

Summary
Osteoporosis is a multifactorial genetic disease characterized by reduction of bone mass due to dysregulation of osteoclast differentiation or maturation. Herein, we identified a novel regulator of osteoclastogenesis, the murine homologue of inositol polyphosphate 4-phosphatase type IIa (Inpp4bα). Expression of Inpp4bα is detected from early osteoclast differentiation to activation stage. Targeted expression of native Inpp4bα ex-vivo repressed whereas phosphatase-inactive Inpp4ba stimulated osteoclast differentiation. Inpp4bα acts on intracellular calcium level that modulates NFATc1 nuclear translocation and activation. In vivo mice deficient in Inpp4b displayed increased osteoclast differentiation rate and potential resulting in decreased bone mass and osteoporosis. Importantly, INPP4B in human was identified as a susceptibility locus for osteoporosis. This study defined Inpp4b as a major modulator of the osteoclast differentiation and as a gene linked to variability of bone mineral density in mice and humans.
Introduction
Maintenance of bone mass is a dynamic process regulated by two cellular mechanisms, bone formation and bone resorption. While osteoclasts control bone formation, the osteoclasts are responsible for bone matrix resorption: these functions are tightly regulated via reciprocal crosstalk to maintain a constant bone mass. Disruption of this strict balance leads to osteoporosis or bone depletion and osteopetrosis or increase bone mass, both of which are often linked to abnormal differentiation or maturation of the osteoclast.
Osteoclasts are derived from hematopoietic precursors that undergo multiple differentiation steps shared with the monocyte-macrophage lineage (Boyle et al., 2003; Teitelbaum, 2000; Teitelbaum and Ross, 2003; Zaidi, 2007). Critical for differentiation and survival of early monocyte-osteoclast precursors is expression of the M-CSF cytokine produced and secreted by osteoblasts. Subsequently, the monocyte-osteoclast precursors are differentiated by the RANK/RANKL (receptor activator of NF-kB/ligand) signaling pathway into committed preosteoclasts (Lacey et al., 1998) that migrate to the bone matrix where they fuse to become mature multinucleated osteoclasts (Ishii et al., 2009). Concomitantly, several intracellular signaling cascades are triggered by RANKL/RANK. Two of these RANK cascades implicate first recruitment of the TNF receptor associated factor 6 (TRAF6) leading to activation of two main differentiation effectors, MAP kinases (p38, JNK, ERK) and NFkB. In another cascade, RANK independently of TRAF6 can cooperate with ITAM adaptors, FcRγ and DAP12 linked to the immune receptors OSCAR and TREM2, to activate PLCγ. PLCγ activation will in turn triggers the release of intracellular Ca++ ER stores. Calcium via calcineurin phosphatase activity will cause NFATc1 dephosphorylation and nuclear translocation (Koga et al., 2004) that will lead to NFATc1 autoamplification and full activation (Asagiri et al., 2005). NFATc1 like NFkB in the preosteoclast promote transcription of a set of specific genes (calcitonin receptor, integrin beta3 and tartrate resistant acid phosphatase) resulting in osteoclast maturation (Takayanagi, 2005; Takayanagi et al., 2002). TRAF6 in addition to a role in differentiation also promote cell survival signal through activation of the phosphatidylinositol 3-phosphate kinase (PI3kinase) and subsequently, the serine/threonine kinase AKT/PKB (Wong et al., 1999). PI3Kinase signaling can be antagonized by two known lipid phosphatases, Pten (phosphatase and tensin homolog deleted on chromosome 10) and Ship1 (SH2-containing inositol 5-phosphatase) that modulate the level of PI3Kinase second messengers (Carracedo and Pandolfi, 2008; Golden and Insogna, 2004). Consistently, Pten overexpression in RAW osteoclast-derived cells reduces the anti-apoptotic function of Akt (Sugatani et al., 2003). In contrast, Ship1 null mice display increased osteoclast proliferation and growth resulting in decreased bone mass (Takeshita et al., 2002). While these studies identified genes implicated in osteoclast survival, key phosphatidyl inositol phosphate signaling regulators in osteoclast differentiation remain to be identified.
To characterize novel molecular effectors involved in osteoclast differentiation/maturation and bone mass regulation, bone gene expression profiles from an osteopetrotic mouse mutant were performed and identified the inositol polyphosphate 4-phosphatase type IIα (Inpp4bα), a member of the PI3Kinase signaling pathway markedly downregulated. We demonstrated by ex-vivo and in vivo analysis that Inpp4b is a phosphatase-dependent repressor of osteoclast differentiation. Mice deficient for Inpp4b displayed bone loss and osteoporosis. Inpp4b intracellular mechanism acts by controlling calcium signaling and the NFATc1 osteoclast differentiation pathway. The importance of Inpp4b in human was revealed through genetic analysis, as a novel determinant of bone mineral density or osteoporosis susceptibility locus in pre-menopausal women.
Results
Downregulation of Inpp4bα in bone resorption defect
To identify critical determinants of osteoclast differentiation, bone expression profiles from the osteopetrotic gl/gl homozygous mice with enhanced osteoclast differentiation and maturation (Rajapurohitam et al., 2001) were compared to wild type control mice. One of the identified transcripts, downregulated by ∼80% in quantitative PCR from gl/gl bone tissue, encodes the murine homologue of the rat inositol phosphatase Inpp4bα (Ferron and Vacher, 2006). Inpp4bα expression in gl/gl total bone tissue or osteoclasts, showed significant down-regulation of the transcript relative to wild-type controls in semi-quantitative and real time PCR analysis (Figure 1A,B,C). This downregulation appears specific to the gl/gl phenotype, as expression of Ship1 inositol phosphatase with a known role in murine bone homeostasis (Takeshita et al., 2002), was unaffected in gl/gl tissues (Figure 1A,B). In addition, expression of Inpp4bα was unchanged in bone of the c-src and oc/oc null osteopetrotic mutants (Figure 1D; S1). These results suggested that down-regulation of Inpp4bα is not secondary to osteopetrosis or associated with a general alteration in the regulators of PtdInsPs second messengers.
Figure 1. Regulation of Inpp4bα expression in bone and osteoclastogenesis.

(A) Semi-quantitative expression of Inpp4bα in total bone from the gl osteopetrotic mice (gl/gl) was analyzed in comparison to wild type (+/+). Expression of Runx2 and Ship1 served as osteoblast and osteoclast markers respectively and β-actin as internal control. RT stands for Reverse Transcriptase.
(B) Semi-quantitative expression analysis of Inpp4bα in osteoclasts from the gl osteopetrotic mice (gl/gl) relative to wild type (+/+) was performed using Ship1 and β-actin as controls. RT stands for Reverse Transcriptase.
(C) Quantitative real time PCR of Inpp4bα in total bone and osteoclasts from wild type (+/+) (n=3) and osteopetrotic mice (gl/gl) (n=3) relative to the ribosomal protein S16 control. Data are arbitrary units presented as mean ± SD (*** P <0.001).
(D) Semi-quantitative expression of Inpp4bα in total bone from the c-src-/- osteopetrotic mouse was analyzed compared to wild-type (+/+) controls and β-actin as internal control. (see also Figure S1).
(E) Semi-quantitative expression of Inpp4bα and β isoforms was analyzed in wild-type total bone, primary osteoblasts (OB) and osteoclasts (OC). Only Inpp4bα isoform was detected in both total bone and OC but not in OB. Expression of Runx2 and Ship1 served as osteoblast and osteoclast markers, respectively and β-actin as controls. RT stands for Reverse Transcriptase.
(F) Inpp4ba expression pattern throughout ex vivo osteoclast lineage differentiation was determined using the pre-osteoclast/osteoclast Acp5 marker gene and β-actin as controls.
