

Abstract
Background
Fanconi anemia (FA) patients are hypersensitive to DNA alkylating agents and require lower doses than non-FA patients to minimize serious toxicity. The mechanism by which hypersensitivity occurs is thought to be due to the inability of these individuals to effectively repair drug-induced interstrand DNA-DNA crosslinks. We recently developed a highly sensitive assay for cyclophosphamide specific interstrand DNA-DNA crosslinks (G-NOR-G) and are able to quantify and compare formation of these adducts in the blood of patients. Therefore we sought to determine whether FA patients have higher in vivo exposure to the cyclophosphamide specific interstrand DNA crosslink, G-NOR-G, relative to patients without FA.
Procedure
Cyclophosphamide interstrand DNA crosslinks were measured with the first dose of cyclophosphamide in FA and non-FA patients receiving a cyclophosphamide based preparative regimen prior to hematopoietic cell transplantation (HCT). FA patients received a lower cyclophosphamide dose than the non-FA patients (5–10 mg/kg/day vs. 50–60 mg/kg/day).
Results
Despite the lower cyclophosphamide dose and lower plasma concentrations in FA patients, they had G-NOR-G amounts similar to the non-FA patients (area under the curve (AUC)0-∞, 99.8 vs. 144.9 G-NOR-G adducts/10^6 nucleotides*hr, respectively, p=0.47). When G-NOR-G AUC was normalized for cyclophosphamide plasma concentrations, FA study subjects produced 15-fold higher adducts than non-FA patients (p=0.05).
Conclusions
FA patients are hypersensitive to DNA alkylating agents possibly as a result of greater formation of cyclophosphamide specific interstrand DNA crosslinks and/or diminished capacity for DNA repair. Identification and quantification of these adducts may be important determinant of cyclophosphamide related toxicity.
Keywords: Transplantation, Fanconi Anemia, cyclophosphamide, DNA adducts
Introduction
Fanconi anemia (FA) is a rare genetic disorder characterized by congenital malformations, predisposition to cancer and progressive bone marrow failure.(1) The FA genes encode for proteins which are involved in the regulation of the cellular response to DNA damage. Mutations in the FA genes lead to impaired DNA repair and presumably greater formation of interstrand DNA crosslinks or adducts after exposure to alkylating drugs (e.g. mitomycin C, diepoxybutane, melphalan, cyclophosphamide) and/or radiation.(2) As a result, FA patients are more sensitive to chemotherapy involving DNA alkylating agents, are at greater risk of toxicity and therefore lower doses are required. In the case of solid tumors, defects in FA genes with are often associated with greater tumor sensitivity to alkylating agents.(3,4)
Hematopoietic cell transplant (HCT) is the only curative therapy for FA patients with severe bone marrow failure. Pre-transplant conditioning chemotherapy usually includes the DNA alkylating agent, cyclophosphamide.(5) Because of excess toxicities observed, patients with FA receive substantially lower total doses of cyclophosphamide (20–40 mg/kg) compared to doses used in non-FA patients (100–120 mg/kg).(6)
Cyclophosphamide is metabolically activated by CYP2B6 enzyme and induces cellular death by forming DNA adducts, in particular, the interstrand DNA crosslink, [G-NOR-G], through formation of the active metabolite, phosphoramide mustard (Figure 1). Cyclophosphamide primarily alkylates the N-7 position of guanine and forms 67% phosphotriester monoadducts, 26% N-7-guanine monoadducts, and 6.7% N-7-guanine-N-7 guanine interstrand crosslinks.(7) Even though interstrand crosslinks comprise only 5–10% of all adducts formed by alkylating agents, these adducts are the most physiologically relevant because of their ability to block DNA replication and their association with cytotoxicity.(8–11)
Figure. 1.
