization were unaffected by DHPG treatment (Fig. 2e, f).
Figure 2. DHPG increases single-channel conductance of AMPARs in CG4 OPCs.
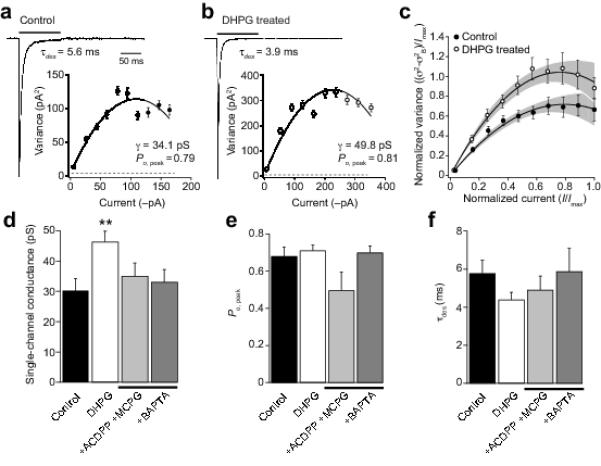
(a) Representative averaged current response to 10 mM glutamate (100 ms, −60 mV) recorded form an outside-out patch taken from an untreated CG4 OPC (average of 80 responses). Weighted time constant of desensitization (see Methods) was 5.6 ms. Inset shows current-variance plot for the same patch as a (fitted with equation 1, see Methods). Symbols indicate mean values and error bars the S.E.M. Dotted line indicates background variance. For this cell, the weighted mean single-channel conductance was 34.1 pS and the peak open probability was 0.79. (b) Same as a, for a representative DHPG-treated cell (average of 110 responses). (c) Global averaged current-variance traces for control (n = 14) and DHPG-treated cells (n = 12). Filled areas indicate 95% confidence intervals for the fits. (d-f) Pooled data showing the effect of DHPG treatment on single-channel conductance, Po,peak and τdes. Note the DHPG-induced increase in conductance was blocked by the mGluR antagonists ACDPP and MCPG (10 μM and 1 mM) and by pre-treatment with BAPTA-AM (20 μM).
The fact that the magnitude of the change in surface AMPARs appeared less when assessed with biotinylation, as opposed to current density, can be ascribed in part to the larger single-channel conductance of CP-AMPARs. Current density will also be influenced by any change in deactivation kinetics, or in sensitivity to cyclothiazide, of the AMPAR/TARP combination that is present following DHPG treatment. This increase in the proportion of CP-AMPARs contrasts with the DHPG-induced decrease in CP-AMPARs described at certain central synapses 20,23, where it is thought to reflect a change in protein synthesis and/or intramembrane movement of AMPARs, consequent to elevation of intracellular calcium ([Ca2+]i).
Mechanisms underlying mGluR-induced changes in CP-AMPARs
We next investigated potential mechanisms that might contribute to the mGluR-induced AMPAR changes in CG4 OPCs, by examining the effects of various treatments on the ability of DHPG to alter either RI or channel conductance. In OPCs, DHPG is known to trigger an increase in [Ca2+]i that is prevented by the selective mGluR5 antagonist MPEP 24, and that results from IP3-mediated Ca2+ release from intracellular stores. To determine whether such Ca2+ mobilization might trigger the AMPAR subunit changes we observed, CG4 OPCs were pretreated with the membrane-permeable Ca2+ chelator BAPTA-AM (20 μM), to rapidly buffer any rise in [Ca2+]i. Following such treatment, the effect of DHPG on AMPAR channel conductance was prevented (34.9 ± 4.5 pS, n = 10; P = 0.61 compared to control) (see Fig. 2d). To confirm that BAPTA-AM did indeed block the increase in [Ca2+]i induced by activation of mGluRs, cultured optic nerve OPCs were loaded with the cell-permeant AM ester of fluo-4 (5 μm; Invitrogen) and somatic fluorescence monitored. Bath application of DHPG (100 μM) produced a roughly 2-fold increase in relative fluorescence, which was fully blocked by BAPTA-AM (20 μM) (n = 10 cells with DHPG; n = 11 cells with BAPTA-AM + DHPG) (data not shown).
In separate experiments, we examined the involvement of potential downstream pathways in the mGluR-mediated insertion of CP-AMPARs. In neurons, mGluR-mediated long-term depression (LTD) has been shown to depend on an increase in [Ca2+]i that is suggested to promote association of the calcium sensing protein (NSC-1), protein interacting with C kinase (PICK-1) and protein kinase C (PKC), resulting in phosphorylation of GluA2 and receptor endocytosis 25. mGluR-triggered phosphorylation of GluA2 can also involve a calcium/calmodulin-dependent protein kinase (CaMK)/c-Jun N-terminal kinase (JNK) pathway 26.
When we pretreated OPCs with a selective cell-permeable PICK-1 inhibitor peptide (TAT-pep2-EVKI; 25 μM), or with a membrane-permeable antagonist for JNKs (SP600125; 100 μM), the effect of DHPG on RI was blocked (0.55 ± 0.05 and 0.61 ± 0.09, n = 7 and 8; P = 0.52 and 0.31 versus control). Activation of phosphoinositide-3 kinase (PI3K) initiates an increase in protein synthesis via the phosphoinositide-3 kinase-Akt-mammalian target of rapamycin (PI3K-Akt-mTOR). This pathway has been implicated in AMPAR plasticity following mGluR activation 23. We found that the PI3K inhibitor wortmannin (100 nM) blocked the effects of DHPG on RI in OPCs (0.50 ± 0.04, n = 7; P = 0.27 versus control). Of note, the effect of SP600125 (above) could also reflect a reduction in the level of phosphorylation of JNK substrates p-ATF2 and p-c-Jun, which has been shown to regulate mGluR mediated hippocampal LTD 27.
Consistent with the view that the targeting of GluA2-lacking AMPARs involves synthesis of new CP-AMPARs or associated proteins, the effects of DHPG on RI were blocked by pre-treatment with the inhibitor of protein translation, cyclohexamide (25 μM) (0.66 ± 0.04, n = 7; P = 0.7317). Together, these experiments suggest that the pathways implicated in mGluR-mediated AMPAR plasticity in certain neurons also play a role in OPCs, albeit to trigger an increase rather than a decrease in the proportion of CP-AMPARs.
Development regulates mGluR effects on optic nerve OPCs
Does mGluR-activation induce AMPAR plasticity in native OPCs? To address this, we purified OPCs from optic nerve 28 and examined their responses at two developmental stages. To preserve OPCs in an immature state they were grown in the presence of growth factors (bFGF and PDGF-AA added once every 24 hours; see Methods) and maintained for 6 DIV prior to patch-clamp recording. At this stage OPCs produced relatively few processes and were immunoreactive for the marker O4 (Fig. 3a). In these cells, the glutamate-evoked whole-cell I-V relationship exhibited modest rectification, similar to that seen in CG4 OPCs (0.64 ± 0.03, n = 14) (Fig. 3a), suggesting the presence of a population of CP-AMPARs. To confirm this, we examined the effect of the selective CP-AMPA receptor blocker philanthotoxin-433 (PhTx-433; 5 μM) on glutamate-evoked currents (100 μM, plus 50μM cyclothiazide). At −100 mV, the response to glutamate was reduced by 46.2 ± 5.2 % (n = 6; P = 0.0031), consistent with the idea that a significant proportion of AMPARs were calcium permeable. Next, to promote differentiation into premyelinating OPCs, growth factors were withdrawn and the medium supplemented with the thyroid hormone (T3); cells were then examined ~48 hrs later (see Methods). In this condition the cells elaborated multiple processes characteristic of pre-myelinating OPCs, and while functional AMPARs were retained 29, unlike those of immature OPCs they exhibited a linear I-V relationship (RI = 0.94 ± 0.02, n = 5; P = 0.025) (Fig. 3b). This loss of rectification in maturing cells implied a loss of CP-AMPARs. Treatment of immature OPCs with DHPG (100 μM; 30 minutes at 37°C) caused the glutamate I-V relationship to become more rectifying (RI = 0.38 ± 0.01, n = 10; P = 0.0012) (Fig. 3a, c). However, DHPG was without effect in pre-myelinating OPCs (RI = 1.01 ± 0.07, n = 5, P = 0.56) (Fig. 3b, c). Thus, the mGluR-induced increase in CP-AMPARs that we observed in immature OPCs was lost by the pre-myelinating stage.