Differential expression of the Inpp4bα isoform in osteoclasts
Because the mouse Inpp4b gene encodes two isoforms (α and β), the expression of each isoform was evaluated. Analysis of Inpp4bα expression from wild-type bone tissues, containing both osteoclast (OC) and the bone-forming osteoblast (OB) cells, demonstrated that only the Inpp4bα isoform is expressed in vivo in association with expression of Ship1 an inositol phosphatase expressed in the monocyte-macrophage/osteoclast lineage and of Runx2 a transcription factor specific of the osteoblast lineage (Figure 1E). Analysis of the two bone cell sub-populations isolated separately from primary cell cultures showed high levels of Inpp4bα expression in OC compared to very low to undetectable level in OB. Interestingly, Inpp4bα expression showed a progressive increase during OC differentiation that paralleled expression of the specific marker gene tartrate resistant acid phosphatase Acp5 (Figure 1F), suggesting that Inpp4bα could play a prime role in the regulation of osteoclast lineage differentiation to maturation.
Inpp4bα is a negative regulator of osteoclast differentiation
To determine the role of Inpp4bα and the involvement of phosphatase activity in the osteoclast lineage differentiation/maturation, we used the monocytic RAW 264.7 cell line. Differentiation potential and kinetics in presence of RANKL were analyzed from three experiments using two stable clones expressing the native Inpp4bα-EGFP, a phosphatase inactive Inpp4bα(C845A)-EGFP mutant protein or the EGFP control (Figure S2). The number and size of fully differentiated/mature osteoclasts from the different transfected clones was markedly reduced by expression of active Inpp4b(x03008)-EGFP compared to inactive Inpp4bα (C845A)-EGFP and control EGFP clones at day 5 (Figure 2A,D). The number of differentiated/mature OC produced from Inpp4b(x03008)-EGFP clones was decreased by ∼5-fold relative to controls and by ∼10-fold relative to inactive Inpp4ba (Figure 2B). Further, expression of mutant Inpp4b(x03008)(C845A)-EGFP caused earlier occurrence of multinucleated TRAP positive OC at day 3 in comparison to controls whereas only mononucleated cells were detected at that stage with active Inpp4bα–EGFP (Figure 2A,B). Since cell density can affect OC differentiation potential, parallel kinetics of differentiation for the transfected clones were carried out using a range of plated cells. As shown in Figure 2C, Inpp4bα (C845A)-EGFP clones exhibited at low cell densities not only increased cell differentiation rate but also a ∼3-fold higher differentiation cell potential in comparison with EGFP and even higher with Inpp4bα-EGFP clones. Associated with OC slower differentiation rate in Inpp4bα-EGFP clones, OC size was also significantly decreased relative to control cells (Figure 2A,D). Accordingly, ∼65% of cells from Inpp4bα-EGFP clones had 3 to 4 nuclei per OC whereas ∼60% of cells from control and ∼70% of cells from mutant Inpp4bα had ≥5-nuclei per osteoclast (Figure 2E). Consistently, resorption activity of Inpp4bα-EGFP was decreased relative to mutant Inpp4bα(C845A)-EGFP clones and to controls by 5- and 3-fold respectively (Figure 2F). Concurrent to Inpp4bα-EGFP effect on osteoclastogenesis, Inpp4bα-EGFP expression also caused increase apoptosis in comparison to mutant Inpp4bα and controls (Figure 2G). These data suggested that the phosphatase function in Inpp4bα modulates not only cell size but also the rate and potential of osteoclast differentiation.
Figure 2. Inpp4bα impairs osteoclastogenesis in RAW cells.
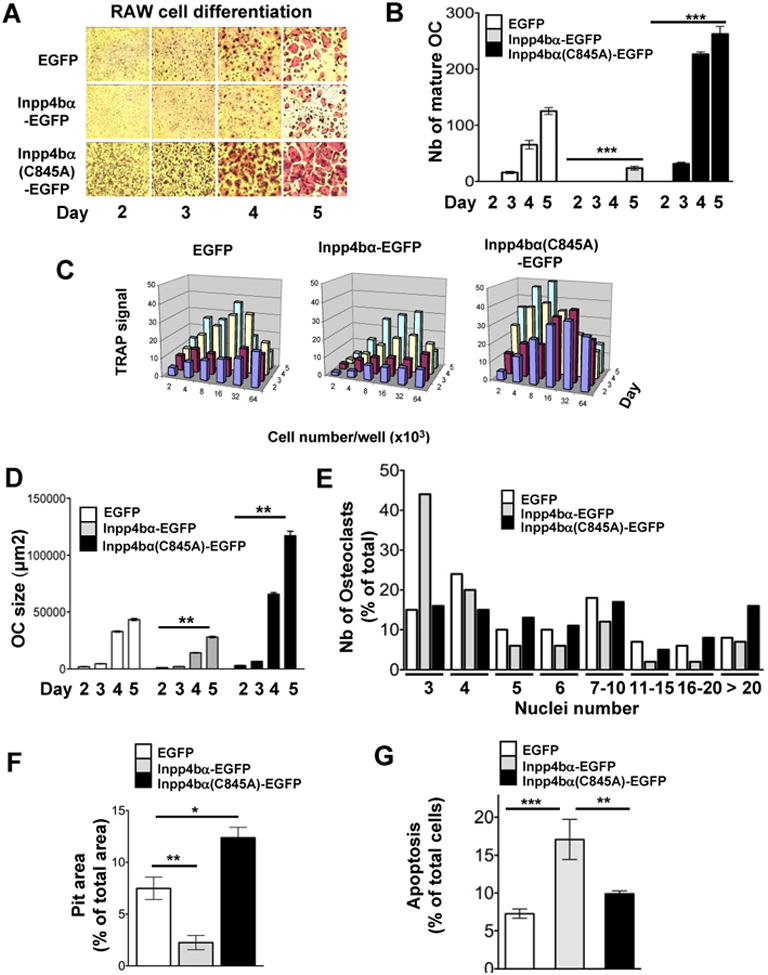
(A) Comparison of RAW cell differentiation kinetic into osteoclast transfected with control (EGFP), native Inpp4bα (Inpp4bα-EGFP) and phosphatase-inactive Inpp4bα (Inpp4bα-C845A-EGFP) monitored upon RANKL stimulation (50ng/ml) for 2-5 days and detected by TRAP staining. (see also Figure S2).
(B) Quantification of mature osteoclast number at various differentiation stages for the three RAW transfected clones (n=4 each) as in (A). Values are in Mean ± SEM (***P<0.001).
(C) Comparison of RAW cell differentiation kinetic with various seeded cell numbers from the three RAW transfected clones as in (A). TRAP signal corresponding to OC appearance was quantified from 2 to 5 days following RANKL stimulation with Image Quant software.
(D) Quantification of osteoclast size at various differentiation stages for the three RAW transfected clones (n=2 each) as in (A). Data are presented as mean cell surface area ± SEM; statistical comparison of OC size was performed between control and active or inactive Inpp4bα differentiation groups (**P <0.01).
(E) Quantification of the number of nuclei in individual osteoclast (subgroups of 3 to >20) at day 5 of differentiation for the three RAW transfected clones upon RANKL stimulation (50ng/ml).
(F) OC resorption activity was quantified by assay of pit area from osteoclast derived of the three RAW transfected clones seeded on Osteologic substrate. Values are in Mean ± SEM (* P<0.05; ** P<0.01).
(G) Quantification of apoptosis in preosteoclast derived from the three RAW transfected clones stained for AnnexinV and analyzed by FACS. Values are in Mean ± SEM (** P <0.01; *** P <0.001).
