

Abstract
BACKGROUND
To clarify the role of angiotensin II (Ang II) in insulin-induced arteriosclerosis, we examined the effects of Ang II on insulin-induced mitogen-activated protein (MAP) kinase activation and cellular hypertrophy in rat vascular smooth muscle cells (VSMCs).
METHODS
Phosphorylated MAP kinases were detected with western blot analysis. Cellular hypertrophy and glucose uptake were evaluated from incorporation of [3H]-labeled-leucine and -deoxy-D-glucose, respectively. Cell sizes were measured by Coulter counter.
RESULTS
While Ang II (100 nmol/l, 18 h) augmented cellular hypertrophy by insulin (10 nmol/l, 24 h), insulin alone did not affect hypertrophy without Ang II pretreatment. Insulin increased p38MAP kinase and c-Jun N-terminal kinase (JNK) phosphorylation; in the presence of Ang II, p38MAP kinase, and JNK were further activated by insulin. Treatment of a p38MAP kinase inhibitor, SB203580 (10 μmol/l), and a JNK inhibitor, SP600125 (20 μmol/l), abrogated the [3H]-leucine incorporation by insulin in the presence of Ang II. Both the Ang II receptor blocker, RNH-6270 (100 nmol/l), and an antioxidant, ebselen (40 μmol/l), inhibited vascular cell hypertrophy. Specific depletion of insulin receptor substrate-1 with small interfering RNA increased [3H]-leucine incorporation by insulin (10 nmol/l, 24 h); pretreatment with Ang II attenuated insulin (10 nmol/l, 30 min)-induced glucose uptake.
CONCLUSIONS
Ang II attenuates insulin-stimulated glucose uptake and enhances vascular cell hypertrophy via oxidative stress- and MAP kinase-mediated pathways in VSMCs. Ang II may also cause insulin signaling to diverge from glucose metabolism into vascular remodeling, affecting insulin-induced arteriosclerosis in hypertension.
Keywords: angiotensin II, blood pressure, hypertension, insulin resistance, oxidative stress, signal transduction, vascular smooth muscle cell
Insulin activates biaxial signaling pathways; the insulin receptor substrate (IRS)-1/Akt pathway, which mainly regulates glucose metabolism, and the mitogen-activated protein (MAP) kinase pathway, which induces cell growth, hypertrophy, and fibrosis in various cell types.1,2 Insulin regulates homeostasis of cellular functions and glucose metabolism by maintaining the balance between these signaling pathways.3 Insulin resistance is considered to occur when insulin signals are not transmitted to the glucose metabolic pathway, resulting in increased insulin concentration to maintain normal blood glucose levels.4 Therefore, patients with type 2 diabetes mellitus, which is often characterized by insulin resistance, usually show hyperinsulinemia. These patients also show proliferative and hypertrophic vascular changes, arteriosclerosis and finally, cardiovascular events.5 The molecular mechanism of insulin resistance is reportedly considered to occur via serine phosphorylation of IRS-1 or its degradation.2,6,7 However, in a state of insulin resistance, effects of insulin on hypertrophic change via MAP kinase activation, independently of IRS-1, have not been clarified yet.
Angiotensin (Ang) II not only regulates vascular constriction, cell proliferation, and hypertrophy, but also is deeply involved in the onset and progression of type 2 diabetes mellitus.8-10 Clinical studies have shown Ang II type 1 (AT1) receptor blockers (ARBs) to reduce the incidence of new-onset type 2 diabetes mellitus and improve insulin resistance,8,9 indicating that Ang II affects insulin signaling. It is also reported that ARBs prevent arteriosclerosis in diabetes patients, independently of systemic glucose metabolism.10
We have evaluated how insulin signaling is affected by Ang II in vasculature, and have partially clarified the molecular mechanism of vascular insulin resistance.6 Ang II induces IRS-1 degradation, subsequently attenuates signal transduction, and finally inhibits glucose metabolism in vascular smooth muscle cells (VSMCs). However, systemic glucose metabolism is mostly regulated in skeletal muscle, adipose tissue and liver.11 Therefore, vasculature’s role in glucose metabolism is considered to be minor, and the role of insulin signaling in vasculature is still controversial. Considering the evidence mentioned above, we hypothesized that Ang II reduces glucose metabolic insulin signals via IRS-1 depletion, causing diversion of insulin signals into the cell hypertrophic pathway mediated by MAP kinase activation in VSMCs. To test this hypothesis, we examined the effects of Ang II on the VSMC insulin signaling pathways, including IRS-1-Akt and MAP kinase, glucose metabolism and hypertrophic changes.
