

Abstract
Background
Mutations in SLC26A4 cause Pendred syndrome (hearing loss with goiter) or DFNB4 (non-syndromic hearing loss with inner ear malformation, such as enlarged vestibular aqueduct or Mondini deformity). The relationship between mutations in SLC26A4 and Mondini deformity without enlarged vestibular aqueduct has not been studied in any Chinese deaf population. The purpose of this study was to assess whether mutations in the SLC26A4 gene cause Mondini deformity without an enlarged vestibular aqueduct (isolated Mondini deformity) in a Chinese population.
Methods
In total, 144 patients with sensorineural hearing loss were included and subjected to high-resolution temporal bone CT. Among them, 28 patients with isolated Mondini dysplasia (MD group), 50 patients with enlarged vestibular aqueduct with Mondini dysplasia (EVA with MD group), 50 patients with enlarged vestibular aqueduct without Mondini dysplasia (EVA group), and 16 patients with other types of inner ear malformations (IEM group) were identified. The coding exons of SLC26A4 were analyzed in all subjects.
Results
DNA sequence analysis of SLC26A4 was performed in all 144 patients. In the different groups, the detection rate of the SLC26A4 mutation differed. In the isolated MD group, only one single allelic mutation in SLC26A4 was found in one patient (1/28, 3.6%). In the EVA with MD group, biallelic and monoallelic SLC26A4 mutations were identified in 46 patients (46/50, 92.0%) and three patients (3/50, 6.0%), respectively. Also, in the EVA group, biallelic and monoallelic SLC26A4 mutations were identified in 46 patients (46/50, 92.0%) and three patients (3/50, 6.0%), respectively. These percentages were identical to those in the EVA plus MD group. Only two patients carried monoallelic mutations of the SLC26A4 gene in the IEM group (2/16, 12.5%). There were significant differences in the frequency of SLC26A4 mutation among the groups (P < 0.001). The detection rate of SLC26A4 mutation in the isolated MD group was significantly lower than in the EVA group (with or without MD; P < 0.001), and there was no significant difference in the detection rate of SLC26A4 between the MD group and IEM group (P > 0.5).
Conclusion
Although mutations in the SLC26A4 gene were frequently found in Chinese EVA patients with and without MD, there was no evidence to show a relationship between isolated MD and the SLC26A4 gene in the Chinese population examined. Hearing impairment in patients with isolated MD may be caused by factors other than mutations in the SLC26A4 gene.
Keywords: SLC26A4, hearing loss, Mondini dysplasia, enlarged vestibular aqueduct, inner ear malformation.
Introduction
Hearing impairment is the most common neurosensory disorder in humans with congenital or prelingual deafness, affecting about 1 in 300 to 1 in 1000 children [1-3]. Approximately half of cases have a genetic etiology, including syndromic and non-syndromic forms, with extraordinary genetic heterogeneity. Non-syndromic deafness accounts for 60-70% of inherited hearing impairment. It involves more than 100 different genes with autosomal dominant (DFNA), autosomal recessive (DFNB), X-linked (DFN), and maternal inheritance [4]; autosomal recessive is the most common. Mutations in the SLC26A4 gene are probably the second most common cause of inherited hearing loss, after GJB2 mutations, and are responsible for Pendred syndrome as well as DFNB4 (non-syndromic hearing loss with inner ear abnormalities, such as enlarged vestibular aqueduct (EVA) with or without Mondini dysplasia (MD)) [5,6].
Enlarged vestibular aqueduct syndrome (EVAS) and MD are the most frequently encountered radiographic anomalies of the inner ear. The most accepted criterion for definition of a large vestibular aqueduct is that suggested by Valvassori [7]: a vestibular aqueduct is considered to be enlarged if its diameter is > 1.5 mm at the midpoint between the common crus and the external aperture of the vestibular aqueduct on CT images. The imaging diagnostic criterion is differet about Mondini malformarion. Mondini first described by Carlo Mondini in 1791[8]detailed a small cochlea completing only one and a half turns and lacking a complete interscalar septum, addtionally, he described a bulbous endolymphatic sac and a dilated vestibular aqueduct. Since then, the term "Mondini" has been used as an umbrella term in clinical practice to describe almost any type of congenital cochlear deformity, included temporal bones with a common cavity and an incomplete interscalar septum of the cochlea. And a Mondini cochlea (isolated Mondini) occurs only when the two and a half turns of the cochlea are replaced by a single cavity in the apical region because of the absence of the interscalar septum [9-11]. It is usually bilateral and frequently associated with other inner ear malformations (IEMs) such as cystic expansion of the vestibules and semicircular canal dysplasia. Temporal bone abnormalities, ranging from isolated EVA to MD, have been causally linked to mutations in the anion transporter gene SLC26A4, which encodes the protein pendrin [5]. Pourova [12] found that 40.9% of Czech EVA/MD patients carried SLC26A4 mutations (27.3% had biallelic mutations and 13.6% had monoallelic mutations), and the detection rate of SLC26A4 mutations in Italian patients was 30% in Bogazzi's research [13]. These observations suggest that SLC26A4 mutations are frequently present in patients with EVA or MD. In the Chinese deaf population, SLC26A4 was also found to have a close relationship with the pathogenesis of DFNB4 [14,15], but no systematic study was applied to examine the role of SLC26A4 in the pathogenesis of isolated Mondini.
To understand the molecular etiology of isolated MD (MD without EVA) and provide possible genetic testing and counseling for hearing loss patients with isolated MD in China, we performed SLC26A4 sequence analysis in 28 patients with isolated MD, 50 patients with EVA and MD, 50 patients with isolated EVA, and 16 patients with other forms of IEM. In this study, the imaging diagnostic criterion of isolated Mondini is a cochlea of one and a half turns instead of the normal two and a half turns, comprising a normal basal turn and a cystic apex.