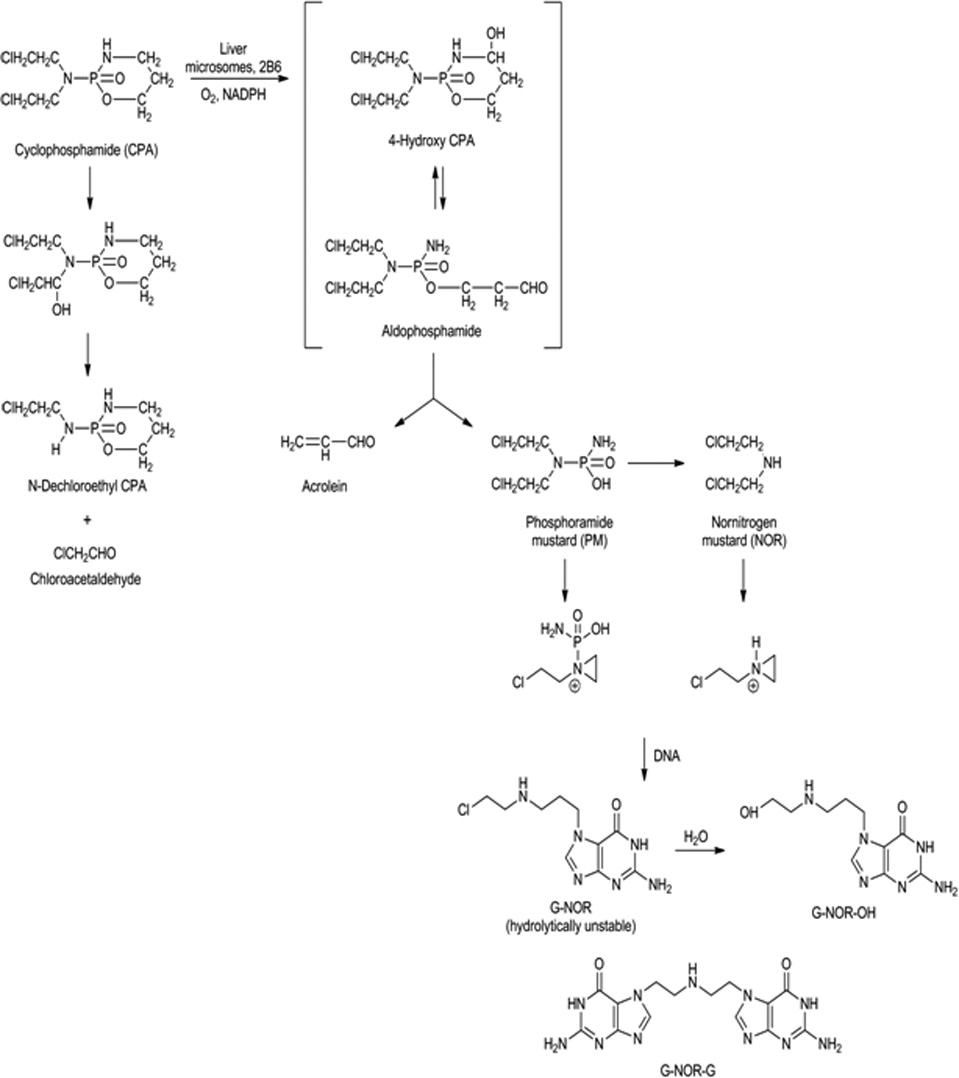
Cyclophosphamide metabolism and formation of guanine specific DNA adducts.
Several theories have been postulated as to the mechanism of cyclophosphamide induced hypersensitivity in FA. Such theories include genetic mutations in the DNA repair pathway thereby preventing the repair of damaged DNA induced by cyclophosphamide;(12–14) altered oxygen metabolism resulting in overproduction in reactive oxygen metabolites and;(15–17) altered drug metabolism resulting in high exposure of cyclophosphamide and/or its metabolites.(18) These theories speculate the accumulation of DNA adducts, in particular the interstrand cross links, in FA patients which induces a greater pharmacologic response. This study sought to verify whether FA patients have higher in vivo exposure to the interstrand DNA crosslink, G-NOR-G, relative to patients without FA.
Methods
Patients
Twenty pediatric and adult patients underwent HCT for a hematologic disorder (n=10 lympho-hematological malignancy and n=10 FA) at the University of Minnesota. All patients and/or guardians signed informed consent which was approved by the University of Minnesota IRB Board (IRB#0610M94328). Two patients were excluded from the final analysis due to missing time points or DNA extraction failure. Patient characteristics are shown in Table I. All subjects received intravenous cyclophosphamide over 2 hours at a constant infusion rate. FA study participants received one of the following conditioning regimens: cyclophosphamide 5 mg/kg/dose IV daily given on days -6, -5, -4, -3, fludarabine 35 mg/m2 daily IV on days -6, -5, -4, -3 plus antithymocyte globulin 30 mg/kg given daily on days -6,- 5, -4, -3, -2 (n=2) for those with sibling donors; cyclophosphamide 10 mg/kg IV daily on days -5, -4, -3, -2, fludarabine 35 mg/m2 –IV daily on days -5, -4, -3, -2, antithymocyte globulin 30 mg/kg given daily on days -5, -4, -3, -2, -1, plus 300 cGy of total body irradiation (TBI) on day -6 (n=8) for those with unrelated donors. Non-FA patients received one of the following disease-specific regimens: cyclophosphamide 60 mg/kg/dose IV daily given on days -6 and -5 plus 165 cGy TBI twice daily on days -4, -3, -2, -1 (n=1); cyclophosphamide 50 mg/kg/dose IV on day -6 plus fludarabine 40 mg/m2 IV daily for 5 days on day -6, -5, -4, -3, -2 and 200 cGY of TBI on day -1 (n=7); cyclophosphamide 50 mg/kg/dose IV on day -7 plus fludarabine 40 mg/m2 IV daily for 5 days on day -6,-5,-4,-3,-2 plus Campath 0.2 mg/kg IV daily for 5 days on day -10,-9,-8,-7,-6 (n=1); cyclophosphamide 60 mg/kg daily on days -7 and -6, fludarabine 25mg/m2 daily IV on days -8,-7,-6 plus 165 cGy TBI twice daily on days -4,-3,-2,-1 (n=1).
Table I.