METHODS
Materials
Anti-IRS-1 antibody was purchased from BD Biosciences (Franklin Lakes, NJ). Antibodies to phospho-extracellular regulated kinase (ERK) 1/2, phospho-p38MAP kinase, phospho-c-Jun N-terminal kinase (JNK), phospho-Akt, ERK 1/2, p38MAP kinase, JNK, and Akt were purchased from Cell Signaling Technology (Beverly, MA). RNH-6270 (an active olmesartan metabolite) was provided by Daiichi Sankyo (Tokyo, Japan). All other chemicals and reagents, including insulin, Ang II and Dulbecco’s modified Eagle’s medium (DMEM) with 25 mmol/l Hepes and 4.5 g/l glucose, came from Sigma-Aldrich (St Louis, MO).
Cell culture
VSMCs were isolated, using enzymatic digestion, from thoracic aortae of 4-week-old, male Sprague–Dawley rats, as previously described.12 All experimental procedures were performed according to the guidelines for the care and use of animals established by Kagawa University. Cells were grown in DMEM supplemented with 10% fetal bovine serum (HyClone, Logan, UT), penicillin (100 U/ml, Life Technologies, Carlsbad, CA) and streptomycin (100 μg/ml, Life Technologies) at 37°C under 5% CO2/95% air in a humidified incubator. VSMCs were used for experiments between their third and eight passages.
Small interfering RNA (siRNA) transfection experiments
The method of transfecting of VSMCs with siRNA was previously described.13 Stealth Select RNAi pre-designed against IRS-1 was synthesized by Life Technologies. The negative control, nontargeting scrambled siRNA was purchased from Life Technologies. The VSMCs were transfected with Lipofectamine 2000 (Life Technologies) and 30 pmol of siRNA for 48 h, according to the manufacturer’s protocol.
Western blot analysis
Cells at 80–90% confluence were made quiescent by incubation with DMEM containing 0.1% fetal bovine serum for 24 h. Cells were stimulated with agonists at 37°C in serum-free DMEM, lysed as described previously;6 solubilized proteins were isolated by centrifugation and quantified by the Bradford assay. Proteins were separated using SDS-polyacrylamide gel electrophoresis (6% polyacrylamide gel for IRS-1 and 10% gel for MAP kinase and Akt), and transferred to nitrocellulose membranes. After blocking, blots were incubated with primary antibodies; proteins were detected by enhanced chemiluminescence (GE Healthcare, Buckinghamshire, UK). To confirm equal protein loading, each membrane was reprobed with anti-β-actin antibody (Sigma-Aldrich). Band intensities were quantified by immunoblot densitometry, using NIH ImageJ software.
Dihydroethidium staining
The oxidative fluorescence dihydroethidium was used to evaluate intracellular O2− levels as described previously.14
[3H]-leucine incorporation and glucose uptake
Protein synthesis was assessed by measuring [3H]-leucine incorporation. In brief, quiescent VSMCs were cultured in 6-well plates, then treated with insulin (2–100 nmol/l) containing 1 μCi/ml [3H]-leucine for 24 h. After labeling, cells were washed twice with phosphate-buffered saline and twice with ice-cold 5% trichloroacetic acid to remove the unincorporated [3H]-leucine, solubilized in 500 μl of 0.25 mol/l NaOH containing 0.1% SDS and neutralized. Aliquots of the samples were added to 10 ml of scintillation fluid and counted in a scintillation counter.
Glucose uptake was determined as previously described.15 Serum-starved VSMCs, with or without Ang II stimulation, were incubated in Krebs-Ringer-HEPES buffer for 2 h. Cells were incubated with insulin (10 nmol/l) for 30 min, and 0.2 mmol/l 2-deoxy-D-glucose containing 1 μCi/ml 2-deoxy-D-[3H] glucose was added for another 30 min. Cells were disrupted, and amounts of labeled glucose that were taken up were determined by scintillation counting.
Cell volume measurement
Cell volume measurements were performed by electronic sizing with a Coulter Counter Multisizer 3 (Beckman-Coulter, Miami, FL). Briefly, cells were washed in phosphate-buffered saline and lifted by incubation with 0.25% trypsin-EDTA (Life Technologies). Trypsin was inactivated adding of an equal volume of medium; cells were pelleted by brief centrifugation and resuspended in Isoton II Diluent (Beckman-Coulter). The range of cell sizes that could be effectively evaluated using the aperture was 10–100 μm. Cell size measurements were obtained every 10 s, and each measurement contained an average of 2,000 cells.
Statistical analysis
Values are presented as means ± s.e. Multiple-group comparisons were made using one-way or two-way analyses of variance (ANOVA), followed by Bonferroni’s test. Values of P < 0.05 were considered statistically significant.