Inpp4bα as a modulator of NFATc1 in osteoclast differentiation
To gain insight into the second messengers critical for cellular signaling modulation by Inpp4b, we first identified Inpp4b native substrate(s) preference through an in vitro hydrolysis assay. As shown in Figure 3A, Inpp4b can dephosphorylate PI(3,4)P2 as previously reported (Norris et al., 1997) whereas the inactive Inpp4bα(C845A) was incapable of hydrolyzing PI(3,4)P2. Inpp4b was equally efficient (∼20%) at hydrolyzing the native substrate PI(4,5)P2 but was unable to hydrolyse PI(3,4,5)P3 and Ins(1.4.5)P3. The low efficiency in hydrolysing phosphatidylinositol (PIPs) by Inpp4b is consistent with mild or unaffected PIPs levels in the clones (Figure S2D,E). Strikingly, Inpp4b preferred substrate for dephosphorylation was Ins(1,3,4)P3 with ∼5-fold higher efficiency (∼95%) than PI(3,4)P2 or PI(4,5)P2 (Figure 3A). In all cases, none of the potential substrates were hydrolyzed by the mutant Inpp4bα(C845A), showing that this mutation abrogate Inpp4b phosphatase enzymatic activity.
Figure 3. Inpp4bα modulates the NFATc1 signalling pathway during osteoclastogenesis.

(A) Analysis of Inpp4b substrate specificity was carried out on native substrates on a malachite assay using equivalent immunoprecipitates from cell extracts of control EGFP, native Inpp4bα-EGFP and phosphatase-inactive Inpp4bα-C845A-EGFP. This assay showed that the C845 residue is critical for Inpp4b phophatase activity. Values are in Mean ± SEM. (**** P <0.0001).
(B) Comparison of nuclear and cytosolic NFATc1 and NFκB p50 levels in the three RAW transfected cells (control EGFP, native Inpp4bα-EGFP and phosphatase-inactive Inpp4bα-C845A-EGFP) at different time points (0-60min) upon RANKL (100ng/ml) stimulation, relative to β-actin and tubulin as controls. (see also figure S3).
(C) Analysis of Ca2+ oscillations performed on RAW transfected cells (control EGFP, native Inpp4bα-EGFP and phosphatase-inactive Inpp4bα-C845A-EGFP) after 48hr of RANKL treatment (pre-osteoclasts). Maximum fluorescence ratio was obtained by addition of 10 mM ionomycin at the end of tracings (arrow). Data are presented as mean of percentage oscillating cells ± SEM (* P <0.02).
(D) Comparison of quantitative expression kinetics for NFATc1 target genes (Acp5, Itgb3, Ctsk, Nfatc1) during differentiation of the RAW transfected (control EGFP, native Inpp4bα-EGFP and phosphatase-inactive Inpp4bα-C845A-EGFP) clones (n=2 each) upon RANKL stimulation. Values are in Mean ± SEM (* P <0.05; ** P <0.01;*** P <0.001).
To directly determine the intracellular signaling cascade(s) modulated by Inpp4bα upon RANKL activation, we investigated the major signaling effectors implicated in osteoclast survival (PI3K/AKT) and differentiation (MAPK, PLCy). In absence of serum, we observed no AKT activation (t=0) in stable clones expressing Inpp4bα-EGFP, Inpp4bα(C845A)-EGFP or EGFP, but upon addition of RANKL, all clones showed a similar mild increase in AKT phosphorylation (Figure S3A). In presence of serum, P-AKT level appear activated even prior RANKL addition in Inpp4bα(C845A)-EGFP or EGFP clones but remained low in clones expressing Inpp4bα-EGFP, an observation which may account for the increased apoptosis in these cells (Figure S3B, 2G). The MAP kinases p38, ERK, and JNK phosphorylation levels were then monitored independently of serum stimulation and were not significantly altered in the stable clones expressing Inpp4bα-EGFP, Inpp4bα(C845A)-EGFP or EGFP (Figure S3 A). Similarly, no changes in kinetic pattern of activation or in expression levels were detected for these effectors. Since PI(4,5)P2 was mildly hydrolyzed by Inpp4b in vitro, we also analyzed in the three stable clones P-PLCγ (1 and 2) levels relative to total PLCγ. However, active or mutant Inpp4b did not alter PLCγ phosphorylation and kinetic patterns in presence or not of RANKL compare to controls (Figure S3C,D). These data suggest that Inpp4bα does not modulate osteoclast differentiation via the PI3K/AKT, MAPK, or PLCγ pathways.
To examine whether the transcription factors NFκB p50 and NFATc1 were activated by Inpp4bα, total cellular and nuclear expression levels were analyzed following RANKL stimulation. NFκB p50 nuclear level appeared slightly repressed by the active Inpp4bα (Figure 3B). Reciprocally, a sustained but mild increase of nuclear NFκB p50 level independently of RANKL stimulation was detected with the inactive Inpp4bα that may affect OC gene targets (Figure 3B). Concomitantly, expression of active Inpp4bα caused a decrease in NFATc1 nuclear levels whereas total cellular levels of NFATc1 were not reduced (Figure 3B; Figure S3E). In contrast, expression of the inactive Inpp4bα (C845A) resulted in constitutively and markedly elevated nuclear and cytosolic NFATc1 levels compared to controls well above the increase in total cellular NFATc1 (Figure 3B; Figure S3E,F). Quantification of NFATc1 nuclear to cytosolic ratio indicates that expression of the phosphatase inactive Inpp4bα induced a strong nuclear translocation compared to control and active Inpp4bα (Figure S3F). These data suggest an important role for Inpp4bα in regulation of NFATc1 localization that is dependent on phosphatase activity.
Since RANKL signaling activates intracellular calcium levels that promote NFATc1 nuclear translocation (Crabtree and Olson, 2002), we investigated whether Inpp4bα could be a modulator of intracellular calcium by measuring Ca2+ oscillations in the three different RAW clones. In all three experiments, the control clones showed Ca2+ oscillations in ∼45% of cells from 24 to 48 hrs following RANKL stimulation, (Figure 3C). With expression of active Inpp4bα, the percentage of Ca2+ oscillating cells was significantly decreased relative to controls, correlating with reduced NFATc1 nuclear levels. Inversely, the number of Ca2+ oscillating cells expressing the inactive Inpp4bα(C845A) was significantly increased compared to control (Figure 3C). These analyses indicate that Inpp4bα can modulate Ca2+ signaling that can then regulate NFATc1 pathway.
Because active Inpp4bα have a negative effect on NFATc1 nuclear levels and presumably translocation, we quantified expression of NFATc1 known downstream osteoclast-specific target genes (Asagiri et al., 2005; Matsumoto et al., 2004; Takayanagi et al., 2002) during OC differentiation in the three different transfected clones (Figure 3D). In control cells, Nfatc1, Acp5, cathepsin K (Ctsk), β3 integrin (Itgb3) expression gradually increased from 0 to 72hr. In contrast, Nfatc1, Acp5, Ctsk and Itgb3 transcriptional levels in cells expressing active Inpp4bα were markedly decreased by 2-to 4-fold throughout the 72hr treatment relative to control (Figure 3D), consistent with the reduced NFATc1 nuclear levels. In the cells expressing the inactive Inpp4bα(C845A) levels of the target genes Acp5, Ctsk and Itgb3 were elevated during differentiation correlating with increase NFATc1 translocation and auto-amplification. These results showed that Inpp4bα is a major determinant of NFATc1 nuclear translocation and transcriptional activation.