Materials and methods
Patients
In total, 144 patients with sensorineural hearing loss (from unrelated families) were reached through the Otolaryngology Department of Chinese PLA General Hospital and included in this study. High-resolution temporal bone CT was available for all 144 recruited patients. Isolated MD was defined as a complex malformation in which the normal cochlear two and a half turn spirals were replaced by a hypoplastic coil of one and a half turns because of an absence of the interscalar septum. EVA was considered to be enlarged if its diameter was > 1.5 mm at the midpoint between the common crus and the external aperture of the vestibular aqueduct. Based on the image information in these 144 patients, isolated MD was identified in 28 patients (19.4%), EVA with MD was identified in 50 patients (34.7%), EVA without MD was identified in 50 patients (34.7%), and other types of IEM were identified in 16 patients (11.1%). The other types of IEM included six cases of cystic cochlea, dilated vestibule, and enlarged internal auditory canal, two cases of common cavity (a cystic cavity representing the cochlea and vestibule without showing any differentiation into cochlea and vestibule), semicircular canal aplasia, and narrow internal auditory canal, one case of semicircular canal hypoplasia, one case of inner ear ossification, one case of vestibule and semicircular ossification, three cases of common cavity and enlarged internal auditory canal, one case of common cavity, and one case of common cavity with narrow internal auditory canal (Figure 1). Of the 144 patients, 82 were males and 62 were females. Ages ranged from 10 months to 25 years. In isolated MD group, ages ranged from 14 months to 26 years and the average age was 8 years. In EVA with MD group, ages ranged from 8 months to 29 years and the average age was 11 years. In EVA group, ages were from 9 months to 31 years and the average age was 10 years, and in IEM group, ages ranged from 15 months to 27 years and the average age was 10 years.
Figure 1.
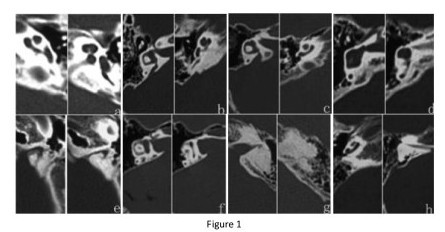
Typical CT scan images of MD, EVA with MD, EVA, and other inner ear malformations. a. MD; b. EVA with MD; c. EVA; d. common cavity (a cystic cavity representing the cochlea and vestibule without showing any differentiation into cochlea and vestibule); e. narrow internal auditory canal; f. internal auditory canal enlarged; g. inner ear ossification; h. cochlear, vestibular, and semicircular canal hypoplasia.
Clinical evaluation
A complete history, physical and otoscopic examinations, and audiological testing, including pure tone audiometry, tympanometry, or auditory brainstem response, were carried out in all patients. No patient was found to have any goiter symptom or sign or syndromic symptoms in other systems. Pure-tone average (PTA) was calculated as the average of the threshold measured at 0.5, 1.0, 2.0, and 4.0 kHz and was used to compare subgroups of patients. The level of hearing loss, in terms of PTA, was described as follows: normal hearing, < 20 dB; mild hearing impairment, 21-40 dB; moderate hearing impairment, 41-70 dB; severe hearing impairment, 71-90 dB; and profound hearing impairment, > 91 dB.
Mutational analysis
Informed consent was obtained from all participants or the parents of minors prior to blood sampling. This study was performed according to the protocol approved by the Ethics Committee of the Chinese PLA General Hospital.
DNA was extracted from peripheral blood leukocytes using a commercially available DNA extraction kit (Watson Biotechnology, Inc., Shanghai, China). DNA sequence analysis of the SLC26A4 gene was performed by polymerase chain reaction amplification of the 21 coding exons plus approximately 50-100 bp of the flanking intron regions with previously published primers [16]. The reaction mixture contained 100 ng DNA, 1.5 units of DNA Taq polymerase(TaKaRa, Dalian, China), 200 uM dNTPs, 3 pmol of each forward and reverse primer, 2.5 ul of 10 × buffer(with 2.5 mM MgCl2) and the final reaction volume was filled to 25ul with ddH2O. The exons in the gene were amplified according to the PCR conditions described previously [17,18]. The PCR amplified products were sequenced and analyzed using an ABI 3130 DNA sequencing machine (ABI, Foster City, CA, USA) and the ABI 3130 Analysis Software (v.3.7 NT) according to the manufacturer's instructions.
Statistical testing
The distribution of SLC26A4 in the different groups belongs to nonparametric randomized block design information. The Friedman rank sum test (M-test) was used to determine whether there was a significance difference in the observed number of mutant SLC26A4 genes in the isolated MD, EVA with MD, EVA, and IEM groups [19]. The chi-squared test(R × C table) was used to compare the hearing level among the four groups[20]. All statistical methods were analyzed on SPSS (statistical package for the Social Sciences)15.0 software. A P-value of 0.05 or less was accepted as statistically significant.
Results
The hearing loss phenotype ranged from moderate to profound. Table 1 shows the distribution of hearing impairment in the isolated MD, EVA with MD, EVA, and IEM groups. Most patients in the four groups had severe to profound hearing loss, but there was no difference in the distribution of hearing level among the four groups.
Table 1.
Hearing levels in the different groups
| Hearing level | MD | EVA with MD | EVA | IEM |
|---|---|---|---|---|
| Moderate | 2 | 5 | 4 | 1 |
| Severe | 6 | 17 | 14 | 5 |
| Profound | 20 | 28 | 32 | 10 |
MD: Isolated Mondini dysplasia, EVA with MD: enlarged vestibular aqueduct with Mondini dysplasia, EVA: enlarged vestibular aqueduct without Mondini dysplasia, IEM: other types of inner ear malformations
DNA sequence analysis of SLC26A4 was performed in all 144 patients. Biallelic, monoallelic, and no SLC26A4 mutations were detected in 92 patients (63.9%), nine patients (6.3%), and 43 patients (29.9%), respectively. In the different groups, the detection rate of the SLC26A4 mutation was discrepant (Table 2).
Table 2.
Distribution of SLC26A4 mutations in the different groups
| Inner ear image | No mutation | Monoallelic mutation | Biallelic mutation | Total |
|---|---|---|---|---|
| MD | 27 (96.4%) | 1 (3.6%) | 0 | 28 (100%) |
| EVA with MD | 1 (2.0%) | 3 (6.0%) | 46 (92.0%) | 50 (100%) |
| EVA | 1 (2.0%) | 3 (6.0%) | 46 (92.0%) | 50 (100%) |
| IEM | 14 (87.5%) | 2 (12.5%) | 0 | 16 (100%) |
| Total | 43 | 9 | 92 | 144 |
MD: Isolated Mondini dysplasia, EVA with MD: Enlarged vestibular aqueduct with Mondini dysplasia, EVA: enlarged vestibular aqueduct without Mondini dysplasia, IEM: Other types of inner ear malformations
In the isolated MD group, only one monoallelic mutation in SLC26A4 (c.919-2A>G, aberrant splicing) was found in one patient (1/28, 3.6%), and no mutation was found in the other 27 patients (27/28, 96.4%). In the EVA with MD group, biallelic, monoallelic, and no SLC26A4 mutations were identified in 46 patients (46/50, 92.0%), three patients (3/50, 6.0%), and one patient (1/50, 2.0%), respectively. In the EVA groups, biallelic, monoallelic, and no SLC26A4 mutations were identified in 46 patients (46/50, 92.0%), three patients (3/50, 6.0%), and one patient (1/50, 2.0%), respectively. Similar to the isolated MD group, only two patients were identified to carry monoallelic mutations of the SLC26A4 gene in the IEM group (2/16, 12.5%). Among them, one patient carried the c.919-2A>G mutation, and the other carried the c.1286C>A(p.A429E) mutation. All variants of SLC26A4 found in the EVA plus MD group and EVA group are listed in Table 3.
