

Abstract
Cystathionine β-synthase catalyzes the condensation of serine and homocysteine to yield cystathionine and is the single most common locus of mutations associated with homocystinuria. In this study, we have examined the kinetic consequences of a pair of linked patient mutations, P78R/K102N, that are housed in the catalytic core of the protein and compared it to the effects of the corresponding single mutations. The P78R mutation affords purification of a mixture of higher order oligomers, P78R-I, which resembles the mixed quaternary state associated with wild type enzyme. However, unlike wild type enzyme, P78R-I converts over time to P78R-II, which exists predominantly, as a full-length dimer. The specific activities of the K102N, P78R-I and P78R-II mutants in the absence of AdoMet are ~3-, 9- and 3-fold lower than of wild-type enzyme and are stimulated 2.9-, 2.5- and 1.4-fold respectively by AdoMet. However, when linked, the specific activity of the resulting double mutant is comparable to that of wild-type enzyme but it is unresponsive to AdoMet, revealing that interactions between the two sites modulate the phenotype of the enzyme. Steady-state kinetic analysis for the double mutant reveals a sigmoidal dependence on homocysteine that is not observed with wild-type enzyme, which is ascribed to the mutation at the K102 locus and indicates changes in subunit interactions. Hydrogen-deuterium mass spectrometric analyses reveals that even in the absence of AdoMet, the double mutant is locked in an activated conformation that is observed for wild-type enzyme in the presence of AdoMet, providing a structural rationale for loss of this allosteric regulation. To our knowledge, this is the first example of mutations in the catalytic core of cystathionine β-synthase that result in failure of AdoMet-dependent regulation. Furthermore, analysis of individual single mutations has permitted for the first time, partial kinetic characterization of a full-length dimeric form of human cystathionine β-synthase.
Cystathionine β-synthase is a pyridoxal phosphate (PLP)1-dependent protein that catalyzes the condensation of serine and homocysteine to give cystathionine and is activated by the allosteric effector, AdoMet (1–3). It catalyzes the first step in the transsulfuration pathway connecting the methionine cycle to cysteine production. Mutations in cystathionine β-synthase are the single most common cause of hereditary hyperhomocysteinemia and over one hundred pathogenic mutations have been described so far (4). They represent a roadmap of residues that are important for the structural and/or functional integrity of the enzyme and characterization of these mutants have begun to provide invaluable insights into the organization and regulation of this unusual enzyme (5–9). The pathogenic mutations are dispersed over all three domains of the modular protein: the N-terminal heme domain, which is found in some but not all eukaryotic cystathionine β-synthases, the central active site domain where PLP is bound and the C-terminal regulatory domain, which is required for allosteric activation of the enzyme by AdoMet.
The purified human enzyme is a homooligomer with a subunit mass of 63 kDa and exists in multiple higher order oligomeric states ranging from a dimer to a 16-mer (10), which have not been purified and characterized separately to date (Figure 1). A hypersensitive site leads to the facile proteolytic separation between the catalytic core and the regulatory domain (11). The resulting truncated enzyme is a well-behaved dimer that is not prone to aggregation unlike the full-length enzyme. It has a subunit mass of 45 kDa and exhibits an ~4-fold higher kcat than the full-length form (9) but is unresponsive to AdoMet (12). The regulatory domain thus appears to be autoinhibitory and its cleavage alleviates inhibition of cystathionine β-synthase activity (11–13). Furthermore, truncation is accompanied by a change in the oligomerization state of the protein from tetramer to dimer. These results reveal a complex role for the C-terminal domain in mediating regulation of cystathionine β-synthase including intrasteric and allosteric regulation and in the oligomerization of the protein beyond the dimeric state.
Fig. 1.
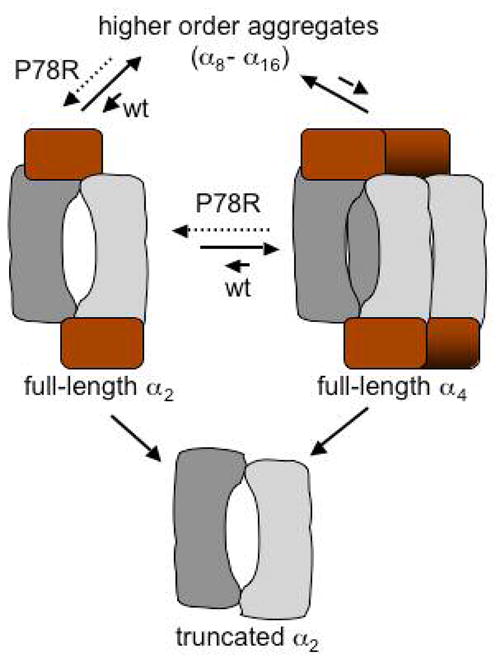
Model of the oligomeric heterogeneity seen with human cystathionine β-synthase. It is not known whether the equilibrium between the different oligomeric states involves dissociation into monomers and has been shown for simplicity, to involve either the dimer or the tetramer. The P78R mutation shifts the equilibrium towards the full-length dimer compared to wild type enzyme. Deletion of the regulatory domain leads to a truncated dimer.