Figure 3. mGluR-induced AMPAR plasticity is developmentally regulated in native OPCs.
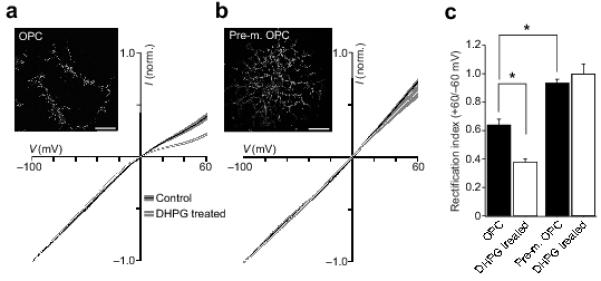
(a) Global averaged normalised whole-cell I-V plots for OPCs (6 DIV; n = 14 and 10) traces show means and shaded areas denote S.E.M. Note the increase in rectification following DHPG treatment. Inset shows an immature OPC labelled with antibody against O4 (permeabilized). Scale bar 25 μm. (b) Same as a, for a pre-myelinating OPC developed from OPCs starved of growth factors. I-Vs from 5 cells each. Note the linear I-V in control, and the lack of change following DHPG treatment. (c) Pooled data showing the effect of DHPG treatment on RI in immature OPCs, and the lack of effect in pre-myelinating OPCs.
We next examined the single-channel properties of AMPARs in OPCs maintained with and without growth factors. Rapid application of glutamate to excised patches (10 mM, 100 ms, −60 mV; see Methods) gave responses that desensitized with a weighted mean time constant (τdes) of 5.1 ± 0.1 ms (immature OPCs) and 4.6 ± 0.3 ms (pre-myelinating OPCs) (n = 7 and 6). NSFA of these macroscopic patch responses gave an estimate for the weighted mean single-channel chord conductance of 35.4 ± 2.8 pS in immature OPCs, compared with 21.6 ± 1.3 pS in pre-myelinating OPCs (P = 0.0003). These values are consistent with the view that pre-myelinating cells in optic nerve express predominantly CI-AMPARs. An age-dependent change in the AMPAR subtype repertoire has been described previously for OPCs (NG2+-OPCs), however the overall AMPAR density appears to decrease rapidly during cell differentiation 7.
Activation of P2Y receptors decreases CP-AMPARs in OPCs
Our results, thus far, suggest that activation of mGluRs by the glutamate released from axons in optic nerve or white matter 13,30 could produce AMPAR plasticity in immature OPCs. Indeed, it is possible that this underlies the neuron-glia long-term potentiation (LTP) triggered in hippocampal NG2+-OPCs by the evoked release of glutamate from nerve terminals 16 (see below). However, axons, and neighboring astrocytes in the white matter 17 also release ATP, which acts on P2Y and P2X7 receptors in OPCs 31, triggering many of the intracellular cascades linked with mGluR activation. To investigate whether ATP receptors might induce a similar form of AMPAR plasticity, we treated immature OPCs with ATP (1 mM for 10 min at 37°C). Unexpectedly, such treatment led to a significant decrease in rectification of the glutamate I-V relationship (RI changed from 0.51 ± 0.03 to 0.98 ± 0.12, n = 10 and 7; P = 0.0037) (Fig. 4a, b), indicating that following ATP treatment the current was carried mainly by CI-AMPARs. In agreement with this view, the AMPAR single-channel conductance was significantly decreased from 35.6 ± 3.0 to 19.2 ± 2.4 pS (n = 10 and 6, P = 0.0014), without affect on Po, peak (Fig. 4c-e). In addition, the rate of desensitization was slowed from 4.1 ± 0.2 to 6.9 ± 0.6 ms (P = 0.0005) (Fig. 4e).
Figure 4. ATP reduces AMPAR rectification in native OPCs.
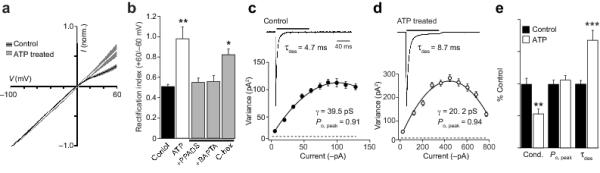
(a) Global averages of normalized I-V plots obtained from untreated OPCs (n = 10) and OPCs treated with ATP (1 mM; n = 7). Filled areas indicate S.E.M. ATP treatment decreased AMPAR rectification. (b) Pooled data showing the effect of ATP on RI. The effect of ATP was blocked by the P2 antagonist PPADS (100 μM) or the calcium chelator BAPTA-AM (20 μM) but not by cyclohexamide (C-hex; 25 μM). (c) Representative averaged response from an outside-out patch from an untreated OPC to fast application of 10 mM glutamate (100 ms, −60 mV; mean of 88 traces). Inset shows the current-variance plot for this cell. (d) Same as c, for an OPC treated with ATP (mean of 40 traces). (e) Pooled normalized data showing the effect of ATP treatment on weighted mean single-channel conductance (Cond.), Po, peak and τdes.
ATP activates P2Y and P2X7 receptors and is rapidly degraded into ADP, AMP and adenosine, which could in principle activate adenosine receptors. To determine whether the ATP-induced AMPAR plasticity was mediated via purinergic or adenosine receptors (A1, A2 and A3), we used the purinergic receptor antagonist PPADS, and the adenosine receptor agonist 2-chloroadenosine. We found that 2-chloroadenosine (100 μM) did not alter AMPAR RI in OPCs (0.54 ± 0.06 versus 0.61 ± 0.01, n = 6, and n = 4, respectively; P = 0.98). However, PPADS (100 μM) blocked the effect of ATP on RI (0.52 ± 0.05, n = 5; P = 0.951 compared to control) (Fig. 4b), indicating that ATP-induced plasticity was mediated via the activation of purinergic receptors. PPADS has been shown to inhibit the activation of both P2X7 and P2Y receptors 32. To investigate whether the effects of ATP could be ascribed to the activation of P2X7 receptors, we tested the P2X7 agonist BzATP (2 μM). This treatment did not alter AMPAR RI (0.59 ± 0.05 in BzATP versus 0.51 ± 0.03 in control, n = 7 and 6, respectively; P = 0.73), suggesting that the effects of ATP on rectification were mediated via P2Y receptor activation.
ATP has recently been shown to increase [Ca2+]i in NG2+-OPCs 17. We found that the decrease in rectification produced by ATP was prevented when cells were pretreated with 20 μM BAPTA-AM (RI = 0.56 ± 0.06, n = 6; P = 0.892), consistent with a role for intracellular Ca2+ in promoting the ATP-induced AMPAR plasticity. However, the effect of ATP was not suppressed by treatment with the protein synthesis inhibitor cyclohexamide (25 μM) (RI = 0.83 ± 0.05, n = 5; P = 0.015) (Fig 4b). It is of note that the whole-cell current was not significantly changed by ATP (83.2 ± 17.7 in control, 102.1 ± 12.6 pA.pF−1 following ATP; n = 10 and 7, respectively; P = 0.732). This suggests that ATP mediated plasticity involves a loss of CP-AMPARs, and the targeting of CI-AMPARs from a pool that does not require new receptor synthesis. This finding is consistent with the view that the fraction of CP-AMPARs expressed in immature OPCs is regulated differentially by mGluRs and P2Y receptors, and that separate pathways are likely to underlie these effects.