Patient Characteristics
| Non-Fanconi Anemia (n=9) |
Fanconi Anemia (n=9) |
|
|---|---|---|
| Gender (M/F) | 5/4 | 6/3 |
| Age median (range) | 52 (25–56) | 7 (1–9) |
| Disease | ||
| Aplastic anemia | 1 | - |
| Acute myelogenous leukemia | 5 | - |
| Chronic myelogenous leukemia | 1 | - |
| Multiple myeloma | 1 | - |
| NonHodgkin lymphoma | 1 | - |
| Race/ethnicity | ||
| Caucasian | 9 | 8 |
| African American | 0 | 1 |
| FA Complementation Group | ||
| A | - | 5 |
| D1/D2 | - | 1 |
| Other | - | 1 |
| Not determined | - | 2 |
| Days to engraftment median (range) | 6.5 (6–25) | 11 (10–12) |
| Day 100 TRM (95% CI) | 20% (0–45%) | 0% |
| Day 180 TRM (95% CI) | 58% (26–905) | 0% |
TRM, treatment related mortality; engraftment is defined as ANC >500 cells µL for three consecutive days
Cyclophosphamide and DNA Adduct Quantification, and Pharmacokinetic Analysis
Pharmacokinetic sampling was conducted with the first dose of cyclophosphamide. Cyclophosphamide and G-NOR-G adducts concentrations were measured on each sample and were obtained at times 0 (prior to the start of infusion), 2, 6, 8 and 22 hours after the end of the 2 hour cyclophosphamide infusion. At each time point, 5 ml of blood was collected into a purple top tube and placed on wet ice. Plasma was separated by centrifugation and DNA was isolated from the remaining component. Plasma was frozen at −80 degrees C° until time of analysis. Plasma cyclophosphamide concentrations were measured by HPLC using a validated assay with UV detection as previously described.(19) Briefly, ifosphamide was used as an internal standard. UV absorption data for the internal standard (ifosphamide) and cyclophosphamide was recorded at 195 nm and eluted at 19.2 and 22.2 minutes, respectively. The cyclophosphamide assay was linear from 0.01–20 ug/ml of cyclophosphamide and the lowest limit of quantification was 0.01 ug/ml. The accuracy and precision of the cyclophosphamide assay was 98.2% and 2.5%, respectively.
Cyclophosphamide specific DNA adduct (G-NOR-G) concentrations were measured in each blood sample after isolating DNA using Qiagen DNA extraction kits (Qiagen, Valencia, CA, USA) by the manufacturer’s instructions. DNA amounts were estimated by UV spectrophotometry and G-NOR-G subsequently determined by HPLC-LSI-MS/MS analysis of deoxygaunine (dG) in enzymatic hydrolysates as described by Malayappan et al.(19) Briefly, DNA samples were spiked with internal standard [15N10]-G-NOR-G and subjected to neutral thermal hydrolysis (70°C for 1 hour) to release G-NOR-G as free base adducts. G-NOR-G and its internal standard were purified by solid phase extraction. They were eluted with 100% methanol (6 mL). Solid phase extraction recovery averaged between 90–95%. Fractions containing G-NOR-G and 15N10- G-NOR-G were dried under nitrogen and the residues were reconstituted in 20 µL of 15 mM ammonium acetate buffer, pH 6.8 for capillary high pressure liquid chromatography electrospray ionization (ESI)+-tandem mass spectroscopy analysis. The lower limit of detection was 50 fmol with an accuracy and precision of 93% and 7.0%, respectively.
Plasma cyclophosphamide and G-NOR-G adduct concentrations were analyzed by noncompartmental analysis using standard software (WinNon Lin Professional 5.2, Pharsight Corp, Mountain View, California). Area under curve (AUC0-∞), and Cmax were determined for cyclophosphamide and DNA adducts. The AUC was estimated by the trapezoidal rule. Cmax was the highest observed concentration. Because the FA and non-FA patients received different doses and consequently different plasma exposures of cyclophosphamide a ratio of G-NOR-G AUC0-∞ to cyclophosphamide AUC0-∞ was determined to normalize the number of adducts formed relative to the plasma exposure.
Statistical analysis
Comparisons of cyclophosphamide AUC0-∞, G-NOR-G adduct AUC0-∞, and concentration normalized DNA adduct exposure (DNA adduct AUC0-∞ /cyclophosphamide AUC0-∞) were assessed between FA and non-FA study subjects. Statistical comparisons between FA and non-FA groups, were made using two-way ANOVA.
Results
Patient Characteristics and Clinical Outcomes
Patient characteristics are given in Table I. FA patients were younger than the non-FA patients [median (range) 7 (1–9) vs 52 (25–56) years]. Time to donor stem cell engraftment was slightly longer in the FA patients. Non-FA patients engrafted at a median (range) of 6.5 (6–25) days posttransplant, whereas the FA patients engrafted at 11 (10–12) days. Treatment related mortality in the non-FA patients was 20% (95% CI: 0–45%) at day 100 and 58% (95% CI: 26–90%) at day 180 posttranplant. There was no treatment related mortality in the FA patients at day 100 or 180.