A class of pathogenic mutations housed in the regulatory domain has been described that display a common and seemingly paradoxical phenotype (6, 9, 14, 15). When mimicked in vitro, these regulatory domain mutants are unresponsive to activation by AdoMet but exhibit robust enzyme activity comparable to or greater than that of wild type enzyme (6, 9). Mathematical modeling of methionine metabolism provides a rationale for the build up of homocysteine by the mere loss of allosteric activation of cystathionine β-synthase, particularly under conditions of methionine excess, confirming an important role for AdoMet-dependent regulation in intracellular homocysteine homeostasis (16).
The crystal structure of the catalytic core of cystathionine β-synthase has been reported and reveals the relative juxtaposition of the heme- and PLP-domains (17, 18). However, the structure of the regulatory domain and its interaction with the catalytic core are unknown. The C-terminal domain is comprised of a tandem repeat of two CBS domains, a β–α–β–β–α secondary structure motif that is found in diverse proteins in all three kingdoms of life and believed to function as an energy sensor (19, 20). A homology-modeled structure of the catalytic core has been generated based on the crystal structure of a CBS-domain containing protein from Methanobacter thermoautotrophicus (21). The only structural insights that are presently available on the mechanism of allosteric signal transduction between the regulatory and the catalytic domain derives from hydrogen exchange MS studies, which identified a single peptide in the CBS2 domain extending from residues 511–531 that exhibits a lower propensity for deuterium incorporation in the presence of AdoMet (21). In combination with other hydrogen exchange MS data, these studies have been used to build a docked model of the regulatory and catalytic domains (Figure 2) (21), which in the absence of a crystal structure for the full-length enzyme, provides a useful framework for interpreting the functional data (22). Although the regulatory domain is believed to house the binding site for AdoMet (20), the role of the CBS domains in cystathionine β-synthase, the protein from which this motif derives its name, is unknown.
Fig. 2.

Modeled structure of full-length cystathionine β-synthase dimer showing locations of the P78 and K102 residues. The structure was generated by docking the crystal structure of the catalytic core of cystathionine β-synthase (in tan) with the homology modeled structure of the regulatory domain (in scarlet) as described previously (21). The locations of P78 and K102 (in blue ball and stick representation) are indicated as are of the heme (in red) and PLP (in blue).
In this study, we report on the characterization of a linked patient nucleotide mutation, C233G/G306C, corresponding to P78R/K102N in the polypeptide, which are located in the catalytic core of the protein (Figure 2). These paternally inherited cis mutations have been described in three siblings who, like most homocystinurics, are compound heterozygotes, and have a maternally inherited E239K mutation in the other allele (23). Interestingly, the siblings display a wide range of clinical phenotypes from severe (in the two sisters) to benign (in the brother) despite sharing an identical genotype at this locus, which indicates possible gender-dependent modulation of the expression of cystathionine β-synthase-dependent homocystinuria. Previous studies of the P78R/K102N mutation were limited to cell lysates of Escherichia coli expressing recombinant cystathionine β-synthase (23). We have analyzed, using purified recombinant human cystathionine β-synthase, the biochemical penalties associated with the individual mutations to elucidate the contribution of each locus to the phenotype of the double mutant. The P78R mutation allowed for the first time, limited kinetic characterization of a full-length dimeric form of cystathionine β-synthase. Interestingly, interactions between the K102N and P78R mutations result in a compound phenotype that is similar to but also distinct from those of the single mutants. The double mutant is as active as the wild-type enzyme but is unresponsive to the allosteric activator, AdoMet. The loss of allosteric regulation is explained by hydrogen exchange MS data, which revealed that the linked mutation locks the protein in a conformation achieved by wild-type enzyme in the presence of AdoMet. To our knowledge, this is the first example of mutations in the catalytic core that confer AdoMet insensitivity, exhibit high basal activity and thus resemble a subset of regulatory domain mutations that have been described in homocystinuric patients.