Inpp4b regulates bone mass in vivo
To determine whether Inpp4b regulates bone remodeling in vivo, mice deficient in Inpp4b were generated and adult mice obtained at the expected mendelian ratio with loss of Inpp4b protein in osteoclasts (Figure S4). Analysis of 8 weeks old Inpp4b-/- male mice showed a decrease in femur trabecular bone density (Figure 4A) associated with a 2-3 fold increase in TRAP+ osteoclast number and size compared to controls (Figure 4B,C). Accordingly, bone resorption, as determined by urinary deoxypyridinoline (DPD) cross-links excretion, was significantly increased in adult Inpp4b-/- mice in comparison to control mice (Figure 4D). Because elevated bone resorption is coupled to bone formation (Martin and Sims, 2005), we examined bone formation in the Inpp4b-/- mice. Importantly, our analysis showed enhanced osteoblast cell population in Inpp4b-/- mice compared to controls resulting in a significant increase in the mineral apposition rate (MAR) in vivo and mineralization potential ex-vivo (Figure 4E; S4K,L). Since we did not detected Inpp4b expression in osteoblasts (Figure 1E), this result indicated that Inpp4b does not impede the osteoclast/osteoblast coupling mechanism as revealed by the BFR (Figure S4F). We then investigated whether loss of Inpp4b could have an impact on long bones growth and detected small but consistent bone length decrease in Inpp4b-/- compared to controls (Figure S4J). Further characterization of the bone phenotype of Inpp4b-/- mice showed from micro-CT and histomorphometry analyses of the femur at 8 weeks and 4 months of age, a major decrease in bone volume fraction (BV/TV) (Figure 4F, S4G-I; Table S1). Interestingly, in young 8-week old animals, this decreased bone mass is associated with important reduction in trabeculae number whereas in 4-month old mice it was correlated with major decrease in trabecular thickness (Table S1); such difference is most likely due to intense bone structure remodeling in young animals (Bearmer et al., 1996). In addition, histomorphometric quantification of bone volume fraction in vertebra, acknowledged as a complementary and highly reliable method for bone mass analysis (Ducy et al., 2000b), revealed as well a ∼25% reduction in Inpp4b-/- bone volume relative to controls (Figure 4G). Hence, Inpp4b-/- mice display net bone loss, reduced bone growth and osteoporosis. These data revealed that Inpp4bα is a negative modulator of osteoclast differentiation with a major physiologic impact on bone mass.
Figure 4. Ablation of Inpp4b in mice causes decreased bone mass.
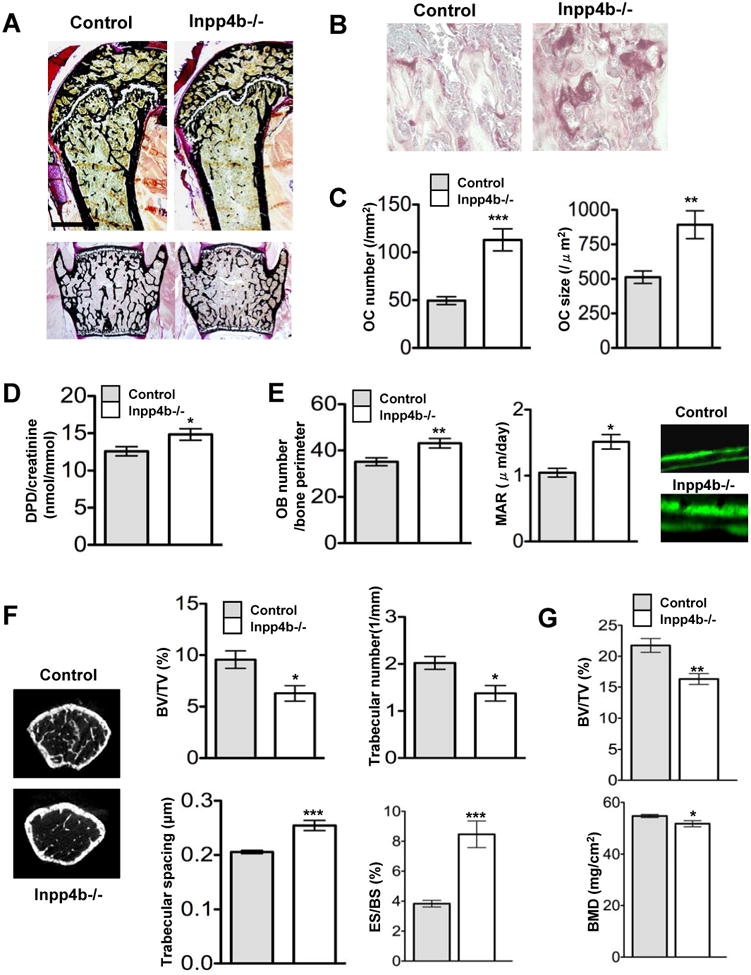
(A) Undecalcified sections of femur (upper panel) and vertebra (lower panel) from control and Inpp4b-/- mice (8-weeks old) were stained with Von Kossa. (see also figure S4 and Table S1).
(B) Femur sections of control and Inpp4b-/- mice were monitored for trabecular bone structure, osteoclast size and number by TRAP staining.
(C) Osteoclast number and size in Inpp4b-/- mice (n=6) were compared to controls (n=6). Data are presented as Mean ± SEM (** P <0.01; *** P <0.001).
(D) Bone resorption was evaluated by excretion of urinary deoxypyridinoline (DPD) cross-links in Inpp4b-/- mice compared to controls (n=11 each). Values are in Mean ± SEM (* P <0.05).
(E) In vivo osteoblast number (OB number/bone perimeter) was quantified following bone staining with alkaline phosphatase (** P <0.01). Osteoblast activity was monitored by mineralization apposition rate (MAR). Quantification was carried out following calcein double labeling in Inpp4b-/- mice relative to control. Values are in Mean ± SEM (* P <0.05).
(F) Microcomputed tomography analysis of male femur from control and Inpp4b-/- mice (8-weeks old) was carried out to quantify relative bone volume (BV/TV), trabecular numbers, trabecular spacing and erod surface/bone surface (ES/BS). Values are in Mean ± SEM (* P <0.05; *** P <0.001).
(G) Histomorphometrical analysis of male vertebrae from control and Inpp4b-/- mice (8-weeks old) quantify relative bone volume (BV/TV) and Bone Mineral density (BMD). Values are in Mean ± SEM (* P <0.05; ** P <0.01).
Inpp4b modulates osteoclast differentiation through NFATc1 signaling
To assess the differentiation potential of Inpp4b-/- pre-osteoclasts ex vivo, osteoclasts were derived from bone marrow cell cultures. A significant 1.7-fold increase in OC production in Inpp4b-/- culture (232±39; n=8) compared to controls (137±23; n=8) was measured (Figure 5A) that correlated with the increased number of TRAP+ cells in vivo (Figure 4B,C). OC size were also increased by ∼3-fold in Inpp4b-/- culture (Figure 5B) and as illustrated in Figure 5A. Moreover, the kinetic of OC differentiation from Inpp4b-/-bone marrow cells was significantly accelerated compared to controls (Figure 5B). Analysis and quantification of Inpp4b-/- osteoclast activity demonstrated significant increase in pit area formation by ∼50% relative to controls (Figure 5C). This increased resorption is likely due to the expanded osteoclast cell population (Figure 4C, S4H), and not to increase in individual osteoclast activity (Figure 5C). Together, these results confirmed that Inpp4b is a negative modulator of osteoclast differentiation.
Figure 5. Inpp4b modulates osteoclast differentiation ex vivo.

(A) Ex vivo osteoclast differentiation kinetic from Inpp4b-/- bone marrow monocytes relative to control was monitored upon M-CSF (10 ng/ml) and RANKL (50 ng/ml) stimulation for 7 days and detected by TRAP staining. (see also Figure S5).
(B) Quantification of osteoclast size ex-vivo at various differentiation stages for Inpp4b-/-(n=3) and controls (n=3). Data are mean ± SEM and statistical comparison of OC size was performed between control and null Inpp4b differentiation groups (*** P <0.001).
(C) Quantification of bone resorption was performed from Inpp4b-/- and control osteoclasts on dentine subs trate and pit resorption area quantified by toluidine blue staining. Values are in Mean ± SEM (**P <0.01).