RESULTS
Effect of IRS-1 depletion using siRNA on Akt phosphorylation and protein synthesis induced by insulin
To investigate the contribution of the IRS-1 depletion on insulin signaling pathway in VSMCs, we knocked down IRS-1 using siRNA. Reduction in the IRS-1 protein levels attenuated insulin (10 nmol/l, 5 min)-induced Akt phosphorylation in VSMCs (Figure 1a). In addition, siRNA of IRS-1 augmented insulin-induced p38MAP kinase phosphorylation (see Supplementary Figure S1 online). We also measured [3H]-leucine incorporation as an index of protein synthesis in VSMCs. Insulin (10 nmol/l, 24 h) significantly increased [3H]-leucine incorporation in IRS-1 siRNA-transfected VSMCs (Figure 1b). However, in VSMCs transfected with scrambled siRNA, insulin had no significant effect on leucine incorporation. These results suggest that IRS-1 affects the insulin signaling pathway, and IRS-1 depletion augments insulin-induced hypertrophic alterations in VSMCs.
Figure 1.
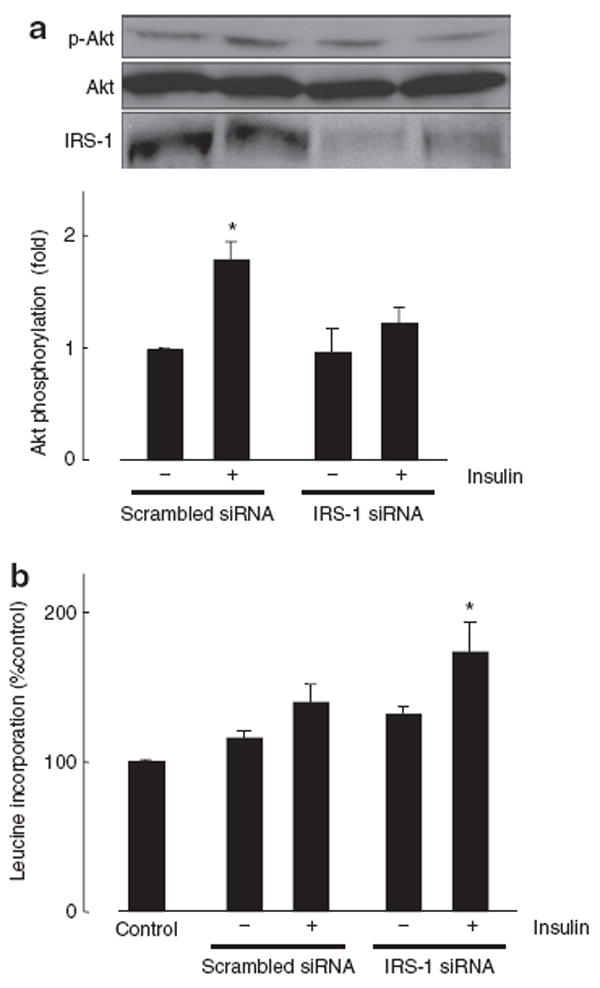
Effect of IRS-1 siRNA on Akt phosphorylation and protein synthesis. (a) VSMCs were transfected with IRS-1 siRNA or scrambled siRNA for 48 h and then exposed to vehicle or 10 nmol/l insulin for 5 min. Western blotting with anti-IRS-1, phospho-Akt or Akt antibody was performed. (b) VSMCs transfected with IRS-1 siRNA or scrambled siRNA were exposed to vehicle or 10 nmol/l insulin for 24 h. Protein synthesis was measured by [3H]-leucine incorporation. Data represent mean ± s.e. (n = 6), expressed as fold change compared with unstimulated cells. *P < 0.05 vs. control VSMCs. IRS-1, insulin receptor substrate-1; siRNA, small interfering RNA; VSMCs, vascular smooth muscle cells.
Effects of Ang II pretreatment on insulin-induced phosphorylation of MAP kinase
To evaluate effects of Ang II pretreatment on the insulin signaling pathway, we measured MAP kinase phosphorylation in VSMCs using western blot analysis. Concentration and pretreatment duration of Ang II (100 nmol/l, 18 h) were determined on the basis of results from previous studies in VSMCs.6 Insulin (10 nmol/l, 5 min) significantly increased p38MAP kinase phosphorylation in Ang II (100 nmol/l, 18 h)-pretreated VSMCs (Figure 2a). Pretreatment with Ang II augmented insulin-induced p38MAP kinase phosphorylation (Figure 2b), and RNH-6270 (100 nmol/l), an ARB, completely inhibited Ang II-augmented p38 MAP kinase phosphorylation induced by insulin (Figure 2c). Similarly, insulin-induced JNK phosphorylation was higher in Ang II-pretreated VSMCs (Figures 2d,e). However, insulin-induced ERK 1/2 phosphorylation was not affected by Ang II pretreatment (Figures 2f,g). On the other hand, pretreatment with Ang II (100 nmol/l, 18 h) attenuated Akt phosphorylation induced by insulin (10 nmol/l, 5 min) in VSMCs (see Supplementary Figure S2 online). In addition, we used dihydroethidium fluorescence to evaluate reactive oxygen species (ROS) formation. We confirmed that Ang II augmented ROS formation in a time-dependent manner (see Supplementary Figure S3 online).