Cyclophosphamide Pharmacokinetics
Pharmacokinetic parameters are given in Table II. Non-FA patients received approximately ~ 10 fold higher daily cyclophosphamide dose (mg/kg) than FA patients (p<0.001) and cyclophosphamide plasma exposure was significantly higher in the non-FA patients (Figure 2). Median (range) cyclophosphamide plasma concentrations in the non-FA patients at 2, 4, 8 and 22 hours after the end of the infusion were 60.9 (29.3–86.7), 36 (21.2–50.7), 31.2 (14.4–42.9), 27.3 (0.65–11.4) mcg/mL, respectively. Median (range) cyclophosphamide plasma concentrations in the FA patients at 2, 4, 8, and 22 hours after start of infusion were 5.8 (0.35–9.234), 2.89 (0.26–6.04), 2.0 (0.22–5.03) and 0.16 (0–2.8) mcg/mL, respectively. The median (range) cyclophosphamide AUC0-∞ was approximately13-fold lower in the FA patients compared to non-FA (659.6 vs. 49.3 mcg/mL*hr) (p<0.001).
Table II.
| Patient | Total daily cyclophosphamide dose (mg) |
Cyclophosphamide AUC (mcg/ mL * hr) |
DNA adduct AUC (adducts/ 10^6 nucleotides * hr) |
Ratio of DNA adduct AUC /cyclophosphamide AUC |
|---|---|---|---|---|
| Non-FA n=9 | ||||
| 1 | 5640 | 515.3 | 96.7 | 0.19 |
| 2 | 4000 | 529.2 | 142.8 | 0.27 |
| 3 | 4600 | 659.6 | 425.3 | 0.64 |
| 4 | 3700 | 908.7 | 276.0 | 0.30 |
| 5 | 2750 | 868.0 | 19.0 | 0.02 |
| 6 | 3000 | 647.4 | 33.5 | 0.05 |
| 7 | 4450 | 463.0 | 50.8 | 0.11 |
| 8 | 3400 | 961.0 | 147.0 | 0.15 |
| 9 | 3000 | 300.6 | 177.4 | 0.59 |
| Median (range) | 3850 (2750–5640) | 659.6 (300.6–908.7) | 144.9 (19.0–425.3) | 0.20 (0.02–0.64) |
| FA n=9 | ||||
| 1 | 115 | 4.2 | 64.3 | 15.16 |
| 2 | 195 | 8.1 | 36.1 | 4.43 |
| 3 | 210 | 82.3 | 26.3 | 0.32 |
| 4 | 206 | 167.5 | 171.3 | 1.02 |
| 5 | 100 | 134.9 | 148.7 | 1.10 |
| 6 | 95 | 39.7 | 141.2 | 3.55 |
| 7 | 165 | 81.6 | 271.5 | 3.33 |
| 8 | 93 | 49.3 | 72.4 | 1.47 |
| 9 | 190 | 33.6 | 99.8 | 2.97 |
| Median (range) | 140 (93–210) | 49.3 (4.2–167.5) | 99.8 (26.3–271.5) | 3.0 (0.32–15.16) |
| p-value* | <0.001 | <0.001 | 0.47 | 0.05 |
P-values are comparison of non-FA and FA pharmacokinetic measures.
Figure. 2.
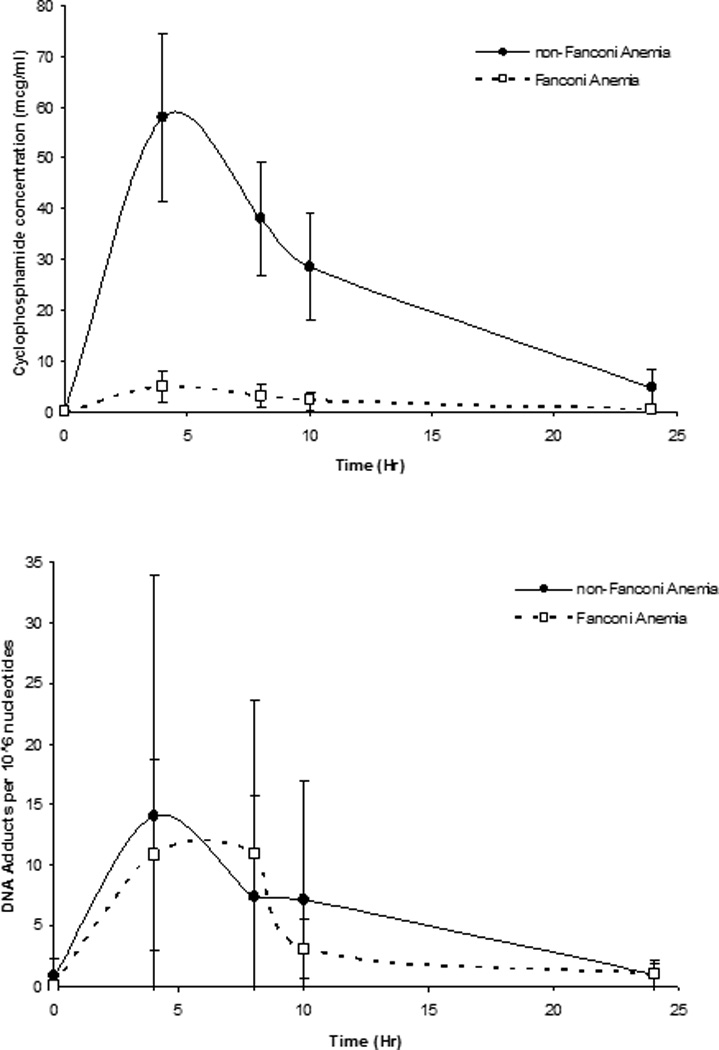
Time concentration profiles for cyclophosphamide (top panel) and G-NOR-G DNA adducts per 106 nucleotides (bottom panel). FA profiles are denoted with open squares (□) and non-FA profiles are denoted with closed circles (●). Times are hours after the end of a 2 hour infusion. Zero hour designates the time immediately before the start of infusion. Data are mean ± S.D.