TARPs regulate mGluR-mediated plasticity in OPCs
In neurons, transmembrane AMPAR regulatory proteins (TARPs) have been implicated in various forms of AMPAR plasticity 33,34. TARPs have been identified in glia 35,36 and we have recently reported that CP-AMPAR in cerebellar Bergmann glia have channel properties indicative of TARP association 37. Although gene expression studies have identified TARPs in purified OPCs acutely isolated from forebrain (Cacng4, -5 and -8 – i.e. γ-4, -5 and -8; Ref. 6), it was not known whether TARPs associate with, and regulate, AMPARs in OPCs. Of note, oligodendrocyte lineage cells express GluA1-4 AMPAR subunits 6, yet the single-channel conductance of ~35 pS that we observed in immature OPCs is higher than would be expected for any combination of AMPAR subunits expressed without a TARP 37. Consistent with the view that TARPs associate with AMPARs in immature OPCs to functionally modify their properties, we found that the partial agonist kainate displayed a relatively high efficacy. The ratio of kainate- and glutamate-evoked peak currents (IKA/IGlu; both 1 mM) was 41.2 ± 4.1% (n = 6). This is much greater than the ratio obtained with GluA1 or GluA1/GluA2 receptors in the absence of a TARP (~10%), but similar to that of receptors co-expressed with TARPs 38.
As a first step in assessing the possible contribution of TARPs, we extracted mRNA from optic nerves of P7 rats and carried out RT-PCR using primers for all known TARPs (γ-2, -3, -4, -5, -7 and -8) and the related proteins γ-1 and -6 (see Methods). We detected the presence of γ-2, -3, -4, -5 and -6 (Fig. 5a). To verify the presence of TARP proteins in oligodendrocyte lineage cells, immature- and pre-myelinating OPCs (identified with antibody against O4 or NG2) and oligodendrocytes (identified with antibody against O1), were co-labeled with either a TARP antibody (‘pan-TARP’) that recognized γ-2, -3, -4 and -8, an antibody against γ-5, or an antibody against γ-7. Although we did not detect labeling with anti-γ-5 or anti-γ-7, both immature- and pre-myelinating OPCs, as well as oligodendrocytes, were readily labeled with the pan-TARP antibody (Fig. 5b-e).
Figure 5. TARPs are expressed in OPCs.
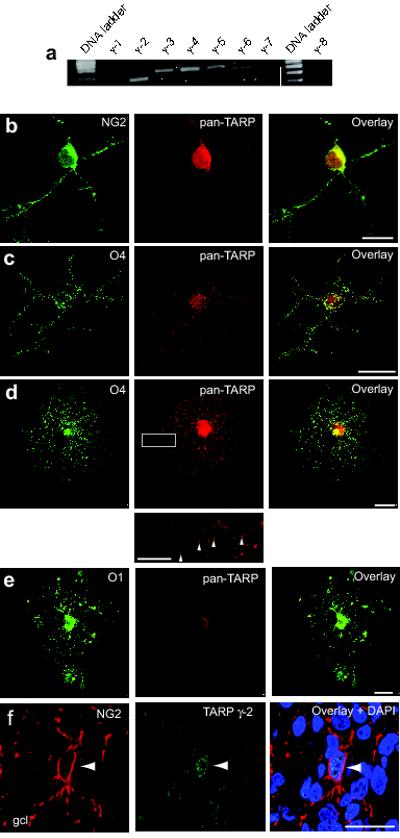
(a) RT-PCR analysis of TARP expression in the rat optic nerve. mRNA for γ-2, γ-3, γ-4, γ-5 and the TARP-related protein γ-6 was detected. (b-d) Representative confocal images showing labeling of (b) NG2+ cells, (c) O4+ cells, and (d) pre-myelinating OPCs, with anti-pan-TARP (red), anti-NG2 (green), anti-O4 (green) antibodies. Note the punctate TARP labeling (indicated by arrowheads) along the processes of the pre-myelinating OPC (inset, from white rectangle in d). (e) Representative confocal images of oligodendrocytes identified with anti-O1 (green). The cells exhibited reduced TARP immunoreactivity compared with that seen with pre-myelinating OPCs. Labeling similar to that shown in b-e was seen in 8-20 cells of each type across 12 separate cultures. Scale bars 25 μm (10 μm, inset). (f) Confocal images of a representative sagittal section of cerebellar cortex from a P7 rat, showing labeling of the granule cell layer (gcl) with anti-NG2 (red) and anti-pan-TARP (green) antibodies and nuclear staining with DAPI (blue). Scale bar 25μm. Arrowhead indicates presumptive NG2+ OPC.
To establish whether TARPs are present in OPCs and oligodendrocytes in vivo, we cut cryostat sections (30 μm) of cerebellar cortex (P7 rat) and labeled them with antibodies against TARP γ-2 39 and NG2 or myelin basic protein (MBP) (Fig. 5f and Supplementary Fig. 2). NG2+-OPCs in the white matter and granule cell layer exhibited TARP immunoreactivity; by comparison labeling of TARPs in white matter MBP-positive cells (oligodendrocytes) was relatively low (Supplementary Fig. 2).
We next considered whether TARPs participate in the surface delivery and regulation of AMPARs in OPCs. Cells were transfected either with full-length wild-type γ-2, or a C-terminal truncated form (γ-2ΔC308) that lacked the ‘TTPV’ PDZ binding domain (common to γ-2, -3 -4 and -8). In neurons, this domain is required for TARP interaction with the synaptic protein PSD-95 40, and is necessary for effective AMPAR targeting to the synapse 34. Although all cells displayed similar glutamate-evoked current densities, transfection with γ-2ΔC308 led to markedly more linear I-V relationships indicating that in these conditions CI-AMPARs predominated in the membrane (RI was 1.10 ± 0.07 with γ-2ΔC308 versus 0.53 ± 0.03 with γ-2; n = 7 and 6, respectively; P = 0.0012) (Fig. 6a). This suggested a likely requirement for TARPs that contain the ‘TTPV’ motif in the trafficking of CP-AMPARs to the membrane surface. Consistent with this view, OPCs that were transfected with truncated γ-2 failed to show a DHPG-induced increase in rectification (RI = 0.83 ± 0.11; n = 5, P = 0.33) (Fig. 6b, c). Furthermore, these cells also exhibited a reduced single-channel conductance (from 40.5 ± 4.2 pS to 21.1 ± 3.3 pS; n = 6 and n = 5, P = 0.0043) (Fig. 6d-f) with no change in Po, peak or desensitization rate (Fig. 6f), as would be expected with an increase in the proportion of CI-AMPARs. Our data thus suggest that TARPs are required for delivery of CP-AMPARs in OPCs, and that this interaction involves the distal (TTPV) region of their C-tail.
Figure 6. TARPs control mGluR-induced AMPAR plasticity.
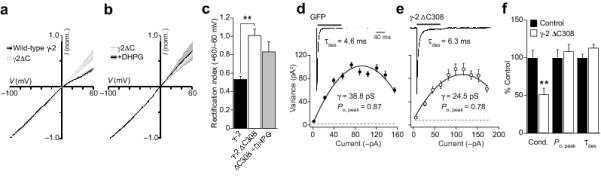
(a) Global averages of normalized I-V plots obtained from OPCs transfected either with full-length γ-2 (n = 6) or γ-2ΔC308 (n = 7). Filled areas indicate S.E.M. (b) DHPG did not alter AMPAR rectification in OPCs transfected with γ-2ΔC308 (n = 5). (c) Pooled data showing RI values. (d) Averaged glutamate-evoked response (10 mM, 100 ms, −60 mV) recorded in a patch excised from an OPC transfected with GFP alone (mean of 60 responses). Inset shows current-variance plot for this patch. (e) Same as d, for an OPC transfected with γ-2ΔC308 (mean of 68 responses). (f) Pooled normalized data showing the effect of γ-2ΔC308 expression on the weighted mean single-channel conductance (Cond.), Po, peak and τdes.
mGluR activation increases synaptic CP-AMPARs in OPCs
To determine whether mGluR activation can trigger an increase of CP-AMPARs at neuron-glia synapses, we examined the effects of DHPG on transmission at climbing fiber (CF) inputs to NG2+-OPCs in cerebellar slices from NG2-DsRed BAC mice 30 (Fig. 7a). OPCs were identified from their morphology 30 and by the presence of characteristic inward sodium current in response to a depolarizing step (Fig. 7b), as previously described 7. Climbing fiber stimulation evoked fast EPSCs that exhibited paired pulse depression typical of CF-EPSCs in neurons and NG2+ cells 41 (Fig. 7c). The rectification index (+60/−80 mV) of these currents was ~0.5 (Fig. 7d, f, g), suggesting that they were mediated by a mixture of CP- and CI-AMPARs. Following treatment with the mGluR agonist DHPG (100 μM for 10 minutes), the EPSCs displayed more marked inward rectification. On average the RI was halved, from 0.48 ± 0.05 to 0.25 ± 0.02 (n = 10 and 5, respectively; P = 0.0080) (Fig. 7e-g).