Figure 2.
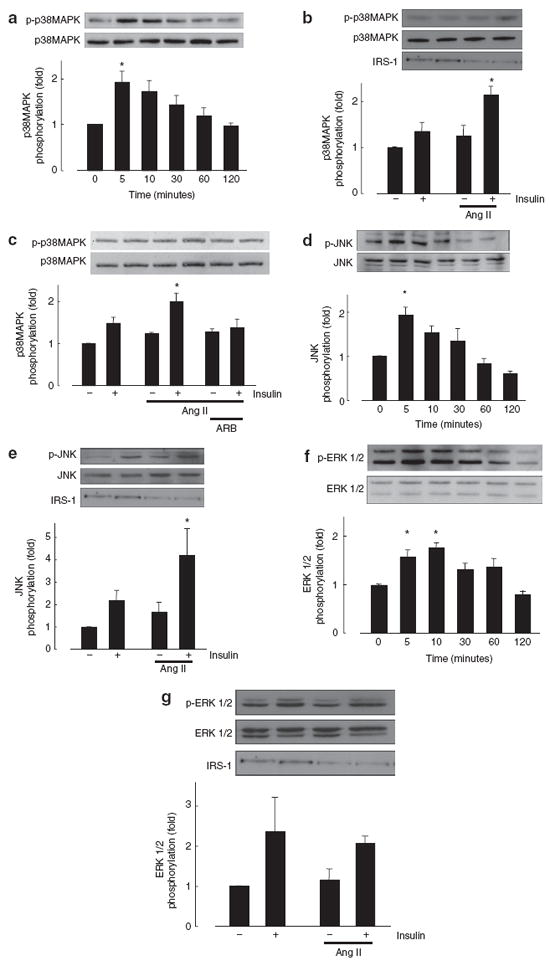
Effect of Ang II on insulin-induced MAP kinase phosphorylation. (a,d,f) VSMCs were pretreated with Ang II (100 nmol/l) for 18 h and then exposed to 10 nmol/l insulin for the indicated times. (b,c,e,g) VSMCs were pretreated with vehicle or Ang II (100 nmol/l, 18 h) and then exposed to insulin (10 nmol/l, 5 min). Western blotting was performed with anti-IRS-1, phospho-p38MAP kinase, p38MAP kinase (a,b,c), phospho-JNK, JNK (d,e), and phospho-ERK 1/2, ERK 1/2 (f,g) antibodies. Data represent mean ± s.e. (n = 5), expressed as fold change compared with unstimulated cells. *P < 0.05 vs. control. Ang II, angiotensin II; ERK, extracellular regulated kinase; IRS-1, insulin receptor substrate-1; JNK, c-Jun N-terminal kinase; MAP, mitogen-activated protein; VSMCs, vascular smooth muscle cells.
Effects of Ang II pretreatment on insulin-induced protein synthesis and cell volume
The MAP kinases, such as p38MAP kinase and JNK, are involved in protein synthesis and cell hypertrophy.16 We therefore measured [3H]-leucine incorporation in VSMCs pretreated with Ang II. Insulin (2–100 nmol/l) alone did not affect [3H]-leucine incorporation in VSMCs (Figure 3a). Pretreatment with Ang II (100 nmol/l, 18 h) significantly augmented insulin (10–100 nmol/l, 24 h)-induced leucine incorporation in VSMCs. To determine the role of MAP kinase activation in insulin-induced protein synthesis, we investigated effects of inhibitors of p38MAP kinase, JNK, and ERK kinase on insulin-induced [3H]-leucine incorporation in VSMCs pretreated with Ang II. Preincubation before insulin (10 nmol/l) treatment with SB203580 (10 μmol/l) and SP600125 (20 μmol/l)—inhibitors of p38MAP kinase and JNK, respectively—abolished insulin-induced leucine incorporation in VSMCs pretreated with Ang II (Figure 3b); however, PD98059 (10 μmol/l), an ERK kinase inhibitor, decreased the leucine incorporation slightly but not significantly. Wortmannin (100 nmol/l), a phosphatidylinositol 3-kinase inhibitor, did not affect leucine incorporation induced by insulin. Use of RNH-6270 (100 nmol/l) or ebselen (40 μmol/l), an antioxidant, completely abolished the insulin-induced leucine incorporation in Ang II-pretreated VSMCs.
Figure 3.