Table 3.
Distribution of SLC26A4 genotypes in patients with different CT scan phenotype
| CT phenotype | Patient number | Genotype of SLC26A4 | |||
|---|---|---|---|---|---|
| allele 1 | allele 2 | ||||
| nucleotide change | amino acid change | nucleotide change | amino acid change | ||
| Isolated MD | 1 | c.919-2 A>G[21] | aberrant splicing | - | - |
| (1-28) | 2-28 | - | - | - | - |
| IEM | 29 | c.919-2 A>G | aberrant splicing | - | - |
| (29-44) | 30 | 1286C>A (this study) | A429E | - | - |
| 31-44 | - | - | - | - | |
| EVA | 45 | c.917insG(6) | Frameshift | - | - |
| (45-94) | 46-57 | c.919-2 A>G | aberrant splicing | c.919-2 A>G | aberrant splicing |
| 58 | c.1226G>A[22] | R409H | c.1226G>A | R409H | |
| 59 | c.919-2 A>G | aberrant splicing | c.1520delT(this study) | Frameshift | |
| 60 | c.919-2 A>G | aberrant splicing | c.1343C>A[23] | S448* | |
| 61 | c.1174A>T[24] | N392Y | c.1975G>C[25] | V659L | |
| 62-65 | c.919-2 A>G | aberrant splicing | c.2168A>G[21] | H723R | |
| 66 | c.1229C>T[26] | T410M | c.1079C>T[27] | A360V | |
| 67 | c.2027T>A[23] | L676Q | c.ivs15+5G>A[26] | aberrant splicing | |
| 68,69 | c.919-2 A>G | aberrant splicing | c.1229C>T | T410M | |
| 70 | c.919-2 A>G | aberrant splicing | c.1829C>A[21] | S610* | |
| 71,72 | c.919-2 A>G | aberrant splicing | c.1343C>T[23] | S448L | |
| 73 | c.1229C>T | T410M | c.1594AG>T (this study) | SF532, 545* | |
| 74,75 | c.919-2 A>G | aberrant splicing | c.1975G>C | V659L | |
| 76,77 | c.919-2 A>G | aberrant splicing | c.946G>T(6) | G316X | |
| 78 | c.919-2 A>G | aberrant splicing | c.ivs9+1G>A (this study) | aberrant splicing | |
| 79,80 | c.919-2 A>G | aberrant splicing | c.1586T>G[23] | I529S | |
| 81 | c.2168A>G | H723R | - | - | |
| 82 | c.2168A>G | H723R | c.2168A>G | H723R | |
| 83 | - | - | - | - | |
| 84 | c.919-2 A>G | aberrant splicing | c.281C>T[23] | T94I | |
| 85 | c.919-2 A>G | aberrant splicing | - | - | |
| 86 | c.919-2 A>G | aberrant splicing | c.1318A>T[23] | K440* | |
| 87 | c.919-2 A>G | aberrant splicing | c.563T>C(this study) | I188T | |
| 88 | c.919-2 A>G | aberrant splicing | c.496delA (this study) | Frameshift | |
| 89 | c.919-2 A>G | aberrant splicing | c.1318A>T | K440* | |
| 90 | c.812A>G[23] | D271G | c.1225C>T (this study) | R409C | |
| 91 | c.919-2 A>G | aberrant splicing | c.1548insC[26] | Frameshift | |
| 92 | c.919-2 A>G | aberrant splicing | c.1173C>A (this study) | S391R | |
| 93 | c.919-2 A>G | aberrant splicing | c.1174A>T | N392Y | |
| 94 | c.2168A>G | H723R | c.2T>C[16] | M1T | |
| EVA with MD | 95 | - | - | - | - |
| (95-140) | 96-99 | c.919-2 A>G | aberrant splicing | c.2168A>G | H723R |
| 100-102 | c.919-2 A>G | aberrant splicing | c.589G>A[23] | G197R | |
| 103 | c.2168A>G | H723R | c.1594AG>T | SF532, 545* | |
| 104-115 | c.919-2 A>G | aberrant splicing | c.919-2 A>G | aberrant splicing | |
| 116 | c.919-2 A>G | aberrant splicing | c.1238A>G[28] | Q413R | |
| 117 | c.919-2 A>G | aberrant splicing | c.414delT (this study) | Frameshift | |
| 118 | c.1229C>T | T410M | c.1343C>A | S448* | |
| 119 | c.2168A>G | H723R | c.754T>C[26] | S252P | |
| 120,121 | c.919-2 A>G | aberrant splicing | c.ivs13+9C>T[29] | aberrant splicing | |
| 122,123 | c.919-2 A>G | aberrant splicing | c.1229C>T | T410M | |
| 124 | c.1174A>T | N392Y | c.2027T>A | L676Q | |
| 125-127 | c.919-2 A>G | aberrant splicing | c.1975G>C | V659L | |
| 128 | c.2168A>G | H723R | c.1079C>T | A360V | |
| 129,130 | c.919-2 A>G | aberrant splicing | c.2027T>A | L676Q | |
| 131 | c.919-2 A>G | aberrant splicing | c.1340delA(this study) | Frameshift | |
| 132 | c.1174A>T | N392Y | - | - | |
| 133 | c.919-2 A>G | aberrant splicing | c.218delA(this study) | Frameshift | |
| 134 | c.2168A>G | H723R | c.2086C>T(this study) | Q696* | |
| 135 | c.ivs15+5G>A | aberrant splicing | c.1520delT | Frameshift | |
| 136,137 | c.919-2 A>G | aberrant splicing | c.1226G>A | R409H | |
| 138,139 | c.919-2 A>G | aberrant splicing | - | - | |
| 140 | c.919-2 A>G | aberrant splicing | c.1991C>T(this study) | A664V | |
| 140 | c.2168A>G | H723R | c.439A>G[21] | M147V | |
| 140 | c.919-2 A>G | aberrant splicing | c.1594A>C[23] | S532R | |
| 140 | c.919-2 A>G | aberrant splicing | c.ivs15+5G>A | aberrant splicing | |
| 140 | c.1174A>T | N392Y | c.1174A>T | N392Y | |
Isolated MD group: Patient 1 had only one mutation and patient 2-28 had no mutation in SLC26A4. IEM group: Patient 29, 30 had one mutation and patient 31-44 had no mutation in SLC26A4. EVA group: Most patients had bi-allelic mutations except Patient 45, 81, 85 had one mutation and patient 83 had no mutation in SLC26A4. EVA with MD group: Most patients had bi-allelic mutations except Patient 132, 138, 139 had one mutation and patient 95 had no mutation.