Figure 7. mGluR activation increases synaptic CP-AMPARs in cerebellar NG2+-OPCs.

(a) Representative confocal images of a sagittal cerebellar slice from an NG2-DsRed mouse (P11), labeled with anti-calbindin (CB; green) to identify Purkinje cells, and stained with DAPI (blue). NG2+-OPCs (red) are readily identified in the Purkinje cell layer (arrowhead). In the middle panel, the molecular layer (ml), Purkinje cell layer (Pcl), granule cell layer (gcl) and white matter (wm) are indicated. Scale bar 25 μm. (b) Representative records from an OPC showing a voltage-gated Na+ current that was blocked by TTX (1 μM). All cells identified as OPCs exhibited such voltage-gated Na+ currents; bar graph shows Na+ current density. (c) Paired-pulse depression of evoked climbing fiber-NG2+-OPC EPSCs. Inset shows representative averaged responses from one cell (−80 mV; pulse separation 500 ms). (d, e) Averaged climbing fiber-evoked EPSCs recorded at +60, 0 and −80 mV in a control cell (d) and in a cell following 15 min application of 100 μM DHPG (e). Corresponding I-V relationships are fitted with third-order polynomials (see Supplementary Fig. 1). The treated cell showed greater inward rectification than seen in the control cell. (f) Averaged normalized I-V relationships from 10 control and 5 DHPG treated cells. Error bars denote S.E.M. and are hidden by symbols. (g) Pooled data showing decreased RI values (increased inward rectification) in the DHPG treated cells.
The increase in rectification at CF-inputs to NG2+ cells was similar to that which we observed in OPCs derived from optic nerve, indicating that mGluR activation triggered a rapid increase in the relative proportion of synaptic CP-AMPARS in NG2+ cells. The rectification seen in DHPG treated cells (Fig. 7f, g), suggests that these EPSCs were mediated predominantly by CP-AMPARs.
Discussion
Here we have shown that the CP-AMPARs in OPCs, which are activated during neuron-glia signalling 3, cell proliferation 1,2 and pathological changes 9,10, are subject to differential regulation by modulatory signals. Specifically, our experiments establish that activation of group 1 mGluRs triggers an increase in the proportion of CP-AMPARs in OPCs – as detected by an increase in single-channel conductance and inward rectification of AMPAR-mediated currents – while activation of purinergic P2Y receptors decreases the proportion of CP-AMPARs. We find that this mGluR1-mediated CP-AMPAR plasticity is triggered by a rise in intracellular Ca2+, and requires activation of PI3 kinase, PICK-1 and the JNK pathways. Furthermore, TARPs, which are essential modulators of AMPAR expression in neurons, also regulate AMPAR plasticity in OPCs.
Implications of mGluR mediated CP-AMPAR plasticity
Activation of group I mGluRs promotes increased insertion of GluA4-containing CP-AMPARs in the OPC membrane. What might be the likely physiological or pathological relevance of such regulation? AMPARs regulate a variety of important physiological and developmental processes in OPCs, including cell proliferation, migration and differentiation. Furthermore, it has been demonstrated that in white matter, glutamate released from axons and certain glial cells (astrocytes) activates CP-AMPARs in NG2+-OPCs 3,13. Calcium influx through these receptors potentiates the expression of immediate early genes NGFI-A and c-fos 42, which are markers for elevated protein expression. Increased expression of CP-AMPARs (likely homomeric GluA4 or heteromeric GluA1/4 receptors) would be expected to facilitate this action, influencing OPC migration and differentiation into myelinating cells 2. The fact that this plasticity mechanism is down regulated in pre-myelinating cells, and in differentiated oligodendrocytes, is therefore consistent with a role in early developmental processes.
In the hippocampus, evoked release of glutamate from nerve terminals onto NG2+-OPCs triggers a form of neuron-glia ‘LTP’, that involves a switch from CI- to CP-AMPARs 16. Although the precise mechanism underlying this change is uncertain, it can be blocked by intracellular BAPTA, suggesting that it requires a rise in intracellular Ca2+. Accordingly, it is possible that the activity-dependent changes seen at neuron-NG2+-OPC synapses could involve an mGluR1-mediated increase in CP-AMPARs of the type we have identified. Indeed, our experiments with OPCs in cerebellar slices indicate that mGluR activation can increase synaptic CP-AMPAR expression. Furthermore, the mGluR-mediated regulation of CP-AMPARs that we describe here is reminiscent of the plasticity seen at certain neuronal synapses, including those between parallel fibers and cerebellar stellate cells 20 and at dopaminergic neurons of the ventral tegmental area (VTA) 23. While these synapses also exhibit an mGluR-induced switch in AMPAR subunit composition, triggered by a rise in intracellular Ca2+, mGluR activation initiates a loss rather than an increase in postsynaptic CP-AMPARs. This could reflect neuronal/glial differences in the expression of AMPAR subtypes, auxiliary proteins or signaling pathways.
Previous work has identified high expression of group 1 mGluRs in CG4 cells and brain derived OPCs 11,24, although, like AMPARs, mGluRs are down regulated in mature oligodendrocytes. It is of note that earlier experiments did not detect an mGluR-mediated change in the surface expression of AMPAR subunits in oligodendrocyte lineage cells 11. While the reason for this is unclear, it could reflect differences in the maturity of the OPCs used.
ATP mediated plasticity
Our experiments demonstrate that the neurotransmitter ATP also regulates AMPAR subunit expression in OPCs – triggering an increase in the proportion of CI-AMPARs. ATP is released from axons and astrocytes in the brain (including the optic nerve) and is known to activate metabotropic P2Y1 receptors to trigger release of calcium from intracellular stores 17,43. Although ionotropic calcium permeable P2X7 receptors are also present in NG2+-OPCs in situ 17, our experiments suggest that P2Y1 receptors are the ones involved in regulation of AMPAR subunit targeting. In addition, the degradation product of ATP – adenosine – can act on adenosine receptors, which have previously been implicated in OPC differentiation 44. In the present study we found that mature pre-myelinating OPCs have reduced expression of CP-AMPARs. It is therefore possible that ATP could prime OPCs for differentiation into myelinating cells by both reducing expression of CP-AMPARs, and promoting differentiation via activation of adenosine receptors.
CP-AMPAR regulation depends on TARP interaction
Our previous studies established that CP-AMPARs in cerebellar Bergmann glia are associated with TARP γ-5, which modifies their channel properties and shapes the time course of AMPAR-mediated quantal events underlying neuron-glia signaling 37. In the present study, we find evidence for TARP expression in OPCs, and that the AMPAR channels display single-channel and kinetic features typical of TARP-associated receptors. Furthermore, as described in neurons 33,34,40, delivery and regulation of AMPAR in OPCs appears to be dependent on TARP interaction. It is of note that a transcriptome database of gene expression in cultured forebrain OPCs 6 identified γ-4 and -5 but not γ-2 or -3. Although we found antibody labeling that may suggest the possible presence of γ-2, -3, -4 or -8, we did not detect γ-5 labeling; however, we do not exclude that it is expressed at a low level.
Transfection of OPCs with a C-terminal truncated form of γ-2 (lacking the last sixteen residues, including the TTPV motif), suppressed insertion of CP-AMPARs, and left a current that was mediated entirely by CI-AMPARs. Thus, the distal region of the TARP C-tail appears critical for surface delivery of CP-AMPARs in OPCs. This, and the fact that our experiments also demonstrated that the mGluR mediated increase in CP-AMPAR was lost when OPCs were transfected with truncated γ-2, suggests that TARP interaction with AMPARs is required for both constitutive and mGluR-mediated insertion of GluA2-lacking CP-AMPARs in these cells.