Cyclophosphamide Interstrand DNA Crosslink (G-NOR-G) Pharmacokinetics
The absolute number of G-NOR-G adducts present at each time point post-dose in the non-FA and FA patients was similar (Figure 2). The median (range) concentrations at 2, 4, 8 and 22 hours after the end of cyclophosphamide infusion were 3.6 (0.1–58.7), 4.65 (0.4–26.5), 2.97 (0.42–26.4), 0.33 (0.03–4.05) G-NOR-G adducts/106 nucleotides, respectively in the non-FA patients. In FA subjects at 2, 4, 8, and 22 hours, concentrations were 8.45 (3.66–29.32), 5.45 (0.53–40.84), 2.19 (0.32–6.58), 0.58 (0–2.99) G-NOR-G adducts/106 nucleotides, respectively. As expected, no adducts were detected before cyclophosphamide administration in either group. Pharmacokinetic results are shown in Table II. The median G-NOR-G adduct AUC0-∞ was similar in the FA and non-FA patients (99.8 vs. 144.9, p=0.47), despite the lower dose of cyclophosphamide in the FA patients. However, after normalization of G-NOR-G adducts for cyclophosphamide plasma concentrations FA patients had 15-fold more G-NOR-G adducts present than the non-FA patients (3.0 vs. 0.20 G-NOR-G to cyclophosphamide AUC ratio, p= 0.05). Maximum observed G-NOR-G concentrations occurred between 4 and 8 hours after completion of infusion. There was high (10–22 fold) interpatient variability in G-NOR-G AUC. Cyclophosphamide plasma exposure was poorly correlated with G-NOR-G adducts (Figure 3).
Figure. 3.
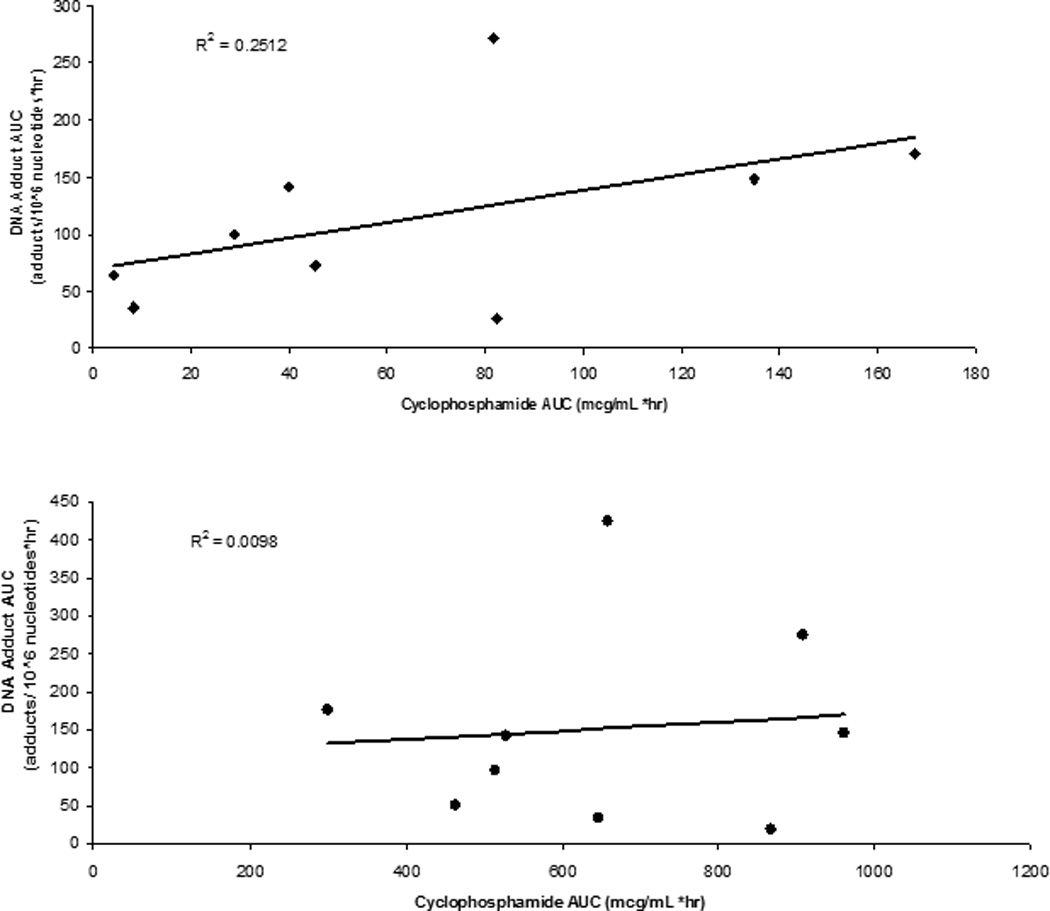
Correlation between cyclophosphamide exposure and G-NOR-G adduct formation in FA (top panel) and non-FA (bottom panel) study subjects.