In this study, in total, 39 SLC26A4 mutations were found among the 144 patients. Most SLC26A4 mutations (97%, 38/39) were found in the EVA group and the EVA with MD group, with the exception of one single allelic mutation (c.919-2A>G) in the isolated MD group and two single allelic mutations (c.919-2A>G and c.1286C>A) in the group with other IEM. Some mutations observed in present work, as indicated in Table 3 were previously reported to be pathogenic. Among these previously reported mutations, c.919-2A>G (51%, 102/200) was the most prevalent in the EVA with MD and EVA groups. The mutations p.H723R, p.T410M, p.V659L, and p.N392Y were also more common than the others in both groups (Table 3). Thirteen variants had not been previously reported (Table 3), consisting of seven missense variants (c.1286C>A(p.A429E), c.1594AG>T(SF532,545*), c.563T>C (p.I188T), c.1225C>T(p.R409C), c.1173C>A(p.S391R), c.2086C>T(p.Q696*), and c.1991C>T (p.A664V)), five deletions (c.1520delT, c.496delA, c.414delT, c.1339-1340delA, and c.218delA), and one splice site variant (c.ivs9+1G>A). Among the novel variants, c.1594AG>T(SF532,545*), c.2086C>T(p.Q696*), c.1520delT(SF507,507*), c.496delA(SF166, 171*), c.414delT(SF138, 144*), c. 1340delA(SF447, 454*), and c.218delA(SF73, 96*) encode either frameshift or nonsense mutations, resulting in early translational termination, severe truncation of the pendrin polypeptide, and, very likely, non-functional and/or unstable polypeptide products. The novel splice site (c.ivs9+1G>A) and five missense mutations (p.A429E, p.I188T, p.R409C, p.S391R, p.A664V) affect residues that are conserved among SLC26A4 orthologs [Mus (mouse), Macaca, Rattus, Bos, Danio; Figure 2]. These mutations were not present in a screen of 50 patients without IEM and 200 normal Chinese controls in Yuan's research [6], suggesting that these five mutations are also likely to be pathogenic.
Figure 2.
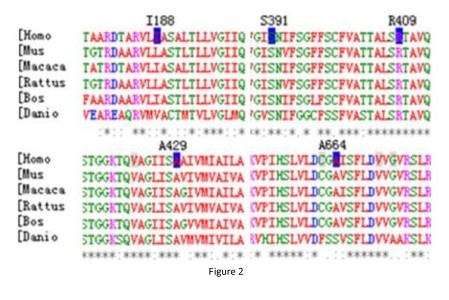
Sequence alignment of partial amino acid sequence of human SLC26A4 and its homologs. Positions of mutations are indicated in blue.
The statistical analysis showed that there were significant differences in the frequency of SLC26A4 mutation among the groups (P < 0.001). In the isolated MD group, only one single allelic mutation in SLC26A4 was found in one patient (1/28, 3.6%). In the EVA with MD group, 46 patients (46/50, 92.0%) carried biallelic mutations, and three patients carried single allelic mutations (3/50, 6.0%). In the EVA group, 46 patients (46/50, 92.0%) had biallelic mutations and three patients with one mutation (3/50, 6.0%). Only two patients carried single allelic mutations of the SLC26A4 gene in the IEM group (2/16, 12.5%). The distribution of SLC26A4 mutation in the isolated MD group was significantly lower than that in the EVA with or without MD patients (P < 0.001), suggesting that mutations in the SLC26A4 gene are more frequently associated with the EVA phenotype. There was no significant difference between the isolated MD and IEM groups (P > 0.5) and no difference in mutation distribution between the EVA with MD group and EVA group (P > 0.5). Yuan's research [6] identified four carriers of heterozygous c.919-2A>G among 200 Chinese individuals with normal hearing. There was no significant difference in mutation distribution between isolated MD patients and Yuan's research (P > 0.5).
Discussion
DNA sequence analysis for SLC26A4 mutations was performed in 144 patients with sensorineural hearing loss in this study. Inner ear malformation was present presented in 144 patients, including isolated MD, EVA with MD, isolated EVA (without MD), and other IEM identified on temporal bone CT scan images. The mutation detection rates of SLC26A4 were 3.6%, 98%, 98%, and 12.5% in the isolated MD group, EVA with MD group, EVA group, and IEM group, respectively. The prevalences of biallelic mutations in SLC26A4 were 0%, 92%, 92%, and 0%, and prevalences of monoallelic mutations were 3.6%, 6.0%, 6.0%, and 12.5% in the four respective aforementioned groups. The detection rate of SLC26A4 mutations in patients with EVA with or without MD in this study were consistent with the frequencies in other Asian studies, which are 87%, 97.9%, 92%, and 78% in Taiwanese, Chinese, Japanese, and Korean EVA patients, respectively [6,21,26,30]. Meanwhile, the mutation detection rate of this gene in Caucasian EVA patients is much lower, at 53% and 40%, respectively, in the UK and Europe [31,32]. In the US population, mutations in SLC26A4 account for about one-third of EVA patients [33]. In these studies, however, the researchers did not clearly define the specific concept of Mondini.