Relevance to excitotoxic damage
The Ca2+-permeability of AMPARs in the developing and adult brain modulates OPC susceptibility to ischemic damage – a major type of pathological insult that causes direct injury to oligodendrocyte lineage cells, myelin and developing/adult white matter. Our experiments identify various features of AMPAR plasticity in OPCs, which may be relevant in understanding the vulnerability of these cells to excitotoxic damage. Specifically, our data suggest that the mGluR-induced changes can be ascribed to an increased surface delivery of GluA4, triggered by an increase in intracellular Ca2+, and that this is dependent on PI3 kinase, PICK-1 and the Jun N-terminal kinase (JNK) pathway. There is precedent for the involvement of these pathways in neuronal AMPAR plasticity. Thus, PI3-kinase is known to play an important role in AMPAR insertion during LTP 45 and it has been suggested that PICK-1 facilitates the surface expression of CP-AMPARs, by causing GluA2-containing AMPARs to be internalized or retained within intracellular compartments 46. Furthermore, GluA4 subunits (and GluA2L) are reported to be JNK substrates in both heterologous cells and neurons, and that phosphorylation of the JNK site on the AMPAR subunit regulates subunit trafficking on a rapid timescale, promoting AMPAR re-insertion into the membrane 47. There is also compelling evidence that the JNK/c-Jun signalling pathway is particularly important in cell death induced by ischemia and related excitotoxic stimuli 48. Furthermore, excitotoxicity via CP-AMPARs requires Ca2+-dependent JNK activation 49. Cells expressing GluA4 subunits are known to be highly susceptible to excitotoxic damage, a process that involves activation of the AP-1 transcription factor 50 the activity of which is high in immature OPCs 28. It therefore seems likely that an mGluR-mediated increase in GluA4 CP-AMPARs would itself contribute to the vulnerability of OPCs to excitotoxic damage. In keeping with this view, the decrease in vulnerability of oligodendrocytes during development appears to coincide with the loss of mGluR-mediated CP-AMPAR plasticity in the older oligodendrocyte lineage cells.
Methods
CG4 OPC cells
CG4 cells (passages 13 – 22) were grown in modified Sato medium containing 30% (v/v) B104 conditioned medium. The modified Sato medium consisted of Dulbecco’s modified Eagle’s medium (DMEM), 0.1% (w/v) bovine serum albumin fraction V, 60 μg/l progesterone, 16.1 mg/l putrescine, 5 μg/l sodium selenite, 50 mg/l holo-transferrin, 5 mg/l insulin and 2 mM l-glutamine. Cells were cultured in a humidified atmosphere at 37°C with 5% (v/v) CO2. Coverslips and flasks were coated with poly-l-lysine (100 mg/l; Sigma). 72 hrs prior to electrophysiological studies cells were treated every 24 hours with medium containing 10 ng/ml each of platelet-derived growth factor-AA (PDGF-AA) and basic fibroblast growth factor (bFGF) (R&D Systems, Inc.). To prepare B104 conditioned medium, B104 cells were grown in DMEM containing 10% (v/v) heat-inactivated fetal calf serum (FCS; Gibco, Invitrogen Ltd) and 2 mM l-glutamine until 70% confluent, and were then conditioned with modified Sato medium for 4 days.
Optic nerve OPC purification
OPCs were purified by immunopanning 28. Briefly, optic nerves were obtained from postnatal day 7 (P7) male and female rats in accordance with the UK Animals (Scientific Procedures) Act 1986. Tissue was diced, digested in trypsin 0.05 % EDTA then gently dissociated for 30 minutes at 37°C. Dissociated tissue was sequentially immunopanned on Ran-2 (LGC Standards; T1B-119), anti-galactocerebrosidase (GalC) (Millipore; AB142), and then O4 antibody plates (R&D Systems, Inc.; MAB1326) to select GalC− O4+ OPCs. Purified OPCs were transferred to poly-l-lysine coated 24-well tissue culture plates containing proliferation medium. All cells were cultured at 37°C, 5% CO2 in DMEM (Invitrogen Ltd) containing Sato medium; human transferrin (100 μg/ml), bovine serum albumin (100 μg/ml), putrescine (16 μg/ml), progesterone (60 ng/ml), sodium selenite (40 ng/ml), N-acetyl-l-cysteine (6.3 μg/ml), bovine insulin (5 μg/ml) (Sigma), glutamine (2 mM), sodium pyruvate (1 mM), penicillin–streptomycin (100 U each) (Sigma). Proliferation medium also contained OPC mitogens PDGF-AA and bFGF (both 10 ng/ml) (R&D Systems, Inc.).
OPC transfection
Optic nerve OPCs (6 DIV) were transfected with GFP-γ-2 (rat), GFP-γ-2ΔC308 or GFP alone (2 μg of cDNA) using calcium phosphate transfection (Invitrogen Ltd), and used for experiments 24 hours later. GFP-γ-2ΔC308 was created using PCR site-directed mutageneis with mismatch primers (Sigma Genosys) γ-2 (F: ACTACGAGGCTGACACCG R: ACTTAGACCTGCAGACACGAAG). The following complementary primers changed K308 to a stop codon: F: CAGAAGGACAGCTAGGACTCTCTCCAC; R: GTGGAGAGTCCTAGCTGTCCTTCT.
OPC differentiation
Optic nerve OPCs were differentiated into pre-myelinating OPCs by the withdrawal of mitogens bFGF and PDGF-AA. The DMEM+Sato medium was supplemented with 3,3′,5-triiodo-l-thyronine (T3) (40 ng/ml; Sigma, T6397) for 48 hours. Cells were immunolabeled with anti-O4 to confirm their mature status. Pre-myelinating OPCs were maintained in this medium for an additional 48 hours to promote their differentiation into oligodendrocytes.
Immunofluorescence
OPC cultures on poly-l-lysine-coated coverslips were fixed with 4% PFA for 10 minutes at 25°C and washed with 1× phosphate-buffered saline (PBS). Cells were then incubated with in 1× PBS containing 250 mg BSA (Sigma), 10% horse serum (Invitrogen UK), and permeabilized with 0.5% Triton for 20 minutes. Cells were labeled with primary antibodies for 1 hour at 25°C: anti-O4 (mouse, Millipore, MAB345; 1:100), pan-TARP (rabbit, Millipore, AB9876; 1:50), anti-TARP γ-5 (rabbit, Sigma, C6488; 1:50), and anti-TARP γ-7 (rabbit, Abnova, H00059284D01; 1:50). Secondary antibodies – goat anti-rabbit Alexa-568 (Invitrogen, A-11011; 1:1000) and goat anti-mouse Alexa-488 (Invitrogen, A-21042; 1:1000) – were applied for 1 hour at 25°C. Coverslips were mounted using anti-fade medium (Invitrogen UK).
P7 rat cerebellar tissue was immersed in 4% paraformaldehyde (wt/vol, PFA, TAAB labs) and PBS overnight. Tissue was subsequently cryo-protected overnight at 4°C in diethylpyrocarbonate-treated 30% sucrose (wt/vol) in PBS, embedded in OCT compound (VWR), frozen on dry ice and stored overnight at −80 °C. Sagittal cerebellar slices (30 μm) were prepared using a vibratome and collected onto gelatin-coated slides (VWR) and dried overnight at 20–25 °C.
Sections were treated with blocking solution (10% normalized goat serum (vol/vol), 0.5% Triton X-100 (vol/vol Sigma) in PBS) for 6 hrs at 20–25 °C, and then incubated in primary antibody overnight at 4°C followed by incubation with secondary antibody for 4 hours in blocking solution at 22-25°C and finally for 10 mins in PBS containing 4′,6-diamidino-2-phenylindole, dihydrochloride (DAPI) (Invitrogen, 300 nM). Slides were prepared with vectashield mounting medium (Vector labs) and examined using a confocal microscope (LEICA-LSM). The following primary antibodies were used: anti-NG2 (mouse, Millipore, MAB5384; 1:200), MBP (mouse, Abcam, ab62631; 1:200), anti-TARP γ-2 (rabbit, 1:50; gift from Masahiko Watanabe). Secondary antibodies were as described above. The anti-TARP antibody was designed to detect γ-2 (Ref. 39). In our hands, using Western blotting, the antibody labeled γ-2 protein isolated from tsA201 cells. In the same experiments, it also labeled γ-3 and γ-4 protein. In fixed brain slices the antibody strongly labeled cerebellum from an adult wild-type C57BL/6 mouse and this labeling was markedly reduced in cerebellum from an adult stargazer mouse (consistent with the previous findings with γ-2 KO tissue 39).