Since Inpp4b-deficient pre-osteoclasts showed increase differentiation potential, main lipid phosphatases and downstream RANKL signaling effectors were analyzed. Expression of the lipid phosphatase Pten and Ship1 was unaffected in Inpp4b-/-osteoclasts, indicating no indirect impact of Inpp4b on PI3K signaling (Figure S6A). As observed in RAW-transfected cells, RANKL-induced Ca2+ oscillation in Inpp4b-/- pre-osteoclasts was increased by ∼28% relative to controls (Figure 6A). Consequently, NFATc1 nuclear levels were significantly elevated in Inpp4b-/- cells as determined by nuclear to cytosolic ratio relative to controls (Figure 6B; S6B,C) whereas activation of AKT, ERK and p38 following stimulation was similar in pre- and mature Inpp4b-/-osteoclasts when compared to controls (Figure S5). Consistently, NFATc1 transcriptional target genes, Nfatc1, Acp5, Ctsk, Itgb3, Itgav, and Ctr showed a significant increase and similar induction kinetic of expression in Inpp4b-/- cells (Figure 6C). These results indicate that Inpp4b acts as a negative regulator of osteoclast differentiation downstream of RANK/RANKL signaling via NFATc1.
Figure 6. Inpp4b modulates osteoclast differentiation through NFATc1.

(A) Analysis of Ca2+ oscillations performed on the control and Inpp4b-/- pre-osteoclasts after 24hr of RANKL treatment. Maximum fluorescence ratio was obtained by addition of 10 mM ionomycin at the end of tracings (arrow). Data are presented as mean of percentage oscillating cells ± SEM. (* P <0.05). (see also Figure S6).
(B) Nuclear NFATc1 levels in Inpp4b-/- and in control bone marrow monocytes derived osteoclasts was evaluated by immunoblot upon stimulation with RANKL (100ng/ml) for various times (0-30min); Lamin C served as internal nuclear control.
(C) Comparison of quantitative expression kinetics for NFATc1 target genes (Acp5, Itgb3, Ctsk, Nfatc1, Itgav, Ctr) during osteoclast lineage differentiation of Inpp4b-/- and control. Data are presented as Mean ± SEM; statistical comparison was performed between control and null Inpp4bα differentiation groups (*P <0.05; **P <0.01; ***P <0.001).
Inpp4b, a candidate gene for human bone mineral density variability
Considering that Inpp4b modulates bone mass in mice and that INPP4B maps to chromosome 4q (Ferron and Vacher, 2006) where quantitative trait loci for bone mineral density (BMD) have been localized (Deng et al., 2002; Ralston et al., 2005), we hypothesized that INPP4B could be a candidate gene for association with this trait in humans. Screening of the Utah residents with ancestry from Northern and Western Europe population from HapMap detected 493 single nucleotide variant (SNP) within the INPP4B genomic locus. Eighty-three Tag SNPs were analyzed for an association with femoral neck bone mineral density (FN BMD) or lumbar spine BMD (LS BMD) in a Quebec pre-menopausal women sample (n=677) (Giroux et al., 2007). Three SNPs, rs2627804, rs13104269 and rs2636683, were significantly associated with BMD variability (p-value <0.05). We then tested these 3 SNPs for replication in a second sample of premenopausal women (Toronto, n=660). Since both the Quebec and Toronto samples were established on the same physiologic characteristics, genotyping results from both samples were combined to increase the number of individuals and statistical power. The three SNPs remained associated in the combined samples with lower p-value indicating that the gene effect was in the same direction. For the SNP rs2627804, the genotypes GG and GA were associated with similar mean BMD in both FN and LS, whereas the AA genotype was significantly associated with higher mean BMD for these sites. Rs2636683 which was in high linkage disequilibrium with rs2627804 and strongly correlated with it (r =0.61) was also associated with FN but to a lesser extent. Rs13104269 showed a weak association with LS BMD and a trend with FN.
Discussion
Characterization of molecular regulators is critical to dissect signaling pathways that control osteoclastogenesis and modulate bone remodeling, in both normal and pathologic conditions. Herein, our study identified a modulator of osteoclast differentiation, the Inositol polyphosphate 4-phosphatase type IIα (Inpp4bα) via its phosphatase function. In vivo ablation of Inpp4b showed that the primary function of Inpp4bα is to repress osteoclastogenesis. Inpp4bα regulation acts specifically on intracellular calcium to inhibit NFATc1 nuclear localization and transcription of osteoclast-specific target genes. Importantly, in humans INPP4B is associated with bone mineral density variability. Together, these results showed that Inpp4bα is a regulator of osteoclastogenesis and bone mass in both mouse and human.
The opposite roles played by the targeted native and phosphatase-inactive Inpp4bα in OC differentiation rate and potential as well as on OC size pointed to the phosphatase catalytic activity as a critical determinant in this differentiation process. Interestingly, the reduced OC cell number and size caused by Inpp4bα overpression is analogous to the negative effect of the isoenzyme Inpp4aα on megakaryocytes and fibroblasts growth (Vyas et al., 2000). Importantly, this analysis provided the first evidence that Inpp4bα can act as a negative regulator of osteoclast differentiation. This finding is further supported in the osteopetrotic gl/gl mice with concomitant Inpp4bα reduced expression and osteoclast differentiation and number stimulation, most likely as a compensatory response mechanism.
Our data identified NFATc1 as a major downstream target regulated by Inpp4b to modulate RANKL induced osteoclast differentiation pathways. Since Ins(1,3,4)P3 a product of IP3 signaling is the preferred substrate of Inpp4b, it could provide an indirect mechanism by which Inpp4b affect calcium flux downstream of PLCγ activation. Indeed, the reciprocal response by the native and inactive Inpp4bα, on intracellular calcium release and NFATc1 nuclear translocation/levels in the OC differentiation model further supports that Inpp4bα acts on calcium signaling to modulate NFATc1 regulatory pathway. The particularly strong constitutive response induced by the phosphatase-inactive Inpp4bα independently of RANKL activation showed that Inpp4bα could bypass RANKL to modulate the NFATc1 pathway. Consistent with our results, it has been shown that introduction of NFATc1 in null NFATc1 bone marrow monocytes was both essential and sufficient to rescue osteoclastogenesis in absence of RANKL (Takayanagi et al., 2002). Importantly, elevated NFATc1 nuclear translocation during Inpp4b-/-osteoclast differentiation validated Inpp4b as an in vivo modulator of NFATc1 signaling pathway. The transcriptional profile of NFATc1 osteoclast-specific genes in mice and in cells converged to indicate that Inpp4bα has a dominant function on NFATc1 signaling pathway. More precisely, the comparable transcriptional pattern of target genes for the null Inpp4bα and phosphatase-inactive Inpp4bα and the opposite pattern for the native Inpp4b assigned a central function to the Inpp4b phosphatase catalytic activity. Similar functional impact of the phosphatase-inactive Inpp4bα and of Inpp4bα-deficiency on osteoclast differentiation indicates that the phosphatase-inactive mutant is most likely acting as a dominant negative. Consistently, our results showed that Inpp4bα phosphatase function is a major determinant of NFATc1 expression, nuclear localization and transcriptional activation. These findings suggest that Inpp4bα is a key regulator of the NFATc1 osteoclastogenesis pathway.
A crucial role of Inpp4bα was determined in vivo in Inpp4b-deficient mice in which a significant increased in the osteoclast cell population and bone resorption resulted in a net bone loss and osteoporosis. Since this phenotype is reminiscent of the Inositol-5-phosphatase SHIP1 deficient mice (Takeshita et al., 2002), it could be inferred that the levels of PtdInsPs second messengers modulate osteoclast cell population and normal osteoclastogenesis. Indeed, loss of SHIP1 promoted increase mature osteoclast survival, arguing for activation of AKT signaling by PI(3,4,5)P3 modulation. However, loss of Inpp4b in OC differentiation and maturation had very little effect on AKT signaling whereas the downstream PLCγ pathway was activated most likely via IP3 modulation. Our studies highlight a distinct signaling cascade for the inositol phosphatase Inpp4b in OC differentiation from SHIP1 mechanism in mature osteoclasts. This general physiological mechanism of Inpp4b will be dissected cellularly in OC and OB lineages in targeted rescue experiments. Nevertheless together our findings and those of others show that a concerted regulation of several inositol phosphatases in osteoclast differentiation and maturation is critical to maintain normal bone physiology, remodeling and bone mass.