Azaiez [34] presented a comprehensive analysis of 474 patients from the Molecular Otolaryngology Research Laboratories of the University of Iowa Hospital and Clinics (most patients were Caucasians) with temporal bone anomalies to determine the correlation between SLC26A4 genotypic and phenotypic data. In Azaiez's study, Mondini was diagnosed a complex malformation that includes a cochlear dysplasia and EVA (EVA with MD). This study showed that 24.1% (28/116) of patients diagnosed EVA with MD had biallelic mutations in SLC26A4, but only 11.1% (38/341) of patients with isolated EVA had biallelic mutations in SLC26A4. There was a statistically significant difference in mutation distribution between isolated EVA patients and EVA with MD patients, with the presence of mutations in SLC26A4 more frequently associated with the EVA with MD phenotype. Azaiez suggested that mutations in SLC26A4 were more likely to be of a genetic etiology in EVA with MD than in EVA, in the other side of the spectrum, the environment is believed to play a bigger role in isolated EVA patients. In Campbell's research [35], 57% of patients from the US with Mondini (EVA with MD) from simplex families carried SLC26A4 mutations, and he considered that SLC26A4 mutations were present mostly in patients with EVA plus MD. The observations in the present study were not consistent with either Azaiez or Campbell's findings. There was no difference in the distribution or mutation frequency between the EVA group and EVA with MD group in our study. The mutation detection rate in SLC26A4 was 98% in both the EVA with MD group and the EVA group, and the percentage of patients with biallelic mutations was 92% and in both groups.
SLC26A4 encodes pendrin, an anion transporter protein [10]. In the inner ear, pendrin is expressed in cells of the external sulcus, epithelial cells of the endolymphatic duct and sac, and nonsensory cells at the margin of the maculae of the utricle and saccule. In all of these cell types, the apical surface is exposed to endolymph, consistent with pendrin's role as an anion transporter. The variant of pendrin was able to exchange anions between the cytosol and extracellular fluid [36]. Table 3 shows that the SLC26A4 mutations c.919-2A>G and p.H723R were highly prevalent in both in the EVA with MD and EVA groups, and the mutation spectrums were also very similar between the two groups. Our data provides strong evidence that the molecular pathogenesis of EVA with or without MD were identical solely because of the similar ORF mutation spectrum in SLC26A4 in the Chinese population and that the ORF mutations in SLC26A4 were not likely to be the reason for the pathogenesis of isolated MD in this Chinese population. So it is reasonable that there were the same mutation detection rates of SLC26A4 in both the EVA with MD group and the EVA group in this study.
The most significant mechanistic insights into the pathogenesis of hearing loss associated with EVA are based upon the SLC26A4 knockout (SLC26A4-/-) mouse that segregates a targeted deletion of exon 8 of SLC26A4 [37] and the loop mouse line segregating a chemically induced mutation of SLC26A4 [38]. In the mouse inner ear, pendrin is expressed in the cochlea, the vestibular labyrinth and the endolymphatic sac and pendrin functions as a Cl-/HCO3- exchanger. The initial pathologic alterarion in SLC26A4-/- mice includes an enlargement of the endolymphatic sac and cochlea that develops at E14.5 [39]. Studies of an SLC26A4 knockout mouse demonstrate that acidification and enlargement of the scala media are early events in the pathogenesis of deafness. The enlargement is driven by fluid secretion in the vestibular labyrinth and a failure of fluid absorption in the embryonic endolymphatic sac.
Our data showed that the distribution of SLC26A4 mutation in the isolated MD group was significantly lower than that in the EVA with or without MD patients; the one patient from the MD group who carried a monoallelic mutation of SLC26A4 (c.919-2A>G) might have been a coincident carrier of this mutation. There was no significant difference among the MD group, IEM group (P > 0.5), and normal controls (previous data published by Yuan [6]). Our results did not show a relationship between isolated MD and the SLC26A4 gene in this Chinese population. The hearing impairment in our MD group might have been caused by factors other than the SLC26A4 gene in our Chinese population.
The development of the human ear begins around the fourth week of embryonic life. The inner ear consists of two components: a membranous and a bony labyrinth. The membranous labyrinth is derived from the ectoderm, whereas the bony labyrinth/otic capsule is derived from the mesoderm and neural crest surrounding the primordial membranous labyrinth. The primordium of the cochlear duct evaginates from the ventral portion of the otocyst starting at the fifth week. This evagination extends anteromedially and gradually begins to coil. By the eighth week, the cochlea has formed 1.5 turns, and at the tenth week, 2 turns; at the 25th week, the cochlear duct has acquired the mature 2.5 turns. Ossification of the cartilaginous capsule to form the bony labyrinth does not occur until the membranous labyrinth has acquired its adult size. Bone formation starts at the 15th week and ends by the 21st week, with a total of 14 ossification centers [40]. In cochlear Mondini, it is thought that the arrest of development is at the seventh week of gestation. During inner ear development, many genes causing IEM and deafness have been identified, including SLC26A4, EYA1 [41], FGFR3 [42,43], and TCOF1 [44,45]. Pathological analysis performed on hearing impaired patients revealed the presence of Mondini dysplasia, characerized by a reduced number of cochlear turns and a incomplete interscalar sptum [46,47]. Mouse models have made it possible to study the molecular origins of the ear pathology found in both VCFS/DGS and DFN3. Mice homozygous null for Tbx1 die at birth and exhibit severe ear defects due to early arrest of inner ear development [48]. Additionally, some Brn4 null mice display a reduction in cochlear coiling. The normal mouse cochlea consist of 1.75 turns; however, approximately 25% of Brn4 null mice have less than 1.5 turns, indicating a role for Brn4 in epithelial-mesenchymal interactions during ear development[49]. With the exception of the aforementioned genes, it was evident to clinicians that retinoids (vitamin A) or other factors had potent teratogenic effects in isolated MD patients when administered early in pregnancy [50]. These factors increase the complexity of determining the real etiology of isolated MD, and our observations hint that ethnic background is apparently important and may affect the etiologic explanation of isolated MD. We suggest that EVA with or without MD and isolated MD in this Chinese population have totally different pathogenic mechanisms.
Conclusion
We did not review the relationship between SLC26A4 and isolated MD in a Chinese deaf population, but we provided evidence to support the idea that mutations in SLC26A4 were the most important factor in the pathogenesis of EVA with and without MD. This will facilitate genetic testing and counseling in patients with different types of inner ear malformation, and may provide motivation to explore the real etiology of isolated MD by new genomic strategies and platforms, such as capturing and sequencing all possible genes related to the development of the inner ear using next-generation sequencing technology.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
SH carried out the molecular genetic studies, participated in the sequence alignment and drafted the manuscript, and participated in the design of the study and performed the statistical analysis. YY and GW carried out temporal CT scan and thyroid hormone assays. D K, XZ, XM and XY participated in the sequence alignment. MD carried out epidemiological survey. DH and PD conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Contributor Information
Shasha Huang, Email: hssch315@hotmail.com.