Electrophysiology
OPCs were viewed with a fixed-stage upright microscope (Axioskop FS1; Zeiss). Macroscopic currents were recorded at 22–24 °C from excised outside-out membrane patches or from isolated whole cells using an Axopatch 1D amplifier (Digidata 1200 interface and pClamp software; Molecular Devices, Inc.). The ‘external’ solution contained (in mM): 145 NaCl, 2.5 KCl, 1 CaCl2, 1 MgCl2, 10 glucose and 10 HEPES (pH 7.3 with NaOH). The ‘internal’ (pipette) solution contained: (in mM) 145 CsCl, 2.5 NaCl, 1 Cs-EGTA, 4 MgATP and 10 HEPES (pH 7.3 with CsOH). Spermine tetrahydrochloride (100 μM; Tocris Bioscience) was added to the intracellular solution.
Fast agonist application to excised patches
Outside-out patches were obtained using electrodes made from thick-walled borosilicate glass (1.5 mm o.d., 0.86 mm i.d.; Harvard Apparatus) with a resistance of 8–12 MΩ (coated with Sylgard resin; Dow Corning 184). Rapid solution switching at the patch was achieved as described previously 21,37. Both control and agonist solutions contained d-AP5 (20 μM), 6-imino-3-(4-methoxyphenyl)-1(6H)-pyridazinebutanoic acid hydrobromide (SR-95531; 20 μM), strychnine (1 μM) and tetrodotoxin (1 μM). To enable visualization of the solution interface and allow measurement of solution exchange 2.5 mg/ml sucrose was added to the agonist solution and the control solution was diluted by 5%. Recorded currents were low-pass filtered at 10 kHz and digitized at 50 kHz.
Non-stationary fluctuation analysis (NSFA)
To determine channel properties from macroscopic responses, we applied glutamate (10 mM) to outside-out patches (100 ms duration, 1 Hz). The ensemble variance of all successive pairs of current responses was calculated using IGOR Pro 5.05 (Wavemetrics) and NeuroMatic (http://www.neuromatic.thinkrandom.com). The single-channel current (i), total number of channels (N) and maximum open probability (Po, max) were then determined by plotting this ensemble variance (σ2) against mean current (), and fitting this with a parabolic function:
| (1) |
where σB2 is the background variance. The weighted-mean single-channel conductance was calculated from the single-channel current and the holding potential (uncorrected for liquid-junction potential). Po, max was calculated by dividing the average peak current by iN.
Whole-cell recordings
Whole-cell recordings were made from isolated cells using thick-walled electrodes with a resistance 4-7 MΩ. The external solution included d-AP5 (20 μM), SR-95531 (20 μM), strychnine (1 μM), cyclothaizide (50 μM) and TTX (1 μM) (Ascent Scientific). A ramp protocol was used to change the holding potential (−100 mV for 200 ms, then to +60 mV at a rate of 162.5 mV/s). Records were filtered at 2 kHz and sampled at 5 kHz. Receptors were activated by a bath application of 100 μM glutamate. Other compounds were used as indicated: DHPG ((S)-3,5-dihydroxy-phenylglycine), ACDPP (3-amino-6-chloro-5-dimethylamino-N-2-pyridinylpyrazine carboxamide hydrochloride), MCPG ((RS)-α-methyl-4-carboxyphenylglycine), SP600125 (anthra[1-9-cd]pyrazol-6(2H)-one), KN-62 (4-[(2S)-2-[(5-isoquinolinylsulfonyl)methylamino]-3-oxo-3-(4-phenyl-1-piperazinyl)propyl] phenyl isoquinolinesulfonic acid ester), BzATP (2′(3′)-O-(4-benzoylbenzoyl)adenosine-5′-triphosphate tri(triethylammonium) salt) (Tocris Bioscience) and wortmannin and BAPTA-AM (1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis(acetoxymethyl ester) (Sigma). GFP-PICK-1 inhibitor peptide pep2-EVKI (a gift from John Wood, UCL) was conjugated to TAT peptide to allow membrane insertion. The selective CP-AMPA receptor blocker philanthotoxin-433 (5 μM; (S)-N-[4-[[3-[(3aminopropyl)amino] propyl]amino]butyl]-4-hydroxy-a-[(1-oxo- butyl)amino] benzenepropanamide tris(trifluoroacetate) salt) was used in some experiments.
Cerebellar slices
Male and female NG2-DsRed BAC mice (P9–11) were anesthetized with isoflurane and decapitated in accordance with the UK Animals (Scientific Procedures) Act 1986. After brain dissection, parasagittal slices (250 μm) were cut from the cerebellar vermis and paravermis, as described previously 21,37. Slices were transferred to a submerged chamber (perfused at 1.5–2.5 ml/min, 22-24°C), and NG2+-OPCs were visualized using epifluorescence imaging and infrared differential interference contrast optics (Axioskop; Zeiss). Recording pipettes were pulled from thick-walled borosilicate glass tubing, coated with Sylgard and fire polished prior to use. Pipettes were filled with ‘internal’ solution (as described above) and had a resistance of 5–10 MΩ. Series resistance following compensation (40-60%) was 8.6 ± 0.9 MΩ (n = 10). Currents were recorded using an Axopatch-200A amplifier (Molecular Devices, Inc.), filtered at 5 or 10 kHz (low-pass 8-pole Bessel filter) and sampled at 50 or 100 kHz.
Fluorescent cells were identified as NG2+-OPCs when the following criteria were satisfied: 1) lack of obvious contact with neighboring fluorescent cells or blood vessels, 2) characteristic rounded soma with few processes, 3) a relatively small input capacitance (12.4 ± 1.2 pF, n = 10) and, 4) the expression of voltage-gated Na+ channels 7. To test for the latter, a depolarizing step (from −80 mV to −10 mV; 8 ms) was applied within 2 minutes of rupturing the patch, and the resulting current recorded, first in control conditions and then (in most but not all cells) following bath application of 1 μM TTX (see Fig. 7b).
Recordings were made from NG2+-OPCs located close to the Purkinje cell layer, and a pipette containing external solution was positioned in the granule cell layer to stimulate a climbing fiber (CF). The position of the stimulating electrode was adjusted until stable CF-EPSCs were elicited that showed typical paired-pulse depression (20-100 V, 20-100 μs duration at 0.1 Hz; paired pulse interval 500 ms; DS2 stimulator, Digitimer Ltd). A voltage step was applied prior to each stimulus pair to assess series resistance; recordings showing a >20% increase were discarded. CF-EPSCs were recorded at different voltages to generate an I-V plot (minimally −80, 0 and +60 mV) (see Supplementary Fig. 2). The following drugs were used, as indicated: 1μM strychnine hydrochloride, 20 μM SR-95531, 20 μM d-AP5 and 100 μM cyclothiazide. mGluRs were activated by bath application of DHPG (100 μM; 15 minutes), during which time the cell was kept at −80 mV. The effect on CF-EPSCs was tested three minutes after drug washout. DHPG was applied in the presence of the purinergic receptor blocker PPADS (100 μM). In all cases, where tested, CF-EPSCs were fully blocked by 10 μM 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulphonamide (NBQX) at the end of the experiment.