Importantly, a role for INPP4B was also revealed in human bone physiology. We identified in our genetic screen a specific INPP4B SNP significantly associated with BMD variability in healthy individuals. Our results defined INPP4B as a second locus that modulates bone mass with a similar impact as LRP5 in human Caucasian populations (Giroux et al., 2007). Similar to LRP5 set of studies, series of replication in additional cohorts and functional studies should be undertaken to consolidate INPP4B as a major BMD contributor. From a clinical perspective, INPP4B could become a clinical marker to predict osteopenia or even osteoporosis in the general women population.
In summary, this study identified Inpp4b as a regulator of osteoclatogenesis and bone mass in both mouse and human. This regulatory mechanism requires Inpp4bα phosphatase catalytic function that controls intracellular calcium and modulates the NFATc1 signaling pathway. That INPP4B is associated with BMD variability in human reveals a genetic determinant and provides a potential therapeutic target and a biomarker for osteoclast defects and osteoporosis.
Experimental Procedures
Mice
The mouse strains GL/Le dl+/+gl backcrossed for 120 generations and c-src+/- were obtained from The Jackson Laboratory (Bar Harbor, ME). Animal use complied with the guidelines of the Canadian Committee for Animal Protection and was approved by the local institutional animal care committee. Generation of Inpp4b-/- mice was specifically designed to generate a similar mutation to the spontaneous weeble mutation in Inpp4a (Nystuen et al., 2001) (Supplemental Experimental Procedures; Figure S4).
PCR-based differential display screening
PCR-based differential screening was conducted as described (Liang et al., 1993). Long bones (femurs and tibia) were dissected from control, gl/gl and oc/oc mice cleaned from soft tissues and frozen in liquid nitrogen. Bones were grinded into powder with mortar/pestle kept on dry ice and RNA extracted with Trizol reagent (Invitrogen, Burlington, ON). Total bone RNA (0.5 μg) was annealed with individual T12NC, T12NG or T12NA (N = A,T,C or G) primer and reverse transcribed. Radiolabeled PCR amplification was then carried out with one of the arbitrary 16 to 20 primers and either T12MC, T12MG or T12MA primer, 1μl of dATP α35S (1200 Ci/mmol) and 1.5U of Taq polymerase. Amplified cDNAs were separated on 6% sequencing gel and bands were excised from dried gels, re-amplified and sequenced.
Production of fusion protein and transfectants
The mouse full-length Inpp4bα cDNA truncated from the stop codon onwards was amplified and cloned in pEGFP-N1 vector (Clontech, Mountain View, CA). To abolish phosphatase activity the catalytic cysteine, C845, was mutated to an alanine resulting in pInpp4ba(C845A)-EGFP construct (Supplemental Experimental Procedures; Figure S2). RAW 264.7 cell line (TIB-71) (ATCC, Manassas, VA) were electroporated and several individual stable clones expressing EGFP, Inpp4bα-EGFP or Inpp4bα(C845A)-EGFP were selected with G418 (500μg/ml). Osteoclasts were generated from each clone in culture on either plastic or glass in α-MEM medium with 10% FBS and RANKL (50ng/ml).
Osteoclast production
Osteoclasts were generated ex vivo by co-culturing newborn primary calvaria osteoblasts (Sanjay et al., 2001) and bone marrow or spleen cells of either wild type or gl/gl mice (Rajapurohitam et al., 2001). Alternatively, OC were obtained by differentiation of individual stable transfectants in the presence of RANKL (50 ng/ml) or from pre-osteoclasts obtained after incubation with M-CSF (10 ng/ml) for six days. Multinucleated OC were identified by Tartrate Resistant Acid Phosphatase (TRAP) staining (Collin-Osdoby et al., 2003) and pit resorption assay was performed as described previously (Miyazaki et al., 2004) on dentine or using BD BioCoat™ Osteologic™ Bone Cell Culture System.
Apoptosis measurement in RAW 264.7 cells
RAW 264.7 cells were cultured on non-treated plastic culture dish in presence or absence of RANKL (50 ng/ml) for 48 hours. Cells were detached with 0.5 mM EDTA in PBS 1X, wash 3 times with PBS 1X and stained with FITC-Annexin V and propidium iodine (PI) using a commercially available kit (BD Bioscience). Stained cell suspensions were next analyzed by FACS. Only cells positive for Annnexin V but negative for PI were considered as apoptotic.
Gene and protein expression analysis
For kinetics of gene/protein expression, cells were cultured in α-MEM medium with 0.5% FBS and then stimulated with 100 ng/ml of RANKL for various times. Total RNA (500 ng) of OC and various tissues from 3 week-old wild type and gl/gl mice were reverse-transcribed (SuperScriptII transcriptase, Invitrogen, Burlington, ON). Semiquantitative PCR expression analysis of the Inpp4b alpha and beta 3′ splicing isoforms used the same Inpp4b forward primer 5′-GATTAAAGAAGGCCAGTTGC-3′ and two specific reverse Inpp4b primers: alpha reverse 5′-ACTTGGGGAAAGCCATCAGC-3′ and beta reverse 5′-AGAAGTGTCGAGAAGTTG GC-3′. The specific alpha and beta PCR products were 533 bp and 447 bp respectively. All specific band intensities were quantified using ImageQuant software.
Quantitative PCR was performed on DNaseI-treated total RNA reverse transcript with polydT primers using Superscript II first stand cDNA synthesis kit (Invitrogen). The primers used for the analysis were designed with GeneFisher 2 software and are listed in Supplemental Experimental Procedures.
Protein extraction and Western analysis were conducted as previously (Rajapurohitam et al., 2001), using phophospecific antibodies against AKT (phospho T-308 and phospho S-473), p38, ERK and JNK (Cell Signal), or Inpp4ba polyclonal purified antiserum. These blots were reprobed with total NFκB p50 (Santa Cruz), NFATc1, β-actin, tubulin, lamin (BD Biosciences), JNK and AKT antibodies (Cell Signal).
In vitro phosphatase assays
PI(3,4)P2, PI(3,4,5)P3, PI(4,5)P2, Ins(1,3,4)P3 and Ins(1,4,5)P3 (Echelon Research Laboratories, NJ) at 50 mM were incubated in phosphatase assay BF (50 mM MOPS pH 7.0, 10 mM EDTA) for 1 hrs at 30°C with either Inpp4ba-EGFP, Inpp4bα(C845A)-EGFP (EGFP-immunoprecipitates from transfected COS-7 cells or with a control EGFP-immunoprecipitate. Free Pi generated was measured by malachite green assay (R&D System) using a microplate reader at 620 nm. Data were corrected for background activity by subtracting phosphatase absorbance from absorbance for each reaction.
Intracellular calcium measurement
Transfected cells and pre-osteoclasts were incubated in αMEM with 10% FBS and RANKL (50ng/ml) for 48hr and 24hr respectively. Intracellular calcium concentration was evaluated in cells loaded with 5mM fluo-4 AM and 5mM Fura Red AM (Invitrogen) for 30 min in serum-free DMEM supplemented with 0.1% BSA, 10 mM Hepes pH 7.4, and 50ng/ml RANKL; cells were then washed and resuspended in HANK's salt solution. By confocal microscopy, cells were excited at 488nm and emission signals for fluo-4 (505-530nm) and for Fura Red (600-680nm) were recorded simultaneously at 5 sec intervals. To evaluate the intracellular Ca2+ concentration in single cells, the ratio of the fluorescence intensity from the fluo-4 to Fura Red was calculated. As control, the maximum ratio increase was determined by addition of 10mM ionomycin at the end of each experiment.