Dongyi Han, Email: hdy301@263.net.
Yongyi Yuan, Email: yyymzh@163.com.
Guojian Wang, Email: wjcmu@163.com.
Dongyang Kang, Email: kdy_333@hotmail.com.
Xin Zhang, Email: zhangxin_615@163.com.
Xiaofei Yan, Email: yanxiaofei0614@163.com.
Xiaoxiao Meng, Email: mengxiaoxiao.123@163.com.
Min Dong, Email: donm.728@163.com.
Pu Dai, Email: daipu301@vip.sina.com.
Acknowledgements
This work was supported by grants from the Project of the National Natural Science Foundation of China (Grant No. 30872862, 30801285, 31071099) and the New Star Program of Beijing Science and Technology (Grant No. 2009 B 34). We thank the patients and families for their participation in this study.
References
- Downs MP. Universal newborn hearing screening--the Colorado story. Int J Pediatr Otorhinolaryngol. 1995;32(3):257–259. doi: 10.1016/0165-5876(95)01183-C. [DOI] [PubMed] [Google Scholar]
- Mehl AL, Thomson V. Newborn hearing screening: the great omission. Pediatrics. 1998;101(1):E4. doi: 10.1542/peds.101.1.e4. [DOI] [PubMed] [Google Scholar]
- Mehl AL, Thomson V. The Colorado newborn hearing screening project, 1992-1999: on the threshold of effective population-based universal newborn hearing screening. Pediatrics. 2002;109(1):E7. doi: 10.1542/peds.109.1.e7. [DOI] [PubMed] [Google Scholar]
- Bitner-Glindzicz M. Hereditary deafness and phenotyping in humans. Br Med Bull. 2002;63:73–94. doi: 10.1093/bmb/63.1.73. [DOI] [PubMed] [Google Scholar]
- Hone SW, Smith RJ. Genetic screening for hearing loss. Clin Otolaryngol Allied Sci. 2003;28(4):285–290. doi: 10.1046/j.1365-2273.2003.00700.x. [DOI] [PubMed] [Google Scholar]
- Yuan Yongyi, Yiwen You, Huang Deliang, Jinghong Cui, Wang Yong, Wang Qiang, Fei Yu, Kang Dongyang, Dongyi Han, Pu Dai. Comprehensive molecular etiology analysis of nonsydromic hearing impairment from typical areas in China. J Transl Med. 2009;7:79. doi: 10.1186/1479-5876-7-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valvassori GE, Clemis JD. The large vestibular aqueduct syndrome. Laryngoscope. 1978;88(5):723–728. doi: 10.1002/lary.1978.88.5.723. [DOI] [PubMed] [Google Scholar]
- Mondini C, Anatomica surdi nati sectio. De Bononiensi Scientiarum et Artium Instituto atque Academia Commentarii. 1791. pp. 28–29. 419-431.
- Zheng Y, Schachern PA, Cureoglu S, Mutlu C, Dijalilian H, Paparella MM. The shortened cochlea: its classification and histopathologic features. Int J Pediatr Otorhinolaryngol. 2002;63(1):29–39. doi: 10.1016/S0165-5876(01)00642-5. [DOI] [PubMed] [Google Scholar]
- Deenadayal DS, Kumar MN, Pandaredathil S, Patel SH. Mondini dysplasia with paradoxical cerebrospinal fluid rhinorrhea. Otolaryngol Head Neck Surg. 2010;143(6):851–852. doi: 10.1016/j.otohns.2010.08.029. [DOI] [PubMed] [Google Scholar]
- Mancini P, D'Elia C, Bosco E, De Seta E, Panebianco V, Vergari V, Filipo R. Follow-up of cochlear implant use in patients who developed bacterial meningitis following cochlear implantation. Laryngoscope. 2008;118(8):1467–1471. doi: 10.1097/MLG.0b013e3181758154. [DOI] [PubMed] [Google Scholar]
- Pourova R, Janousek P, Jurovcik M, Dvorakova M, Malikova M, Raskova D, Bendova O, Leonardi E, Murgia A, Kabelka Z, Astl J, Seeman P. Spectrum and frequency of SLC26A4 mutations among Czech patients with early hearing loss with and without Enlarged Vestibular Aqueduct (EVA) Ann Hum Genet. 2010;74(4):299–307. doi: 10.1111/j.1469-1809.2010.00581.x. [DOI] [PubMed] [Google Scholar]
- Bogazzi F, Russo D, Raggi F, Ultimieri F, Berrettini S, Forli F, Grasso L, Ceccarelli C, Mariotti S, Pinchera A, Bartalena L, Martino E. Mutations in the SLC26A4 (pendrin) gene in patients with sensorineural deafness and enlarged vestibular aqueduct. J Endocrinol Invest. 2004;27(5):430–435. doi: 10.1007/BF03345286. [DOI] [PubMed] [Google Scholar]
- Wang QJ, Zhao YL, Rao SQ, Guo YF, He Y, Lan L, Yang WY, Zheng QY, Ruben RJ, Han DY, Shen Y. Newborn hearing concurrent gene screening can improve care for hearing loss: A study on 14,913 Chinese newborns. Int J Pediatr Otorhinolaryngol. 2011;75(4):535–542. doi: 10.1016/j.ijporl.2011.01.016. [DOI] [PubMed] [Google Scholar]
- Reyes S, Wang G, Ouyang X, Han B, Du LL, Yuan HJ, Yan D, Dai P, Liu XZ. Mutation analysis of SLC26A4 in mainland Chinese patients with enlarged vestibular aqueduct. Otolaryngol Head Neck Surg. 2009;141(4):502–508. doi: 10.1016/j.otohns.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad S, Kolln KA, Cucci RA, Trembath RC, Van Camp G, Smith RJ. Pendred syndrome and DFNB4-mutation screening of SLC26A4 by denaturing high-performance liquid chromatography and the identification of eleven novel mutations. Am J Med Genet A. 2004;124A(1):1–9. doi: 10.1002/ajmg.a.20272. [DOI] [PubMed] [Google Scholar]
- Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, Sheffield VC, Green ED. Pendred syndrome is caused by mutations in a putative sulphate transporter gene(PDS) Nat Genet. 1997;17(4):411–422. doi: 10.1038/ng1297-411. [DOI] [PubMed] [Google Scholar]