Cell surface biotinylation of AMPARs
Cell surface biotinylation was performed as described previously 37. Briefly, CG4 OPCs were chilled on ice and washed twice with ice-cold PBS then treated with 1 mg/ml sulfo-NHS-biotin (Pierce). Unreacted biotinylation reagent was quenched by washing with 50 mM glycine in PBS (with Mg2+ and Ca2+). Cells were harvested in RIPA buffer (Perbio). Homogenates were centrifuged (14,000g, 10 min, 4°C) and the input aliquot removed. The remaining supernatant was incubated with 20 μl of 50% UltraLink Immobilized NeutrAvidin Protein (Pierce). After incubation, the NeutrAvidin protein was washed twice with high-NaCl RIPA buffer (500 mM NaCl) and once with low-NaCl RIPA (150 mM NaCl), and bound proteins were eluted with SDS sample buffer by boiling (5 min, 95°C). Western blotting was carried out using an XCell SureLoc Novex Mini-Cell system (Invitrogen). The biotinylated proteins were probed using antibodies to GluA2, GluA3 and GluA4 (Chemicon; MAB397, MAB5416 and AB1508, respectively). Immunoblots were visualized by ECL development (GE Healthcare Life Sciences) and quantified on a calibrated densitometer (Bio-Rad GS-800).
RNA PCR analysis
RNA was isolated from the rat optic nerve using RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. Reverse Transcription (RT reaction) was carried out using Maxima® Reverse Transcriptase (Fermentas). The products were amplified using 30 cycles of 94°C. The following primers were used: CACNG1 – F: GCGGGGGAAAAGAATTG, R: CAGAGCCCTGCAAAGG. CACNG2-OPC – F: ACTACGAGGCTGACACCG, R: ACTTAGACCTGCAGACACGAAG (181 long). CACNG3 – F: GCTGCTTAGAAGGAGCTTTCC, R: GTTGCTTAGCCCTGCAGAG 62.7°C (248 long). CACNG4 – F: CATCGAAGGCATCTACAAGG, R: GATATTACTGAGGCCTGCAGC (256 long). CACNG5 – F: GCTTCCTCGCAGGTGAG, R: AAGAGAGAGGCCGGATAGG 62.2°C (250 long). CACNG6 – F: TGCCAGGAGAAGCAAACTG, R: AGCAGCAGGCCTGAGAG (481, 307, 232). CACNG7 – F: TTCTTTGCAGGTCGGGAG, R: CAAGGACAGGCCTGATAAAATG (248 long). CACNG8 – F: CCTGGAAGGGTTGAAAAGAG, R: TGATGTTGCTCAGGCCTG (247 long).
PCR products were resolved on a 2% agarose gel. To check the RNA product, we repeated the RNA extraction and RT-PCR protocol to determine which TARPs are present in the optic nerve. RT-PCR products were isolated from the agarose gel and further RT-PCR was carried out on the isolated products. These were sequenced and the identity of the products confirmed.
Statistical Analysis
Data are presented as mean ± S.E.M. Statistical significance was examined using a Wilcoxon rank sum test. Group differences were examined using a Kruskal-Wallis rank sum test followed by pair wise Wilcoxon rank sum tests with Holm’s sequential Bonferroni correction (R 2.9.2, The R Foundation for Statistical Computing; http://www.R-project.org). Results were considered significant with P < 0.05. In the text P values are given to two significant figures. In all figures asterisks denote significance, as follows: * P <0.05, ** P < 0.01, *** P < 0.001.
Supplementary Material
Acknowledgements
We thank Ian Coombs, Cecile Bats, Chris Shelley, David Soto and Dorota Studniarczyk for invaluable help and discussion. We are grateful to Elek Molnar (Bristol University) for CG4-OPC cells, David Attwell and Clare Reynell (UCL) for providing NG2-DsRed mice, Roger Nicoll (UCSF) for TARP cDNA (rat; γ-2), John Wood (UCL) for GFP-PICK-1 inhibitor peptide, Masahiko Watanabe (Hokkaido University) for anti-TARP γ-2 antibody, Beverley Clark (UCL) for anti-calbindin antibody, and Dan Cutler and Mark Marsh (LMCB, UCL) for generous help and access to equipment. This work was supported by a Wellcome Trust Programme Grant (SGC-C and MF). MZ was in receipt of an MRC (LMCB, UCL) studentship.
Footnotes
Author contributions M.Z. performed electrophysiology and molecular experiments on cultured cells. M.R and M.Z. performed slice recordings. M.F. and M.Z. analyzed the data. All authors contributed to the design and interpretation of experiments. S.G.C.-C. and M.F. supervised the project. M.Z., M.F., and S.G.C.-C. wrote the paper.
References
- 1.Gallo V, et al. Oligodendrocyte progenitor cell proliferation and lineage progression are regulated by glutamate receptor-mediated K+ channel block. J Neurosci. 1996;16:2659–2670. doi: 10.1523/JNEUROSCI.16-08-02659.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gudz TI, Komuro H, Macklin WB. Glutamate stimulates oligodendrocyte progenitor migration mediated via an alphav integrin/myelin proteolipid protein complex. J Neurosci. 2006;26:2458–2466. doi: 10.1523/JNEUROSCI.4054-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bergles DE, Roberts JD, Somogyi P, Jahr CE. Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature. 2000;405:187–191. doi: 10.1038/35012083. [DOI] [PubMed] [Google Scholar]
- 4.Traynelis SF, et al. Glutamate receptor ion channels: structure, regulation, and function. Pharmacol Rev. 2010;62:405–496. doi: 10.1124/pr.109.002451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geiger JR, et al. Relative abundance of subunit mRNAs determines gating and Ca2+ permeability of AMPA receptors in principal neurons and interneurons in rat CNS. Neuron. 1995;15:193–204. doi: 10.1016/0896-6273(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 6.Cahoy JD, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Biase LM, Nishiyama A, Bergles DE. Excitability and synaptic communication within the oligodendrocyte lineage. J Neurosci. 2010;30:3600–3611. doi: 10.1523/JNEUROSCI.6000-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Etxeberria A, Mangin J-M, Aguirre A, Gallo V. Adult-born SVZ progenitors receive transient synapses during remyelination in corpus callosum. Nat Neurosci. 2010;13:287–289. doi: 10.1038/nn.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fern R, Moller T. Rapid ischemic cell death in immature oligodendrocytes: a fatal glutamate release feedback loop. J Neurosci. 2000;20:34–42. doi: 10.1523/JNEUROSCI.20-01-00034.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Follett PL, Rosenberg PA, Volpe JJ, Jensen FE. NBQX attenuates excitotoxic injury in developing white matter. J Neurosci. 2000;20:9235–9241. doi: 10.1523/JNEUROSCI.20-24-09235.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng W, Wang H, Rosenberg PA, Volpe JJ, Jensen FE. Role of metabotropic glutamate receptors in oligodendrocyte excitotoxicity and oxidative stress. Proc Natl Acad Sci U S A. 2004;101:7751–7756. doi: 10.1073/pnas.0307850101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Volpe JJ. Cerebellum of the premature infant: rapidly developing, vulnerable, clinically important. J Child Neurol. 2009;24:1085–1104. doi: 10.1177/0883073809338067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kukley M, Nishiyama A, Dietrich D. The fate of synaptic input to NG2 glial cells: neurons specifically downregulate transmitter release onto differentiating oligodendroglial cells. J Neurosci. 2010;30:8320–8331. doi: 10.1523/JNEUROSCI.0854-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iino M, et al. Glia-synapse interaction through Ca2+-permeable AMPA receptors in Bergmann glia. Science. 2001;292:926–929. doi: 10.1126/science.1058827. [DOI] [PubMed] [Google Scholar]
- 15.Liu SQJ, Cull-Candy SG. Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature. 2000;405:454–458. doi: 10.1038/35013064. [DOI] [PubMed] [Google Scholar]
- 16.Ge WP, et al. Long-term potentiation of neuron-glia synapses mediated by Ca2+-permeable AMPA receptors. Science. 2006;312:1533–1537. doi: 10.1126/science.1124669. [DOI] [PubMed] [Google Scholar]
- 17.Hamilton N, Vayro S, Wigley R, Butt AM. Axons and astrocytes release ATP and glutamate to evoke calcium signals in NG2-glia. Glia. 2010;58:66–79. doi: 10.1002/glia.20902. [DOI] [PubMed] [Google Scholar]