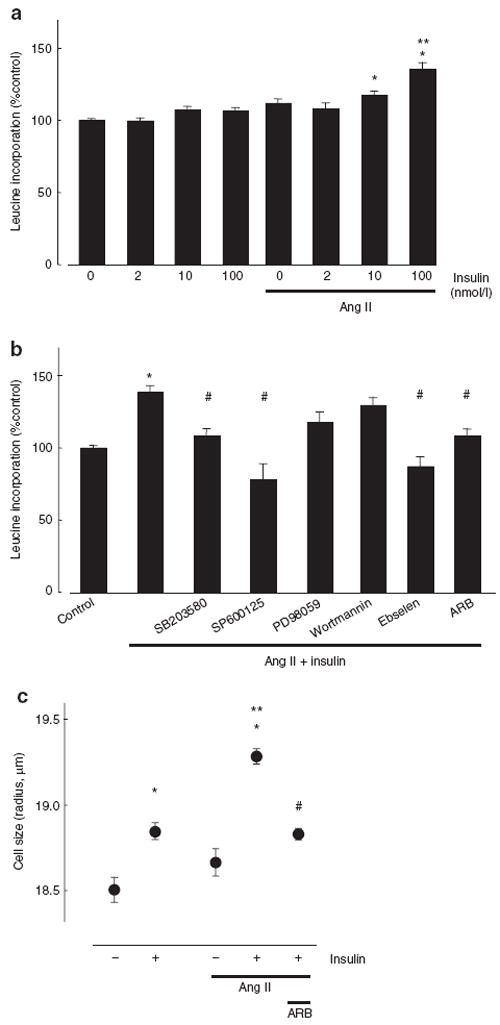
Effect of Ang II on insulin-induced protein synthesis and cell hypertrophy. (a) VSMCs were pretreated with Ang II (100 nmol/l) for 18 h and then exposed to with the indicated concentrations of insulin for 24 h. (b) Cells were pretreated with MAP kinase inhibitors (SB203580; 10 μmol/l, SP600125; 20 μmol/l, PD98059; 10 μmol/l), wortmannin (100 nmol/l) or an antioxidant (ebselen; 40 μmol/l) before insulin (10 nmol/l) incubation (30 min). The ARB (RNH-6270; 100 nmol/l) pretreated before Ang II incubation (30 min). Protein synthesis was measured by [3H]-leucine incorporation. (c) Cell volume was measured by a Coulter Cell Multisizer. Data represent mean ± s.e. (n = 6), expressed as fold change compared with unstimulated cells. Two-way ANOVA showed that the dose-dependent increase of [3H]-leucine incorporation was significantly higher in Ang II-pretreated VSMCs than in control VSMCs. *P < 0.05 vs. control VSMCs. **P < 0.05 vs. Ang II-pretreated VSMCs without insulin. #P < 0.05 vs. insulin treatment in Ang II-pretreated VSMCs. Ang II, angiotensin II; ARB, Ang II type 1 receptor blockers; MAP, mitogen-activated protein; VSMCs, vascular smooth muscle cells.
We measured cell volume using a Coulter Cell Multisizer. Insulin (10 nmol/l, 24 h) significantly increased VSMC size (Figure 3c). Pretreatment with Ang II augmented insulin-induced cell enlargement compared with cells without Ang II pretreatment. The ARB treatment inhibited insulin-induced cell enlargement in Ang II-pretreated VSMCs. These results suggest that Ang II pretreatment augments insulin-induced protein synthesis and cell hypertrophy via AT1 receptors, in oxidative stress-, p38MAP kinase-, and JNK-dependent manners.
Effect of Ang II pretreatment on insulin-induced glucose uptake
We measured the uptake of 2-deoxy-[3H]-glucose into cells as an index of glucose metabolism. Although 10 nmol/l of insulin stimulated glucose uptake in VSMCs, Ang II pretreatment decreased insulin-induced glucose uptake (Figure 4). The ARB completely reversed Ang II-attenuated glucose metabolism. Thus, IRS-1 downregulation by Ang II shifts the insulin-stimulated cellular response from glucose metabolism to cell hypertrophy.
Figure 4.
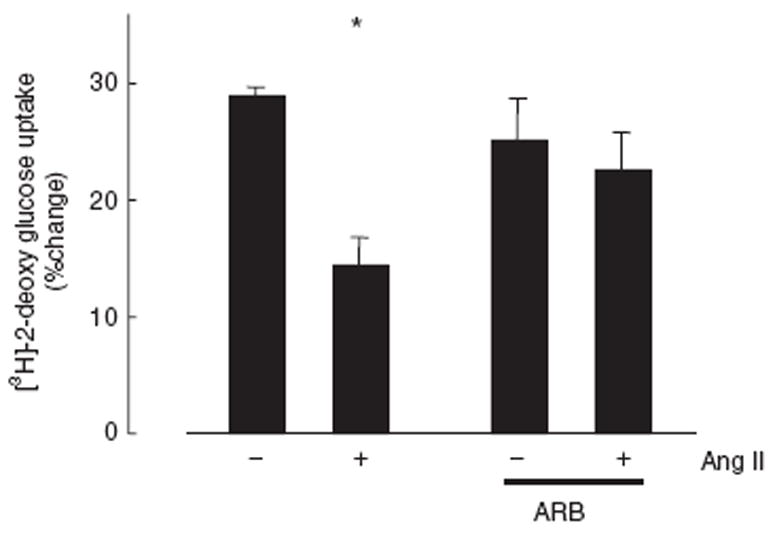
Effect of Ang II on insulin-induced glucose uptake. VSMCs were treated with 100 nmol/l Ang II or vehicle for 18 h in the presence or absence of an ARB, exposed to 10 nmol/l insulin for 30 min, and 2-deoxy-D-[3H]-glucose uptake measured. Data represent mean ± s.e. (n = 10) of percent change in uptake. *P < 0.05 vs. control. Ang II, angiotensin II; ARB, Ang II type 1 receptor blockers; VSMCs, vascular smooth muscle cells.