Materials and Methods
Materials
Serine, IPTG, and D, L-homocysteine were purchased from Sigma. [14C]-Serine (158 mCi/mmol) was purchased from Amersham. GST Sepharose and Superdex™ 200 were purchased from Pharmacia. Deuterium oxide (100.00 atom % D) was purchased from Sigma. AdoMet 1,4-butanedisulfonate was a generous gift from Knoll Farmaceutici Spa (Milano, Italy). Thrombin was purchased from GenTrac Inc. Middleton, WI. Complete tablets (protease cocktail inhibitors) were purchased from Roche Diagnostics (Mannheim, Germany).
Generation of the CBS Mutants and Protein Purification
The single mutants K102N, P78R and the double mutant P78R/K102N, were generated by site-directed mutagenesis using the Quick Change Kit from Stratagene. The template for mutagenesis was pGEX4T1/hCBS containing wild-type human cystathionine β-synthase gene fused in-frame with glutathione S-transferase (24). The forward and the reverse primers that were employed were:
P78R: Sense: 5′:CCAAAATCTTGCGAGATATTCT
Antisense: 5′: AGAATATCTCGCAAGATTTTTGG
K102N: Sense: 5′: AAGTTCGGCCTGAACTGTGAGCTCTTGGC
Antisense: 5′: GCCAAGAGCTCACAGTTCAGGCCGAACTT
The double mutant, P78R/K102N, was constructed by introducing the P78R mutation using the single mutant, K102N as template, and the appropriate set of primers. The mutations were confirmed by nucleotide sequencing at the Genomics Core Facility at the University of Nebraska Lincoln.
Protein purification and enzyme assays
The mutant enzymes were expressed in E. coli and purified as glutathione S-transferase fusion proteins using recombinant expression systems essentially as described for wild-type enzyme (24). The cell extract was prepared and sonicated in 50 mM Tris HCl pH 8, supplemented with protease inhibitor cocktail tablets (Roche Diagnostics). One tablet was used for the 300 ml cell extract prepared from a 6 l culture.
Following partial proteolysis by thrombin (1 U/mg protein) to remove the GST tag, the K102N and P78R/K102N mutants were dialyzed to remove glutathione and purified by anion exchange chromatography as described previously (24). The P78R mutant was purified following thrombin digestion by gel filtration chromatography using a 90 × 3 cm Superdex TM 200 column eluted isocratically with 50 mM Tris HCl containing 0.1 M NaCl. Over 50 mg of ~90% pure protein was obtained from 6 l of culture for each mutant.
The enzyme activity was assayed under aerobic conditions using 30 mM [14C]-serine and 30 mM D, L-homocysteine as described previously (13). One unit of enzyme activity catalyzes the conversion of 1 μmol cystathionine min−1 at 37°C. Protein concentration was determined by the Bradford method using bovine serum albumin as a standard. The concentration of the stock homocysteine solution was determined immediately prior to the experiments by titration with 5,5′-dithiobis 2-nitrobenzoic acid and estimated using a value of ε412 of 13,600 M−1 cm −1.
Steady-state kinetic analysis was performed in the standard radiolabeled assay and initiated by addition of homocysteine. The reaction mixture was incubated for 30 min at 37°C and terminated by the addition of 200 μl of 10% trifluoroacetic acid. The radiolabeled product, [14C]-labeled cystathionine, was separated from [14C]-labeled serine as described previously (13). The kinetic sbehavior of some mutants (indicated in Table I) showed a sigmoidal rather than a hyperbolic dependence on substrate concentration and these data were fitted to the Hill equation to obtain the Hill coefficient.
Table 1.
Comparison of kinetic parameters of the single and double mutants with wild-type cystathionine β-synthase
| Enzyme | Wild type | P78R-I | P78R-II | K102N | K102N/P78R |
|---|---|---|---|---|---|
| S.A.a (+AdoMet) | 378 ± 17 | 47 ± 4 | 84 ± 2 | 185 ± 4 | 169 ± 9 |
| kcatb (+AdoMet) | 6.6 sec−1 | 0.8sec−1 | 1.4 sec−1 | 3.0 sec−1 | 3.0 sec−1 |
| S.A. (−AdoMet) | 178 ± 5 | 19 ± 2 | 62 ± 0.5 | 65 ± 9 | 169 ± 9 |
| kcat (−AdoMet) | 3.0 sec−1 | 0.3 sec−1 | 1.0 sec−1 | 1.0 sec−1 | 3.0 sec−1 |
| KMSer (mM) | 2.0 ± 0.3 | 1.9 ± 0.2 | 8 ± 3 | 3.8 ± 0.8 | 5.1 ± 0.8 |
| Hill Coefficientc | - | 2.4 ± 0.7 | - | - | - |
| KMHcy (mM) | 5.0 ± 0.9 | 5 ± 2 | 2.4 ± 0.5 | 10 ±1 | 4.4 ± 0.3 |
| Hill Coefficient | - | - | - | 3 ±1 | 3.0 ± 0.2 |
Specific activity is in units of μmol cystathionine formed h−1 mg−1 protein.