- Coyle B, Reardon W, Herbrick JA, Tsui LC, Gausden E, Lee J, Coffey R, Grueters A, Grossman4 A, Phelps PD, Luxon L, Kendall-Taylor P, Scherer SW, Trembath RC. Molecular analysis of the PDS gene in Pendred syndrome. Hum Mol Genet. 1998;7(7):1105–1112. doi: 10.1093/hmg/7.7.1105. [DOI] [PubMed] [Google Scholar]
- Lehmann EL, D'Abrera HJM. Nonparametrics. Holden-Day, Inc; 1975. [Google Scholar]
- Marcello Pagano Kimberlee Gauvreau. Principles of Biostatics Second Edition. California: Duxbury Prss; 2002. [Google Scholar]
- Tsukamoto K, Suzuki H, Harada D, Namba A, Abe S, Usami S. Distribution and frequencies of PDS (SLC26A4) mutations in Pendred syndrome and nonsyndromic hearing loss associated with enlarged vestibular aqueduct: a unique spectrum of mutations in Japanese. Eur J Hum Genet. 2003;11(12):916–922. doi: 10.1038/sj.ejhg.5201073. [DOI] [PubMed] [Google Scholar]
- Fugazzola L, Cerutti N, Mannavola D, Crino A, Cassio A, Gasparoni P, Vannucchi G, Beck-Peccoz P. Differential diagnosis between Pendred and pseudo-Pendred syndromes: clinical, radiologic, and molecular studies. Pediatr Res. 2002;51(4):479–484. doi: 10.1203/00006450-200204000-00013. [DOI] [PubMed] [Google Scholar]
- Wang QJ, Zhao YL, Rao SQ, Guo YF, Yuan H, Zong L, Guan J, Xu BC, Wang DY, Han MK, Lan L, Zhai SQ, Shen Y. A distinct spectrum of SLC26A4 mutations in patients with enlarged vestibular aqueduct in China. Clin Genet. 2007;72(3):245–254. doi: 10.1111/j.1399-0004.2007.00862.x. [DOI] [PubMed] [Google Scholar]
- Ishihara K, Okuyama S, Kumano S, Iida K, Hamana H, Murakoshi M, Kobayashi T, Usami S, Ikeda K, Haga Y, Tsumoto K, Nakamura H, Hirasawa N, Wada H. Salicylate restores transport function and anion exchanger activity of missense pendrin mutations. Hear Res. 2010;270(1-2):110–118. doi: 10.1016/j.heares.2010.08.015. [DOI] [PubMed] [Google Scholar]
- Hu H, Wu L, Feng Y, Pan Q, Long Z, Li J, Dai H, Xia K, Liang D, Niikawa N, Xia J. Molecular analysis of hearing loss associated with enlarged vestibular aqueduct in the mainland Chinese: a unique SLC26A4 mutation spectrum. J Hum Genet. 2007;52(6):492–497. doi: 10.1007/s10038-007-0139-0. [DOI] [PubMed] [Google Scholar]
- Park HJ, Shaukat S, Liu XZ, Hahn SH, Naz S, Ghosh M, Kim HN, Moon SK, Abe S, Tukamoto K, Riazuddin S, Kabra M, Erdenetungalag R, Radnaabazar J, Khan S, Pandya A, Usami SI, Nance WE, Wilcox ER, Riazuddin S, Griffith AJ. Origins and frequencies of SLC26A4 (PDS) mutations in east and south Asians: global implications for the epidemiology of deafness. J Med Genet. 2003;40(4):242–248. doi: 10.1136/jmg.40.4.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CC, Chiu CY, Shiao AS, Tso YC, Wu YC, Tu TY, Jap TS. Analysis of the SLC26A4 gene in patients with Pendred syndrome in Taiwan. Metabolism. 2007;56(9):1279–1284. doi: 10.1016/j.metabol.2007.05.013. [DOI] [PubMed] [Google Scholar]
- Pera A, Dossena S, Rodighiero S, Gandia M, Botta G, Meyer G, Moreno F, Nofziger C, Hernandez-Chico C, Paulmichl M. Functional assessment of allelic variants in the SLC26A4 gene involved in Pendred syndrome and nonsyndromic EVA. Proc Natl Acad Sci USA. 2008;105(47):18608–18613. doi: 10.1073/pnas.0805831105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong AM, Goh SS, Zhao Y, Eng PH, Koh LK, Khoo DH. Two Chinese families with Pendred's syndrome--radiological imaging of the ear and molecular analysis of the pendrin gene. J Clin Endocrinol Metab. 2001;86(8):3907–3911. doi: 10.1210/jc.86.8.3907. [DOI] [PubMed] [Google Scholar]
- Choi BY, Stewart AK, Nishimura KK, Cha WJ, Seong MW, Park SS, Kim SW, Chun YS, Chung JW, Park SN, Chang SO, Kim CS, Alper SL, Griffith AJ, Oh SH. Efficient molecular genetic diagnosis of enlarged vestibular aqueducts in East Asians. Genet Test Mol Biomarkers. 2009;13(5):679–687. doi: 10.1089/gtmb.2009.0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert S, Blons H, Jonard L, Feldmann D, Chauvin P, Loundon N, Sergent-Allaoui A, Houang M, Joannard A, Schmerber S, Delobel B, Leman J, Journel H, Catros H, Dollfus H, Eliot MM, David A, Calais C, Drouin-Garraud V, Obstoy MF, Tran Ba Huy P, Lacombe D, Duriez F, Francannet C, Bitoun P, Petit C, Garabedian EN, Couderc R, Marlin S, Denoyelle F. SLC26A4 gene is frequently involved in nonsyndromic hearing impairment with enlarged vestibular aqueduct in Caucasian populations. Eur J Hum Genet. 2006;14(6):773–779. doi: 10.1038/sj.ejhg.5201611. [DOI] [PubMed] [Google Scholar]
- Hulander M, Kiernan AE, Blomqvist SR, Carlsson P, Samuelsson EJ, Johansson BR, Steel KP, Enerback S. Lack of pendrin expression leads to deafness and expansion of the endolymphatic compartment in inner ears of Foxi1 null mutant mice. Development. 2003;130(9):2013–2025. doi: 10.1242/dev.00376. [DOI] [PubMed] [Google Scholar]