Bone physiologic parameters
Bone histology and staining were conducted as described (Rajapurohitam et al., 2001). Bone parameters were quantified using microCT (SkyScan1072) technologies. Urine levels of deoxypyridinoline cross-links were determined using commercial kit (Quidel Corporation) and normalized to urine creatinine following the manufacturer's protocol. Osteoblast number was quantified on bone sections stained with alkaline phosphatase (ALP). Bone histomorphometry was performed as previously (Chappard et al., 1987) (Parfitt et al., 1987). In brief, femur and lumbar vertebrae were dissected, fixed for 24 hr in 10% formalin, dehydrated in graded ethanol series, and embedded in methyl methacrylate resin according to standard protocols. Von Kossa/Von Gieson staining was performed on 7μm sections for bone volume over tissue volume (BV/TV) measurement. Bone formation rate (BFR) and mineralization apposition rate (MAR) were analyzed by the calcein double-labeling method (Ducy et al., 2000a). Calcein (Sigma) was dissolved in buffer (0.15 M NaCl, 2% NaHCO3) and injected twice at 0.125mg/g body weight on days 1 and 4, and mice were sacrificed on day 6. Five μm bone sections were cleared in xylene and used for bone formation rate (BFR) measurements. Histomorphometric analyses were performed using the Osteomeasure Analysis System (Osteometrics, Atlanta, GA).
Human samples and genotyping
Subjects were recruited from two Canadian metropolitan regions, Quebec city metropolitan region for the discovery sample (N=693) and Toronto metropolitan region for the replication sample (N=660) (Giroux et al., 2008). All subjects were premenopausal women and provided written, informed consent, and each institution approved the study through their ethics review board. We used the HapMap results and Tagger pairwise program (Barrett et al., 2005; Consortium, 2007) to select eighty-three single nucleotide polymorphisms (SNPs). These SNPs were chosen as tag common genetic variants with a minor allele frequency (MAF) > 5% and a correlation index r2 of 0.8 within the entire coding region of INPP4B and 40 kb in 5′ of the coding region as well as 10 kb in 3′, covering 452 kb of genomic DNA of chromosome 4. Genotyping was performed by the Sequenom iPlex Gold technology (Genome Quebec Innovation Center) with the DNA from the Quebec sample (n=693) and two controls samples from Centre d'Etude du Polymorphisme Humain (CEPH) were genotyped on each 96-well plate of individual DNAs and compared with HapMap results. The call cluster plot was also evaluated and the Hardy-Weinberg equilibrium was confirmed. FN BMD and LS BMD were tested separately for association with each SNP by ANCOVA analysis adjusted for environmental variables (age, weight, age at menarche, physical activity and smoking). Only three SNPs had a p-value lower than 0.05 with either FN BMD or LS BMD. Rs13104269 was analyzed in two groups because the GG genotype was too rare and thus was combined with GA (Minor allele frequency = 6.9%). The sample from Toronto (n=660) was genotyped with these three SNPs with the same technology and analyzed in combination with the discovery sample as described above (N=1353).
Data analysis
Data are presented as means ± SEM. Statistical analysis was performed by two-tailed Student's t test. The value of P < 0.05 was considered as significant. Simultaneous comparison of groups was performed using ANOVA. The SNPs statistical analyses has been described previously (Giroux et al., 2007).
Supplementary Material
Table 1. Analysis of INPP4B variants in Quebec and Toronto premenopausal samples.
| Quebec Premenopausal Sample | |||||||
|---|---|---|---|---|---|---|---|
| Frequencies | LS BMD means* (g/cm2) | 95% confidence interval | p-value | FN BMD means* (g/cm2) | 95% confidence interval | p-value | |
| rs13104269 | AA = 578 | 1.175 | 1.162 to 1.188 | 0.036 | 0.915 | 0.904 to 0.926 | 0.071 |
| AG/GG = 102 | 1.145 | 1.118 to 1.172 | 0.892 | 0.868 to 0.916 | |||
| rs2627804 | GG = 188 | 1.161 | 1.141 to 1.181 | 0.167 | 0.902 | 0.885 to 0.920 | 0.005 |
| GA = 340 | 1.169 | 1.154 to 1.185 | 0.907 | 0.893 to 0.920 | |||
| AA = 164 | 1.187 | 1.166 to 1.209 | 0.939 | 0.920 to 0.958 | |||
| rs2636683 | CC = 313 | 1.177 | 1.161 to 1.194 | 0.344 | 0.924 | 0.909 to 0.938 | 0.031 |
| CT = 298 | 1.168 | 1.151 to 1.184 | 0.904 | 0.890 to 0.919 | |||
| TT = 65 | 1.152 | 1.118 to 1.185 | 0.888 | 0.859 to 0.917 | |||
| Combined Quebec and Toronto Premenopausal Samples | |||||||
| rs13104269 | AA = 1142 | 1.181 | 1.171 to 1.191 | 0.015 | 0.956 | 0.947 to 0.964 | 0.051 |
| AG/GG = 191 | 1.156 | 1.137 to 1.176 | 0.938 | 0.921 to 0.955 | |||
| rs2627804 | GG = 365 | 1.170 | 1.156 to 1.185 | 0.021 | 0.945 | 0.932 to 0.958 | 0.002 |
| GA = 662 | 1.173 | 1.162 to 1.185 | 0.949 | 0.938 to 0.959 | |||
| AA = 325 | 1.195 | 1.180 to 1.210 | 0.973 | 0.960 to 0.987 | |||
| rs2636683 | CC = 662 | 1.185 | 1.174 to 1.197 | 0.080 | 0.962 | 0.951 to 0.972 | 0.029 |
| CT = 549 | 1.172 | 1.159 to 1.184 | 0.945 | 0.934 to 0.956 | |||
| TT = 121 | 1.163 | 1.139 to 1.186 | 0.944 | 0.923 to 0.965 | |||
LS BMD = lumbar spine bone mineral density, FN BMD = femoral neck bone mineral density
adjusted Means
Acknowledgments
We are grateful to G. Karsenty for providing countless reagents and for his expert support with bone histomorphometry. We thank T. Garcia, P. Mollat, A. Veillette, S. Meloche, P. Ducy, T. Koga and the Centre for Bone and Periodontal Research (Montreal) for MicroCT analyses. This work was supported by Canadian Institutes of Health Research grant 93639 and National Sciences and Engineering Council of Canada grants to J.V., by Canadian Institutes of Health Research grant 86748 to F.R and by NIH grant HL-16634-45 to P.W.M. M.F. was supported by a Doctoral Training Award from the Canadian Institutes of Health Research and M. A. by a Training Program in Skeletal Health Research Award from the Canadian Institutes of Health Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Asagiri M, Sato K, Usami T, Ochi S, Nishina H, Yoshida H, Morita I, Wagner EF, Mak TW, Serfling E, Takayanagi H. Autoamplification of NFATc1 expression determines its essential role in bone homeostasis. J Exp Med. 2005;202:1261–1269. doi: 10.1084/jem.20051150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- Bearmer WG, Donahue LR, Rosen CJ, Baylink DJ. Genetic variability in adult bone density among inbred strains of mice. Bone. 1996;18:397–403. doi: 10.1016/8756-3282(96)00047-6. [DOI] [PubMed] [Google Scholar]
- Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- Carracedo A, Pandolfi PP. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene. 2008;27:5527–5541. doi: 10.1038/onc.2008.247. [DOI] [PubMed] [Google Scholar]
- Chappard D, Palle S, Alexandre C, Vico L, Riffat G. Bone embedding in pure methyl methacrylate at low temperature preserves enzyme activities. Acta Histochem. 1987;81:183–190. doi: 10.1016/S0065-1281(87)80012-0. [DOI] [PubMed] [Google Scholar]
- Collin-Osdoby P, Yu X, Zheng H, Osdoby P. RANKL-mediated osteoclast formation from murine RAW 264.7 cells. Methods Mol Med. 2003;80:153–166. doi: 10.1385/1-59259-366-6:153. [DOI] [PubMed] [Google Scholar]