DISCUSSION
Ang II aggravates insulin resistance and vascular remodeling in various conditions in cardiovascular cells,6 experimental animal models17 and patients;8 however, molecular mechanisms are unclear. In this study, we showed vascular cell hypertrophy induced by insulin in Ang II-pretreated VSMCs. We previously demonstrated that Ang II decreases IRS-1 protein expression, and consequently attenuates glucose metabolism mediated via the phosphatidylinositol 3 kinase–Akt signaling pathway.6 Although we have already demonstrated the crosstalk between MAP kinase and Akt pathways,18 the present study advances these previous findings by showing that Ang II pretreatment shifts the insulin signaling pathway from glucose metabolism to cell hypertrophy and vascular remodeling (see Supplementary Figure S4 online). Hypertrophy of VSMCs is seen in various states of arteriosclerosis;19 and vascular remodeling, including arteriosclerosis and cardiovascular disease, is a major diabetic complication. Our results provide a possible molecular mechanism of vascular remodeling induced by Ang II and insulin in diabetes mellitus.
Insulin receptors are expressed in vasculature, including VSMCs. Although insulin exerts numerous effects via insulin receptors,20 its physiological and pathological effects on vascular function are still controversial. Insulin regulates systemic glucose metabolism through glucose uptake into cells. In vasculature, although insulin increases intracellular glucose uptake, the glucose store in VSMCs is little compared to the whole body, thus, physiological effect of insulin on vascular glucose metabolism might be minimal. In addition, previous studies have revealed that insulin not only regulates glucose levels but also induces proatherosclerotic changes.21 In this study, treatment with insulin alone induced MAP kinase activation, protein synthesis and enlargement of cell size in VSMCs, and more importantly, pretreatment with Ang II augmented proatherosclerotic changes induced by insulin via IRS-1 degradation and MAP kinase activation. Because incubation with Ang II alone did not induce these changes seen in the presence of both insulin and Ang II, IRS-1 degradation, MAP kinase activation and shift of insulin signaling could be necessary for insulin-induced proatherosclerotic changes in Ang II-pretreated VSMCs. Furthermore, as the insulin concentration used in this study was 2–100 nmol/l, while the physiological concentration is considered to be in the low nmol/l range in rodents,22 we assumed that the hypertrophic effects occurred in this hyperinsulinemic setting, which is often seen in patients with insulin resistance. Taken together, these facts suggested that high insulin concentrations augment arteriosclerosis in patients with activation of renin–angiotensin system.
Clinical trials have revealed that angiotensin-converting enzyme inhibitors23 and ARBs10 prevent arteriosclerosis and the onset of cardiovascular diseases in patients with type 2 diabetes mellitus, partially independently of their blood pressure-lowering effects. In addition, it has been already reported that ARBs attenuate arteriosclerotic changes without improving systemic glucose levels.10 Although Ang II regulates blood pressure via vascular tone and modulation of water and electrolytes, it is not a major effector of glucose metabolism. Insulin, on the other hand, mainly regulates blood glucose via intracellular uptake of glucose, and insulin-induced vascular remodeling is still controversial in patients. Based on our previous and present results, we considered that Ang II leads to insulin signal diversion and vascular remodeling.
Oxidative stress is involved in the development of diabetic cardiovascular complications.24 We have demonstrated that Ang II-induced oxidative stress attenuates glucose metabolism in VSMCs.6 As shown in see Supplementary Figure S3 online and Figure 3b, Ang II augmented ROS formation, and an antioxidant completely inhibited insulin-induced hypertrophic effect in Ang II-pretreated VSMCs. In addition, p38MAP kinase and JNK, which were augmented in Ang II-pretreated VSMCs in this study, are known to be ROS sensitive. While oxidative stress directly modulates intracellular signaling pathways, with consequent atherosclerotic changes in diabetes mellitus, insulin diversion mediated by oxidative stress may induce further hypertrophic alterations.