kcat is calculated based on the molecular mass of the monomer. Hill coefficients are reported only when a sigmoidal dependence on substrate concentration was observed. In all other instances, the KM values were obtained by Michaelis-Menten analysis of the data set.
Hydrogen-deuterium exchange mass spectrometry
These experiments were performed with wild type and the P78R/K102N forms of cystathionine β-synthase exactly as described previously (21). To correct for loss of deuterium incurred under the experimental conditions, the peptide mass was determined using equation 1 where D is the adjusted deuterium incorporation, m is the experimentally observed mass at a given time, m0% is the 0 percent or undeuterated
| Equation 1 |
control, m100% is the fully deuterated control and N is the total number of exchangeable amide protons (and excludes the N-terminal proton and any proline residues). To obtain the m0% value, 20 μg of protein was diluted 20-fold in the quench buffer followed rapidly by the addition of an equal amount of exchange buffer in D2O and digested with pepsin as described above. Thus, the final composition of deuterium in the quench and dilution solutions was identical for the samples and the corresponding m0% control.
To obtain the m100% value, a fully deuterated sample was prepared by incubating 20 μg of the protein overnight at room temperature in an 8 M guanidinium hydrochloride solution prepared in D2O. The protein sample was subsequently diluted 10-fold with 100 mM ammonium phosphate, pH 2.2 (at which concentration the denaturant does not interfere with proteolytic digestion), prior to pepsin treatment.
HPLC-ESI Mass Spectrometry
LC/ESI-MS was employed to determine the extent of deuterium incorporation into individual peptides as described previously (21). The masses of individual peptides in hydrogen exchange experiments were determined by ESI-MS on a Quadrupole time-of-flight mass spectrometer (Applied Biosystems). The peak for each peptide was integrated to obtain a centroid value using the Magtran computer program (25). The deuterium incorporation for each peptide was quantified by calculating the difference between the centroid values before and after deuterium exchange at a given time point using equation 1.
Native gel electrophoresis for determining oligomeric state
Protein samples (30 μg each) were separated on a 4–20% gradient gel under nondenaturing conditions. The gels were run at 4°C at 120 V and 15 mA current.
RESULTS
Purification and Oligomeric States of the K102N, P78R and P78R/K102N Mutants
The recombinant single and double mutants were obtained in at least 90% purity as determined by electrophoresis under denaturing conditions and by the ratio of the 280 nm to 428 nm absorption peak (which is ~1 for pure wild type enzyme (24)). The PLP content of both the single and the double mutants was identical to that of wild type enzyme, i.e. one PLP per subunit (data not shown). The P78R single mutant exhibited a shift in the equilibrium distribution of oligomeric states that was detected by analysis of freshly purified enzyme. Gel filtration chromatography resulted in elution of P78R as a broad peak and partial resolution of two species designated hereafter as P78R-I and P78R-II respectively, as revealed by native gel electrophoresis (Figure 3). P78R-I migrated as a mixture of higher-order oligomers, which is also observed with wild-type enzyme, whereas P78R-II is predominantly in the dimeric state. Storage of P78R-I at 4°C for 24–48 h resulted in its conversion to the P78R-II state. In contrast, the K102N and the double mutant behaved like wild-type enzyme by native gel chromatography and exists as a mixture of higher-order quaternary states.
Fig. 3.

The P78R-II mutant behaves like a dimer. Native gel electrophoresis of wild type cystathionine β-synthase and various single and double mutants shows heterogenous oligomeric states. P78R-I* refers to the form obtained by overnight incubation of P78R-I at 4°C. Lane 1 shows migration of molecular weight markers and lane 8, the truncated dimeric form of cystathionine β-synthase (T-α2) which has a molecular mass of ~90 kDa.