Discussion
To the best of our knowledge this is the first study to examine cyclophosphamide specific interstrand DNA crosslinks (G-NOR-G) in a FA pediatric population and an adult population with hematological malignancy.(20,21) Previous studies of interstrand crosslinks have been conducted in cancer patients receiving platinum compounds and/or other alkylating agents.(22,23) However, regardless of the therapeutic agent, DNA adduct formation correlated with therapeutic responsiveness. In seven adults with multiple myeloma receiving high dose melphalan, individuals with the lowest adduct formation exhibited the poorest response to melphalan.(24) Therefore, quantification of in vivo adduct formation may be an important determinant of therapeutic efficacy and toxicity of cyclophosphamide.
Cyclophosphamide alkylates the N-7 position of guanine to form N-7-guanine monoadducts and N-7-guanine-N-7 guanine diadducts interstrand crosslinks.(7) Even though less abundant than the corresponding monoadducts, N-7-guanine-N-7 guanine diadducts interstrand crosslinks (G-NOR-G) are the most physiologically relevant because of their association with cytotoxicity.(8–11) In our study we demonstrated that we could measure the minor yet most physiologically relevant adduct (G-NOR-G) in two distinct populations receiving cyclophosphamide as part of their preparative regimen. Despite the significantly lower cyclophosphamide AUC0-∞ (Figure 2. and Table II) in the FA children, the absolute G-NOR-G exposures were not different from the non-FA group (p=0.47) (Figure 2B and Table II). After correcting the DNA adduct formation for cyclophosphamide exposure, FA patients produced 15-times more G-NOR-G adducts for every unit of cyclophosphamide present in the plasma relative to the non-FA patients (p=0.05, Table II). This supports current clinical practice of lower cyclophosphamide doses in patients with FA.
There are several issues related to cyclophosphamide adduct formation in these populations that deserve discussion. First FA patients may have DNA repair pathway deficiencies and genetic variants (e.g. BER, MMR, NHEJ) leading to the accumulation of G-NOR-G. Although variants may alter G-NOR-G formation no studies have evaluated their importance towards adduct formation. The sequencing of cyclophosphamide and radiation may also affect adduct formation. It is controversial if TBI should precede or follow chemotherapy in preparative regimens and no one clinical protocol has emerged as superior in this regard. TBI causes DNA damage and single strand DNA breaks; however, there are no data to date that suggest that radiation influences the formation of cyclophosphamide-specific DNA adducts in a negative or positive manner. (25) From a practical point, TBI is given first in most FA preparative regimens at our center so that patients are free from nausea and vomiting at the time of TBI administration. Cyclophosphamide is highly emetogenic and when given before TBI makes the delivery of TBI significantly more difficult. Cyclophosphamide is also not a known radiosensitizer in other cancer types. Two of the FA patients (no.’s 1 and 6 in Table 2) received no TBI in their preparative regimen. These two patients had DNA adduct AUC/cyclophosphamide AUC ratios higher than the median. However, should TBI affect adduct formation we would have expected these individuals to have the lowest cyclophosphamide adducts. Conclusions cannot be drawn from two subjects although it argues against TBI enhancement of cyclophosphamide adduct formation. Recent in vitro studies by Meybodi et al. demonstrated in leukocytes isolated from FA and non-FA subjects that initial radiation induced DNA damage was not different between the groups although the FA patients appeared to take longer to DNA repair(26). This study did not address the issue of combination chemotherapy and TBI sequencing. Finally, genetic polymorphisms in CYP2B6, and concomitant administration of drugs that induce or inhibit CYP enzyme activity may also affect adduct formation. Studies by Xie et al. demonstrated that genetic variants in the CYP2B6 enzyme resulted in an increase rate in 4-OH cyclophosphamide formation.(27) These issues should be addressed in future studies.
Despite the apparent differences in adduct formation between our two populations they displayed some commonalities. The maximum observed G-NOR-G concentrations occurred between 4 and 8 hours after the end of the cyclophosphamide infusion. This is consistent with observations for melphalan N-ras and p53 gene specific interstrand crosslink adducts, whereby maximum melphalan induced adducts occurred at 8 hours post exposure.(24) Contradictory to our results, cyclophosphamide induced N-ras specific interstrand cross links occurred at 24 hours in one patient with poluarteritis nodosa.(24) In this study, G-NOR-G concentrations were poorly correlated with plasma cyclophosphamide concentrations (Figure 3). In patients with solid tumor receiving oxaliplatin, DNA adduct formation also did not correlate with platinum blood concentrations.(22) These findings support previous studies where no association was found between cyclophosphamide pharmacokinetics and clinical outcomes after HCT.(28) Cyclophosphamide is a prodrug that must be converted to active metabolites prior to its pharmacologic activity, therefore a better correlation may be observed between phosphoramide mustard (an active metabolite) concentrations and G-NOR-G formation. Further studies are needed to examine the relationships between G-NOR-G, active metabolites, and clinical outcomes.