In conclusion, Ang II attenuates insulin-induced glucose uptake and enhances vascular cell hypertrophy via an oxidative stress-mediated MAP kinase pathway in VSMCs. These results make clear that Ang II induces VSMC insulin resistance. We define VSMC insulin resistance to be an insulin-induced vascular state in which signal recognition required for glucose metabolism is incomplete, accompanying progressive remodeling. In addition, recent reports suggested that vascular insulin resistance is considered to be involved in atherogenic modulation and subsequent cardiovascular events.25 Thus, prevention of vascular insulin resistance may be a therapeutic target for cardiovascular complications in diabetic patients. It is highly probable that Ang II may shift insulin signaling from glucose metabolism to vascular remodeling, which may be involved in high concentration of insulin-induced arteriosclerosis in diabetic patients with hypertension.
Supplementary Material
Acknowledgments
We are grateful to Daiichi Sankyo Co. for supplying RNH-6270, active metabolite of olmesartan. This work was supported in part by a grant-in-aid for scientific research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (20590253 and 22790792) and Sanju Alumni Research Grant.
Footnotes
Disclosure: The authors declared no conflict of interest.
Supplementary material is linked to the online version of the paper at http://www.nature.com/ajh
References
- 1.De Meyts P, Christoffersen CT, Ursø B, Wallach B, Grønskov K, Yakushiji F, Shymko RM. Role of the time factor in signaling specificity: application to mitogenic and metabolic signaling by the insulin and insulin-like growth factor-I receptor tyrosine kinases. Metabolism. 1995;44:2–11. doi: 10.1016/0026-0495(95)90214-7. [DOI] [PubMed] [Google Scholar]
- 2.White MF. IRS proteins and the common path to diabetes. Am J Physiol Endocrinol Metab. 2002;283:E413–E422. doi: 10.1152/ajpendo.00514.2001. [DOI] [PubMed] [Google Scholar]
- 3.Zhang D, Bar-Eli M, Meloche S, Brodt P. Dual regulation of MMP-2 expression by the type 1 insulin-like growth factor receptor: the phosphatidylinositol 3-kinase/Akt and Raf/ERK pathways transmit opposing signals. J Biol Chem. 2004;279:19683–19690. doi: 10.1074/jbc.M313145200. [DOI] [PubMed] [Google Scholar]
- 4.Reaven GM. Banting lecture 1988. Role of insulin resistance in human disease. Diabetes. 1988;37:1595–1607. doi: 10.2337/diab.37.12.1595. [DOI] [PubMed] [Google Scholar]
- 5.Howard G, O’Leary DH, Zaccaro D, Haffner S, Rewers M, Hamman R, Selby JV, Saad MF, Savage P, Bergman R. Insulin sensitivity and atherosclerosis. The Insulin Resistance Atherosclerosis Study (IRAS) Investigators. Circulation. 1996;93:1809–1817. doi: 10.1161/01.cir.93.10.1809. [DOI] [PubMed] [Google Scholar]
- 6.Taniyama Y, Hitomi H, Shah A, Alexander RW, Griendling KK. Mechanisms of reactive oxygen species-dependent downregulation of insulin receptor substrate-1 by angiotensin II. Arterioscler Thromb Vasc Biol. 2005;25:1142–1147. doi: 10.1161/01.ATV.0000164313.17167.df. [DOI] [PubMed] [Google Scholar]
- 7.White MF. Regulating insulin signaling and beta-cell function through IRS proteins. Can J Physiol Pharmacol. 2006;84:725–737. doi: 10.1139/y06-008. [DOI] [PubMed] [Google Scholar]
- 8.Dahlöf B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, Fyhrquist F, Ibsen H, Kristiansson K, Lederballe-Pedersen O, Lindholm LH, Nieminen MS, Omvik P, Oparil S, Wedel H. LIFE Study Group. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet. 2002;359:995–1003. doi: 10.1016/S0140-6736(02)08089-3. [DOI] [PubMed] [Google Scholar]
- 9.Ogihara T, Nakao K, Fukui T, Fukiyama K, Ueshima K, Oba K, Sato T, Saruta T. Candesartan Antihypertensive Survival Evaluation in Japan Trial Group. Effects of candesartan compared with amlodipine in hypertensive patients with high cardiovascular risks: candesartan antihypertensive survival evaluation in Japan trial. Hypertension. 2008;51:393–398. doi: 10.1161/HYPERTENSIONAHA.107.098475. [DOI] [PubMed] [Google Scholar]