- Consortium TIHM. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–862. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree G, Olson E. NFAT signaling: choreographing the social lives of cells. Cell. 2002;109:S67–S79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- Deng HW, Xu FH, Huang QY, Shen H, Deng H, Conway T, Liu YJ, Liu YZ, Li JL, Zhang HT, et al. A whole-genome linkage scan suggests several genomic regions potentially containing quantitative trait loci for osteoporosis. J Clin Endocrinol Metab. 2002;87:5151–5159. doi: 10.1210/jc.2002-020474. [DOI] [PubMed] [Google Scholar]
- Ducy P, Amling M, Takeda S, Priemel M, Schilling AF, Bell FT, Shen J, Vinson C, Rueger JM, Karsenty G. Leptin inhibits bone formation through a hypothalamic relay: a central control of bone mass. Cell. 2000a;100:197–207. doi: 10.1016/s0092-8674(00)81558-5. [DOI] [PubMed] [Google Scholar]
- Ducy P, Schinke T, Karsenty G. The osteoblast: a sophisticated fibroblast under central surveillance. Science. 2000b;289:1501–1504. doi: 10.1126/science.289.5484.1501. [DOI] [PubMed] [Google Scholar]
- Ferron M, Vacher J. Characterization of the murine Inpp4b gene and identification of a novel isoform. Gene. 2006;376:152–161. doi: 10.1016/j.gene.2006.02.022. [DOI] [PubMed] [Google Scholar]
- Giroux S, Elfassihi L, Cardinal G, Laflamme N, Rousseau F. LRP5 coding polymorphisms influence the variation of peak bone mass in a normal population of French-Canadian women. Bone. 2007;40:1299–1307. doi: 10.1016/j.bone.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Giroux S, Elfassihi L, Cole DEC, Rousseau F. Replication of associations between LRP5 and ESRRA variants and bone density in premenopausal women. Osteoporos Int. 2008;19:1769–1775. doi: 10.1007/s00198-008-0617-z. [DOI] [PubMed] [Google Scholar]
- Golden LH, Insogna KL. The expanding role of PI3-kinase in bone. Bone. 2004;34:3–12. doi: 10.1016/j.bone.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Ishii M, Egen JG, Klauschen F, Meir-Schellersheim M, Saeki Y, Vacher J, Proia RL, Germian RN. Sphingosine-1-phosphate mobilizes osteoclast precursors and regulates bone homeostasis. Nature. 2009;458:524–528. doi: 10.1038/nature07713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga T, Inui M, Inoue K, Kim S, Suematsu A, Kobayashi E, Iwata T, Ohnishi H, Matozaki T, Kodama T, et al. Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature. 2004;428:758–763. doi: 10.1038/nature02444. [DOI] [PubMed] [Google Scholar]
- Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Scully S, Hsu H, Sullivan J, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- Liang P, Averbouck L, Pardee AB. Distribution and cloning of eukaryotic mRNAs by means of differential diplay: refinements and optimization. Nucl Acids Res. 1993;21:3269–3275. doi: 10.1093/nar/21.14.3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TJ, Sims NA. Osteoclast-derived activity in the coupling of bone formation to resorption. TIMM. 2005;11:76–81. doi: 10.1016/j.molmed.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Kogawa M, Wada S, Takayanagi H, Tsujimoto M, Katayama S, Hisatake K, Nogi Y. Essential role of p38 mitogen-activated protein kinase in cathepsin K gene expression during osteoclastogenesis through association of NFATc1 and PU.1. J Biol Chem. 2004;279:45969–45979. doi: 10.1074/jbc.M408795200. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, Sanjay A, Neff L, Tanaka S, Horne WC, Baron R. Src kinase activity is essential for osteoclast function. J Biol Chem. 2004;279:17660–17666. doi: 10.1074/jbc.M311032200. [DOI] [PubMed] [Google Scholar]
- Norris FA, Atkins RC, Majerus PW. The cDNA cloning and characterization of inositol polyphosphate 4-phosphatase type II. J Biol Chem. 1997;272:23859–23864. doi: 10.1074/jbc.272.38.23859. [DOI] [PubMed] [Google Scholar]
- Nystuen A, Legare ME, Shultz LD, Frankel WN. A null mutation in inositol polyphosphate 4-phosphatase type I causes selective neuronal loss in webble mutant mice. Neuron. 2001;32:203–212. doi: 10.1016/s0896-6273(01)00468-8. [DOI] [PubMed] [Google Scholar]
- Parfitt AM, Drezner MK, Glorieux FH, Kanis JA, Malluche H, Meunier PJ, Ott SM, Recker RR. Bone histomorphometry: standardization of nomenclature, symbols, and units. J Bone Miner Res. 1987;2:595–610. doi: 10.1002/jbmr.5650020617. [DOI] [PubMed] [Google Scholar]
- Rajapurohitam V, Chalhoub N, Benachenhou N, Neff L, Baron R, Vacher J. The mouse osteopetrotic grey-lethal mutation induces a defect in osteoclast maturation/function. Bone. 2001;28:513–523. doi: 10.1016/s8756-3282(01)00416-1. [DOI] [PubMed] [Google Scholar]
- Ralston SH, Galwey N, MacKay I, Albagha OME, Cardon L, Compston JE, Cooper C, Duncan E, Keen R, Langdahl B, et al. Loci for regulation of bone mineral density in men and women identified by genome wide linkage scan: the FAMOS study. Hum Mol Genet. 2005;14:943–951. doi: 10.1093/hmg/ddi088. [DOI] [PubMed] [Google Scholar]
- Sanjay A, Houghton A, Neff L, DiDomenico E, Bardelay C, Antoine E, Levy J, Gailit J, Bowtell D, Horne WC, Baron R. Cbl associates with Pyk2 and Src to regulate src kinase activity, avp3 integrin-mediated signaling, cell adhesion, and osteoclast motility. J Cell Biol. 2001;152:181–195. doi: 10.1083/jcb.152.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugatani T, Alvarez U, Hruska KA. PTEN regulates RANKL- and osteopontin-stimulated signal transduction during osteoclast differentiation and cell motility. J Biol Chem. 2003;278:5001–5008. doi: 10.1074/jbc.M209299200. [DOI] [PubMed] [Google Scholar]
- Takayanagi H. Mechanistic insight into osteoclast differentiation in osteoimmunology. J Mol Med. 2005;83:170–179. doi: 10.1007/s00109-004-0612-6. [DOI] [PubMed] [Google Scholar]
- Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue JI, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889–901. doi: 10.1016/s1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- Takeshita S, Namba N, Zhao JJ, Genant HK, Silva MJ, Brodt MD, Helgason CD, Kalesnikoff J, Rauh MJ, Humphries RK, et al. SHIP-deficient mice are severely osteopetrotic due to increased numbers of hyper-resorptive osteoclasts. Nat Med. 2002;8:943–949. doi: 10.1038/nm752. [DOI] [PubMed] [Google Scholar]
- Teitelbaum SL. Bone resorption by osteoclasts. Science. 2000;289:1504–1508. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- Teitelbaum SL, Ross P. Genetic regulation of osteoclast development and function. Nat Rev Genet. 2003;4:638–649. doi: 10.1038/nrg1122. [DOI] [PubMed] [Google Scholar]
- Vyas P, Norris FA, Joseph R, Majerus PW, Orkin SH. Inositol polyphosphate 4-phosphatase type I regulates cell growth downstream of transcription factor GATA-1. Proc Natl Acad Sci USA. 2000;97:13696–13701. doi: 10.1073/pnas.250476397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong BR, Besser D, Kim N, Arron JR, Vologodskaia M, Hanafusa H, Choi Y. TRANCE, a TNF family member, activates Akt/PKB through a signaling complex involving TRAF6 and c-src. Mol Cell. 1999;4:1041–1049. doi: 10.1016/s1097-2765(00)80232-4. [DOI] [PubMed] [Google Scholar]
- Zaidi M. Skeletal remodeling in health and disease. Nat Med. 2007;13:791–801. doi: 10.1038/nm1593. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.