A limitation of our study is the lack of the ideal comparison group to the FA patients. The best control group is children with leukemia undergoing HCT of the same age who receives an identical preparative regimen and cyclophosphamide dose. This control group does not exist given the low doses of cyclophosphamide used in FA. Therefore, we chose to compare the FA children to an adult population receiving the most similar preparative regimens possible in our center. To directly address the known differences in metabolism rates between children and adults, and the varying cyclophosphamide doses used in FA vs non-FA protocols we measured parent cyclophosphamide blood levels concurrently with every cyclophosphamide DNA adduct measurement. From these data a cyclophosphamide DNA adduct measurement corrected for plasma concentration (DNA adduct/cyclophosphamide ratio) and indirectly for differences in metabolic rates was calculated.
In conclusion, we have shown for the first time that the interstrand DNA crosslink adduct, G-NOR-G, a minor but toxic adduct, is measurable in the blood of FA and non-FA patients using our highly sensitive and novel assay. FA patients formed significantly more G-NOR-G adducts relative to non-FA patients. Future studies are now feasible to determine the relationship between G-NOR-G formation and engraftment, treatment related mortality, toxicity and disease response in a larger number of subjects. Future studies should evaluate the association between DNA adduct formation and DNA repair variants, and the relationship between adduct formation and toxicity. Identification and quantification of other adducts (e.g. acrolien-DNA) may also be important towards the toxicity of cyclophosphamide.
Acknowledgments
We gratefully acknowledge the dedication and hard work of our coordinators Jill Nagorski and Pat Fidler. This work was supported by Children’s Cancer Research Fund Minneapolis, MN, a Grant in Aid (L.J.) from the University of Minnesota Graduate School, and NCI grant RO1-CA-100670 (N.T.).
References
- 1.Auerbach AD. Fanconi anemia and its diagnosis. Mutat.Res. 2009;668:4–10. doi: 10.1016/j.mrfmmm.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parmar K, D'Andrea A, Niedernhofer LJ. Mouse models of Fanconi anemia. Mutat.Res. 2009;668:133–140. doi: 10.1016/j.mrfmmm.2009.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van der Heijden MS, Brody JR, Dezentje DA, Gallmeier E, Cunningham SC, Swartz MJ, et al. In vivo therapeutic responses contingent on Fanconi anemia/BRCA2 status of the tumor. Clin.Cancer Res. 2005;11:7508–7515. doi: 10.1158/1078-0432.CCR-05-1048. [DOI] [PubMed] [Google Scholar]
- 4.van der Heijden MS, Brody JR, Gallmeier E, Cunningham SC, Dezentje DA, Shen D, et al. Functional defects in the fanconi anemia pathway in pancreatic cancer cells. Am.J.Pathol. 2004;165:651–657. doi: 10.1016/S0002-9440(10)63329-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dalle JH. HSCT for Fanconi anemia in children: factors that influence early and late results. Bone Marrow Transplant. 2008;42(Suppl 2):S51–S53. doi: 10.1038/bmt.2008.284. [DOI] [PubMed] [Google Scholar]
- 6.Gluckman E. Allogeneic bone marrow transplantation in Fanconi anemia. Bone Marrow Transplant. 1996;18(Suppl 3):S33–S35. [PubMed] [Google Scholar]
- 7.Maccubbin AE, Caballes L, Riordan JM, Huang DH, Gurtoo HL. A cyclophosphamide/DNA phosphoester adduct formed in vitro and in vivo. Cancer Res. 1991;51:886–892. [PubMed] [Google Scholar]
- 8.Hemminki K. DNA-binding products of nornitrogen mustard, a metabolite of cyclophosphamide. Chem.Biol.Interact. 1987;61:75–88. doi: 10.1016/0009-2797(87)90020-2. [DOI] [PubMed] [Google Scholar]
- 9.Hemminki K. Binding of metabolites of cyclophosphamide to DNA in a rat liver microsomal system and in vivo in mice. Cancer Res. 1985;45:4237–4243. [PubMed] [Google Scholar]
- 10.O'Connor PM, Kohn KW. Comparative pharmacokinetics of DNA lesion formation and removal following treatment of L1210 cells with nitrogen mustards. Cancer Commun. 1990;2:387–394. doi: 10.3727/095535490820873949. [DOI] [PubMed] [Google Scholar]