- 10.Lindholm LH, Ibsen H, Dahlöf B, Devereux RB, Beevers G, de Faire U, Fyhrquist F, Julius S, Kjeldsen SE, Kristiansson K, Lederballe-Pedersen O, Nieminen MS, Omvik P, Oparil S, Wedel H, Aurup P, Edelman J, Snapinn S. LIFE Study Group. Cardiovascular morbidity and mortality in patients with diabetes in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet. 2002;359:1004–1010. doi: 10.1016/S0140-6736(02)08090-X. [DOI] [PubMed] [Google Scholar]
- 11.Koopmans SJ, Mandarino L, DeFronzo RA. Time course of insulin action on tissue-specific intracellular glucose metabolism in normal rats. Am J Physiol. 1998;274:E642–E650. doi: 10.1152/ajpendo.1998.274.4.E642. [DOI] [PubMed] [Google Scholar]
- 12.Hitomi H, Fukui T, Moriwaki K, Matsubara K, Sun GP, Rahman M, Nishiyama A, Kiyomoto H, Kimura S, Ohmori K, Abe Y, Kohno M. Synergistic effect of mechanical stretch and angiotensin II on superoxide production via NADPH oxidase in vascular smooth muscle cells. J Hypertens. 2006;24:1089–1095. doi: 10.1097/01.hjh.0000226199.51805.88. [DOI] [PubMed] [Google Scholar]
- 13.Moriwaki K, Kiyomoto H, Hitomi H, Ihara G, Kaifu K, Matsubara K, Hara T, Kondo N, Ohmori K, Nishiyama A, Fukui T, Kohno M. Interferon-gamma enhances superoxide production in human mesangial cells via the JAK-STAT pathway. Kidney Int. 2006;70:788–793. doi: 10.1038/sj.ki.5001639. [DOI] [PubMed] [Google Scholar]
- 14.Miyata K, Rahman M, Shokoji T, Nagai Y, Zhang GX, Sun GP, Kimura S, Yukimura T, Kiyomoto H, Kohno M, Abe Y, Nishiyama A. Aldosterone stimulates reactive oxygen species production through activation of NADPH oxidase in rat mesangial cells. J Am Soc Nephrol. 2005;16:2906–2912. doi: 10.1681/ASN.2005040390. [DOI] [PubMed] [Google Scholar]
- 15.Hitomi H, Kiyomoto H, Nishiyama A, Hara T, Moriwaki K, Kaifu K, Ihara G, Fujita Y, Ugawa T, Kohno M. Aldosterone suppresses insulin signaling via the downregulation of insulin receptor substrate-1 in vascular smooth muscle cells. Hypertension. 2007;50:750–755. doi: 10.1161/HYPERTENSIONAHA.107.093955. [DOI] [PubMed] [Google Scholar]
- 16.Yoshizumi M, Tsuchiya K, Suzaki Y, Kirima K, Kyaw M, Moon JH, Terao J, Tamaki T. Quercetin glucuronide prevents VSMC hypertrophy by angiotensin II via the inhibition of JNK and AP-1 signaling pathway. Biochem Biophys Res Commun. 2002;293:1458–1465. doi: 10.1016/S0006-291X(02)00407-2. [DOI] [PubMed] [Google Scholar]
- 17.Henriksen EJ, Jacob S, Kinnick TR, Teachey MK, Krekler M. Selective angiotensin II receptor receptor antagonism reduces insulin resistance in obese Zucker rats. Hypertension. 2001;38:884–890. doi: 10.1161/hy1101.092970. [DOI] [PubMed] [Google Scholar]
- 18.Taniyama Y, Ushio-Fukai M, Hitomi H, Rocic P, Kingsley MJ, Pfahnl C, Weber DS, Alexander RW, Griendling KK. Role of p38 MAPK and MAPKAPK-2 in angiotensin II-induced Akt activation in vascular smooth muscle cells. Am J Physiol, Cell Physiol. 2004;287:C494–C499. doi: 10.1152/ajpcell.00439.2003. [DOI] [PubMed] [Google Scholar]
- 19.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;362:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 20.Nigro J, Osman N, Dart AM, Little PJ. Insulin resistance and atherosclerosis. Endocr Rev. 2006;27:242–259. doi: 10.1210/er.2005-0007. [DOI] [PubMed] [Google Scholar]
- 21.Stolar MW. Atherosclerosis in diabetes: the role of hyperinsulinemia. Metabolism. 1988;37:1–9. doi: 10.1016/0026-0495(88)90180-1. [DOI] [PubMed] [Google Scholar]
- 22.Nagai Y, Ichihara A, Nakano D, Kimura S, Pelisch N, Fujisawa Y, Hitomi H, Hosomi N, Kiyomoto H, Kohno M, Ito H, Nishiyama A. Possible contribution of the non-proteolytic activation of prorenin to the development of insulin resistance in fructose-fed rats. Exp Physiol. 2009;94:1016–1023. doi: 10.1113/expphysiol.2009.048108. [DOI] [PubMed] [Google Scholar]
- 23.HOPE Study Investigators. Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy. Heart Outcomes Prevention Evaluation Study Investigators. Lancet. 2000;355:253–259. [PubMed] [Google Scholar]
- 24.Jay D, Hitomi H, Griendling KK. Oxidative stress and diabetic cardiovascular complications. Free Radic Biol Med. 2006;40:183–192. doi: 10.1016/j.freeradbiomed.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 25.Schulman IH, Zhou MS. Vascular insulin resistance: a potential link between cardiovascular and metabolic diseases. Curr Hypertens Rep. 2009;11:48–55. doi: 10.1007/s11906-009-0010-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.