- Pryor SP, Madeo AC, Reynolds JC, Sarlis NJ, Arnos KS, Nance WE, Yang Y, Zalewski CK, Brewer CC, Butman JA, Griffith AJ. SLC26A4/PDS genotype-phenotype correlation in hearing loss with enlargement of the vestibular aqueduct (EVA): evidence that Pendred syndrome and non-syndromic EVA are distinct clinical and genetic entities. J Med Genet. 2005;42(2):159–165. doi: 10.1136/jmg.2004.024208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azaiez H, Yang T, Prasad S, Sorensen JL, Nishimura CJ, Kimberling WJ, Smith RJ. Genotype-phenotype correlations for SLC26A4-related deafness. Hum Genet. 2007;122(5):451–457. doi: 10.1007/s00439-007-0415-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell C, Cucci RA, Prasad S, Green GE, Edeal JB, Galer CE, Karniski LP, Sheffield VC, Smith RJ. Pendred syndrome, DFNB4, and PDS/SLC26A4 identification of eight novel mutations and possible genotype-phenotype correlations. Hum Mutat. 2001;17(5):403–411. doi: 10.1002/humu.1116. [DOI] [PubMed] [Google Scholar]
- Yoshida A, Hisatome I, Taniguchi S, Sasaki N, Yamamoto Y, Miake J, Fukui H, Shimizu H, Okamura T, Okura T, Igawa O, Shigemasa C, Green ED, Kohn LD, Suzuki K. Mechanism of iodide/chloride exchange by pendrin. Endocrinology. 2004;145(9):4301–4308. doi: 10.1210/en.2004-0048. [DOI] [PubMed] [Google Scholar]
- Everett LA, Belyantseva IA, Noben-Trauth K, Cantos R, Chen A, Thakkar SI, Hoogstraten-Miller SL, Kachar B, Wu DK, Green ED. Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum Mol Genet. 2001;10(2):153–161. doi: 10.1093/hmg/10.2.153. [DOI] [PubMed] [Google Scholar]
- Dror AA, Politi Y, Shahin H, Lenz DR, Dossena S, Nofziger C, Fuchs H, Hrabé de Angelis M, Paulmichl M, Weiner S, Avraham KB. Calcium oxalate stone formation in the inner ear as a result of an Slc26a4 mutation. J Biol Chem. 2010;285(28):21724–21735. doi: 10.1074/jbc.M110.120188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HM, Wangemann P. Epithelial cell stretching and luminal acidification lead to a retarded development of stria vascularis and deafness in mice lacking pendrin. PloS One. 2011;6(3):e17949. doi: 10.1371/journal.pone.0017949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James B, Snow JR, John Jacob Ballenger. Otorhinolaryngology Head and Neck Surgery. Sixteenth. pp. 25–30. (ISBN 1-5009-197-2). [Google Scholar]
- Ito T, Noguchi Y, Yashima T, Ohno K, Kitamura K. Hereditary hearing loss and deafness genes in Japan. J Med Dent Sci. 2010;57(1):1–10. [PubMed] [Google Scholar]
- Wilkes D, Rutland P, Pulleyn LJ, Reardon W, Moss C, Ellis JP, Winter RM, Malcolm S. A recurrent mutation, ala391glu, in the transmembrane region of FGFR3 causes Crouzon syndrome and acanthosis nigricans. J Med Genet. 1996;33(9):744–748. doi: 10.1136/jmg.33.9.744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pannier S, Couloigner V, Messaddeq N, Elmaleh-Berges M, Munnich A, Romand R, Legeai-Mallet L. Activating Fgfr3 Y367C mutation causes hearing loss and inner ear defect in a mouse model of chondrodysplasia. Biochim Biophys Acta. 2009;1792(2):140–147. doi: 10.1016/j.bbadis.2008.11.010. [DOI] [PubMed] [Google Scholar]
- The Treacher Collins Syndrome Collaborative Group. Dixon J, Edwards SJ, Gladwin AJ, Dixon MJ, Loftus SK, Bonner CA, Koprivnikar K, Wasmuth JJ. Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome. Nat Genet. 1996;12(2):130–136. doi: 10.1038/ng0296-130. [DOI] [PubMed] [Google Scholar]
- Teber OA, Gillessen-Kaesbach G, Fischer S, Bohringer S, Albrecht B, Albert A, Arslan-Kirchner M, Haan E, Hagedorn-Greiwe M, Hammans C, Henn W, Hinkel GK, Konig R, Kunstmann E, Kunze J, Neumann LM, Prott EC, Rauch A, Rott HD, Seidel H, Spranger S, Sprengel M, Zoll B, Lohmann DR, Wieczorek D. Genotyping in 46 patients with tentative diagnosis of Treacher Collins syndrome revealed unexpected phenotypic variation. Eur J Hum Genet. 2004;12(11):879–890. doi: 10.1038/sj.ejhg.5201260. [DOI] [PubMed] [Google Scholar]
- Ohtani I, Schuknecht HF. Temporal bone pathology in DiGeorge's syndrome. Ann Otol Rhino Laryngol. 1984;93(3):220–224. doi: 10.1177/000348948409300306. [DOI] [PubMed] [Google Scholar]
- Holden PK, Linthicum FH Jr. Mondini dysplasia of the bony and membranous labyrinth. Otol Neurotol. 2005;26(1):133. doi: 10.1097/00129492-200501000-00025. [DOI] [PubMed] [Google Scholar]
- Vitelli F, Viola A, Morishima M, Pramparo T, Baldini A, Lindsay E. TBX1 is required for inner ear morphogenesis. Hum Mo Genet. 2003;12(16):2041–2048. doi: 10.1093/hmg/ddg216. [DOI] [PubMed] [Google Scholar]
- Phippard D, Lu L, Lee D, Saunders JC, Crenshaw EB. Targeted mutagenesis of the POU-domain gene Brn4/Pou3f4 causes developmental defects in the inner ear. J Neurosci. 1999;19(14):5980–5989. doi: 10.1523/JNEUROSCI.19-14-05980.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JG, Roth CB, Warkany J. An analysis of the syndrome of malformations induced by maternal vitamin A deficiency. Effects of restoration of vitamin A at various times during gestation. Am J Anat. 1953;92(2):189–217. doi: 10.1002/aja.1000920202. [DOI] [PubMed] [Google Scholar]