- 11.Akkari YM, Bateman RL, Reifsteck CA, Olson SB, Grompe M. DNA replication is required To elicit cellular responses to psoralen-induced DNA interstrand cross-links. Mol.Cell.Biol. 2000;20:8283–8289. doi: 10.1128/mcb.20.21.8283-8289.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sobeck A, Stone S, Costanzo V, de Graaf B, Reuter T, de Winter J, et al. Fanconi anemia proteins are required to prevent accumulation of replication-associated DNA double-strand breaks. Mol.Cell.Biol. 2006;26:425–437. doi: 10.1128/MCB.26.2.425-437.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.D'Andrea AD, Grompe M. The Fanconi anaemia/BRCA pathway. Nat.Rev.Cancer. 2003;3:23–34. doi: 10.1038/nrc970. [DOI] [PubMed] [Google Scholar]
- 14.Grompe M, D'Andrea A. Fanconi anemia and DNA repair. Hum.Mol.Genet. 2001;10:2253–2259. doi: 10.1093/hmg/10.20.2253. [DOI] [PubMed] [Google Scholar]
- 15.Pagano G, Manini P, Bagchi D. Oxidative stress-related mechanisms are associated with xenobiotics exerting excess toxicity to Fanconi anemia cells. Environ.Health Perspect. 2003;111:1699–1703. doi: 10.1289/ehp.6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pagano G, Youssoufian H. Fanconi anaemia proteins: major roles in cell protection against oxidative damage. Bioessays. 2003;25:589–595. doi: 10.1002/bies.10283. [DOI] [PubMed] [Google Scholar]
- 17.Pagano G, Korkina LG, Degan P, Del Principe D, Lindau-Shepard B, Zatterale A, et al. In vitro hypersensitivity to oxygen of Fanconi anemia (FA) cells is linked to ex vivo evidence for oxidative stress in FA homozygotes and heterozygotes. Blood. 1997;89:1111–1112. [PubMed] [Google Scholar]
- 18.Yule SM, Price L, Cole M, Pearson AD, Boddy AV. Cyclophosphamide metabolism in children with Fanconi's anaemia. Bone Marrow Transplant. 1999;24:123–128. doi: 10.1038/sj.bmt.1701868. [DOI] [PubMed] [Google Scholar]
- 19.Malayappan B, Johnson L, Nie B, Panchal D, Matter B, Jacobson P, et al. Quantitative High-Performance Liquid Chromatography-Electrospray Ionization Tandem Mass Spectrometry Analysis of Bis-N7-Guanine DNA-DNA Cross-Links in White Blood Cells of Cancer Patients Receiving Cyclophosphamide Therapy. Anal.Chem. 2010 doi: 10.1021/ac902923s. [DOI] [PubMed] [Google Scholar]
- 20.Sasaki MS, Tonomura A. A high susceptibility of Fanconi's anemia to chromosome breakage by DNA cross-linking agents. Cancer Res. 1973;33:1829–1836. [PubMed] [Google Scholar]
- 21.Auerbach AD, Wolman SR. Susceptibility of Fanconi's anaemia fibroblasts to chromosome damage by carcinogens. Nature. 1976;261:494–496. doi: 10.1038/261494a0. [DOI] [PubMed] [Google Scholar]
- 22.Pieck AC, Drescher A, Wiesmann KG, Messerschmidt J, Weber G, Strumberg D, et al. Oxaliplatin-DNA adduct formation in white blood cells of cancer patients. Br.J.Cancer. 2008;98:1959–1965. doi: 10.1038/sj.bjc.6604387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Veal GJ, Dias C, Price L, Parry A, Errington J, Hale J, et al. Influence of cellular factors and pharmacokinetics on the formation of platinum-DNA adducts in leukocytes of children receiving cisplatin therapy. Clin.Cancer Res. 2001;7:2205–2212. [PubMed] [Google Scholar]
- 24.Souliotis VL, Dimopoulos MA, Sfikakis PP. Gene-specific formation and repair of DNA monoadducts and interstrand cross-links after therapeutic exposure to nitrogen mustards. Clin.Cancer Res. 2003;9:4465–4474. [PubMed] [Google Scholar]
- 25.Franssen C, Boekema P, De Witte T, Wessels J, Van der Kogel A, Haanen C. DNA strand breaks in human leukocytes induced by chemotherapy and total body irradiation. Leuk.Res. 1990;14:91. doi: 10.1016/0145-2126(90)90151-x. [DOI] [PubMed] [Google Scholar]
- 26.Mohseni Meybodi A, Mozdarani H. DNA damage in leukocytes from Fanconi anemia (FA) patients and heterozygotes induced by mitomycin C and ionizing radiation as assessed by the comet and comet-FISH assay. Iranian biomedical journal. 2009;13:1. [PubMed] [Google Scholar]
- 27.Xie H, Griskevicius L, Stahle L, Hassan Z, Yasar U, Rane A, et al. Pharmacogenetics of cyclophosphamide in patients with hematological malignancies. Eur.J.Pharm.Sci. 2006;27:54–61. doi: 10.1016/j.ejps.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 28.McCune JS, Batchelder A, Deeg HJ, Gooley T, Cole S, Phillips B, et al. Cyclophosphamide following targeted oral busulfan as conditioning for hematopoietic cell transplantation: pharmacokinetics, liver toxicity, and mortality. Biol.Blood Marrow Transplant. 2007;13:853–862. doi: 10.1016/j.bbmt.2007.03.012. [DOI] [PubMed] [Google Scholar]