- 18.Domercq M, et al. P2X7 receptors mediate ischemic damage to oligodendrocytes. Glia. 2010;58:730–740. doi: 10.1002/glia.20958. [DOI] [PubMed] [Google Scholar]
- 19.Haberlandt C, et al. Gray matter NG2 cells display multiple Ca2+-signaling pathways and highly motile processes. PLoS One. 2011;6:e17575. doi: 10.1371/journal.pone.0017575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelly L, Farrant M, Cull-Candy SG. Synaptic mGluR activation drives plasticity of calcium-permeable AMPA receptors. Nat Neurosci. 2009;12:593–601. doi: 10.1038/nn.2309. [DOI] [PubMed] [Google Scholar]
- 21.Soto D, Coombs ID, Kelly L, Farrant M, Cull-Candy SG. Stargazin attenuates intracellular polyamine block of calcium-permeable AMPA receptors. Nat Neurosci. 2007;10:1260–1267. doi: 10.1038/nn1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swanson GT, Kamboj SK, Cull-Candy SG. Single-channel properties of recombinant AMPA receptors depend on RNA editing, splice variation, and subunit composition. J Neurosci. 1997;17:58–69. doi: 10.1523/JNEUROSCI.17-01-00058.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mameli M, Balland B, Lujan R, Luscher C. Rapid synthesis and synaptic insertion of GluR2 for mGluR-LTD in the ventral tegmental area. Science. 2007;317:530–533. doi: 10.1126/science.1142365. [DOI] [PubMed] [Google Scholar]
- 24.Luyt K, Varadi A, Molnar E. Functional metabotropic glutamate receptors are expressed in oligodendrocyte progenitor cells. J Neurochem. 2003;84:1452–1464. doi: 10.1046/j.1471-4159.2003.01661.x. [DOI] [PubMed] [Google Scholar]
- 25.Jo J, et al. Metabotropic glutamate receptor-mediated LTD involves two interacting Ca(2+) sensors, NCS-1 and PICK1. Neuron. 2008;60:1095–1111. doi: 10.1016/j.neuron.2008.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ahn SM, Choe ES. Alterations in GluR2 AMPA receptor phosphorylation at serine 880 following group I metabotropic glutamate receptor stimulation in the rat dorsal striatum. J Neurosci Res. 2010;88:992–999. doi: 10.1002/jnr.22275. [DOI] [PubMed] [Google Scholar]
- 27.Li XM, et al. JNK1 contributes to metabotropic glutamate receptor-dependent long-term depression and short-term synaptic plasticity in the mice area hippocampal CA1. Eur J Neurosci. 2007;25:391–396. doi: 10.1111/j.1460-9568.2006.05300.x. [DOI] [PubMed] [Google Scholar]
- 28.Barres BA, Lazar MA, Raff MC. A novel role for thyroid hormone, glucocorticoids and retinoic acid in timing oligodendrocyte development. Development. 1994;120:1097–1108. doi: 10.1242/dev.120.5.1097. [DOI] [PubMed] [Google Scholar]
- 29.Wyllie DJ, Mathie A, Symonds CJ, Cull-Candy SG. Activation of glutamate receptors and glutamate uptake in identified macroglial cells in rat cerebellar cultures. J Physiol. 1991;432:235–258. doi: 10.1113/jphysiol.1991.sp018383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ziskin JL, Nishiyama A, Rubio M, Fukaya M, Bergles DE. Vesicular release of glutamate from unmyelinated axons in white matter. Nat Neurosci. 2007;10:321–330. doi: 10.1038/nn1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.James G, Butt AM. P2X and P2Y purinoreceptors mediate ATP-evoked calcium signalling in optic nerve glia in situ. Cell Calcium. 2001;30:251–259. doi: 10.1054/ceca.2001.0232. [DOI] [PubMed] [Google Scholar]
- 32.Pankratov YV, Lalo UV, Krishtal OA. Role for P2X receptors in long-term potentiation. J Neurosci. 2002;22:8363–8369. doi: 10.1523/JNEUROSCI.22-19-08363.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rouach N, et al. TARP gamma-8 controls hippocampal AMPA receptor number, distribution and synaptic plasticity. Nat Neurosci. 2005;8:1525–1533. doi: 10.1038/nn1551. [DOI] [PubMed] [Google Scholar]
- 34.Tomita S, Stein V, Stocker TJ, Nicoll RA, Bredt DS. Bidirectional synaptic plasticity regulated by phosphorylation of stargazin-like TARPs. Neuron. 2005;45:269–277. doi: 10.1016/j.neuron.2005.01.009. [DOI] [PubMed] [Google Scholar]
- 35.Fukaya M, Yamazaki M, Sakimura K, Watanabe M. Spatial diversity in gene expression for VDCCgamma subunit family in developing and adult mouse brains. Neurosci Res. 2005;53:376–383. doi: 10.1016/j.neures.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 36.Tomita S, et al. Functional studies and distribution define a family of transmembrane AMPA receptor regulatory proteins. Journal of Cell Biology. 2003;161:805–816. doi: 10.1083/jcb.200212116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Soto D, et al. Selective regulation of long-form calcium-permeable AMPA receptors by an atypical TARP, gamma-5. Nat Neurosci. 2009;12:277–285. doi: 10.1038/nn.2266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shi Y, Lu W, Milstein AD, Nicoll RA. The stoichiometry of AMPA receptors and TARPs varies by neuronal cell type. Neuron. 2009;62:633–640. doi: 10.1016/j.neuron.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamazaki M, et al. TARPs gamma-2 and gamma-7 are essential for AMPA receptor expression in the cerebellum. European Journal of Neuroscience. 2010;31:2204–2220. doi: 10.1111/j.1460-9568.2010.07254.x. [DOI] [PubMed] [Google Scholar]
- 40.Bats C, Groc L, Choquet D. The interaction between Stargazin and PSD-95 regulates AMPA receptor surface trafficking. Neuron. 2007;53:719–734. doi: 10.1016/j.neuron.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 41.Lin SC, et al. Climbing fiber innervation of NG2-expressing glia in the mammalian cerebellum. Neuron. 2005;46:773–785. doi: 10.1016/j.neuron.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 42.Pende M, Holtzclaw LA, Curtis JL, Russell JT, Gallo V. Glutamate regulates intracellular calcium and gene expression in oligodendrocyte progenitors through the activation of DL-alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors. Proc Natl Acad Sci U S A. 1994;91:3215–3219. doi: 10.1073/pnas.91.8.3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ishibashi T, et al. Astrocytes promote myelination in response to electrical impulses. Neuron. 2006;49:823–832. doi: 10.1016/j.neuron.2006.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stevens B, Porta S, Haak LL, Gallo V, Fields RD. Adenosine: a neuron-glial transmitter promoting myelination in the CNS in response to action potentials. Neuron. 2002;36:855–868. doi: 10.1016/s0896-6273(02)01067-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Man HY, et al. Activation of PI3-kinase is required for AMPA receptor insertion during LTP of mEPSCs in cultured hippocampal neurons. Neuron. 2003;38:611–624. doi: 10.1016/s0896-6273(03)00228-9. [DOI] [PubMed] [Google Scholar]
- 46.Clem RL, Anggono V, Huganir RL. PICK1 regulates incorporation of calcium-permeable AMPA receptors during cortical synaptic strengthening. J Neurosci. 2010;30:6360–6366. doi: 10.1523/JNEUROSCI.6276-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomas GM, Lin DT, Nuriya M, Huganir RL. Rapid and bi-directional regulation of AMPA receptor phosphorylation and trafficking by JNK. Embo J. 2008;27:361–372. doi: 10.1038/sj.emboj.7601969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Borsello T, et al. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med. 2003;9:1180–1186. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- 49.Vieira M, et al. Excitotoxicity through Ca2+-permeable AMPA receptors requires Ca2+-dependent JNK activation. Neurobiol Dis. 2010;40:645–655. doi: 10.1016/j.nbd.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Santos AE, et al. Excitotoxicity mediated by Ca2+-permeable GluR4-containing AMPA receptors involves the AP-1 transcription factor. Cell Death Differ. 2006;13:652–660. doi: 10.1038/sj.cdd.4401785. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.