Kinetic Properties of the K102N, P78R and P78R/K102N Mutants
The steady-state kinetic parameters for the variants were compared with those of wild type enzyme (Table 1). Each of the single mutants diminished the specific activity with the K102N showing a 3-fold lower activity (65 μmol mg−1 h−1) and the P78R-I and P78R-II forms exhibiting 9- (19 μmol mg−1 h−1) and 3-fold (62 μmol mg−1 h−1) lower activity than wild type enzyme (178 μmol mg−1 h−1). Overnight storage of P78R-I at 4°C resulted in its conversion to P78R-II and was accompanied by a 3-fold increase in specific activity to 58 ± 5 μmol mg−1 h−1. Interestingly, the deficits in activity associated with each of the single mutations were negated in the double mutant (169 μmol mg−1 h−1), which was as active as wild-type enzyme in the absence of AdoMet.
Some changes were observed in the kinetic parameters for the substrates. The KM for serine is ~2-fold higher for the K102N and P78R/K102N mutants and ~4-fold higher for the P78R-II mutant compared to wild-type enzyme. A sigmoidal dependence of activity on serine concentration was seen with P78R-I with a Hill coefficient of 2.4. Sigmoidal kinetics are observed with the K102N single and the P78R/K102N double mutants when homocysteine is the variable substrate with a Hill coefficient of 3, indicating positive cooperativity. The KM for L-homocysteine is 2-fold higher for the K102N mutant, lower for the P78R-II mutant, and comparable to wild-type enzyme for the double mutant (Table I).
The P78R/K102N Mutant is Insensitive to Allosteric Regulation
In the presence of AdoMet, the specific activity of wild-type cystathionine β-synthase increases ~2-fold from 178 μmol mg−1 h−1 to 378 μmol mg−1 h−1 (24). The K102N and P78R-I variants exhibit sensitivity to AdoMet and were activated 2.9- and 2.5-fold respectively. In contrast, P78R-II, which exists predominantly in the dimeric state, shows diminished activation (1.4-fold) by AdoMet. Based on the densitometric analysis of the native gel, we estimate that ~30% of P78R-II is present in the higher oligomeric state. Thus, the AdoMet responsiveness of P78R-II indicates that the full-length dimer is also sensitive to allosteric regulation. In contrast, the P78R/K102N double mutant is unresponsive to AdoMet. Thus, interaction between the P78 and K102 positions in the double mutant results in loss of allosteric regulation.
Hydrogen Exchange MS Reveals Conformational Lock in the P78R/K102N Mutant
Hydrogen exchange MS was employed to determine if the insensitivity of the double mutant results from a conformational lock in the kinetically distinguishable “basal” and “activated” states (Figure 4A). Of the ~50 peptides that were identified for the double mutant, a significant difference in the extent of deuterium incorporation was observed in only one peptide, extending from residues 511–531 (Figure 4B) with a mass of 2502.704. The same peptide in wild-type enzyme had previously been shown to exhibit a downshift in the centroid mass from 2503.36 in the absence to 2498.24 in the presence of AdoMet (21). This corresponds to a decrease in maximal deuterium incorporation from 85% in the wild-type enzyme to 51% in the double mutant. The P78R/K102N double mutant thus appears to be conformationally locked in the “activated” state, explaining its insensitivity to AdoMet. The decrease in deuterium concentration upon exposure to AdoMet in wild-type enzyme and in the double mutant as isolated, indicates decreased solvent exposure and/or hydrogen bonding interaction in this conformation.
Fig. 4.

Conformational change induced by AdoMet binding to cystathionine β-synthase. A. Model of an AdoMet-induced conformational change in cystathionine β-synthase. The model correlates differences in kinetic properties associated with the basal and activated states of cystathionine β-synthase with the structural changes revealed by hydrogen-deuterium mass spectrometry. The P78R/K102 patient mutation is locked in the activated conformation even in the absence of AdoMet. B. Mass spectrometric analysis of wild-type and the P78R/K102N mutants of cystathionine β-synthase. Hydrogen-deuterium exchange mass spectrometric analysis of cystathionine β-synthase identified a single peptide extending from residues 511 to 531 that revealed a difference in the centroid mass after 30 s of hydrogen exchange in the presence (m/z = 624.803) versus absence (m/z = 625.676) of AdoMet. The same peptide in the double mutant in the absence of AdoMet, exhibited a centroid mass corresponding to that observed with wild-type enzyme in the presence of AdoMet.
Discussion
Pathogenic mutations represent Nature’s map of residues that are important for the structural and/or functional integrity of proteins. In the case of cystathionine β-synthase, over 100 patient mutations have been described that are dispersed across all three domains of the modular protein (4). Biochemical characterizations of a limited number of missense mutations that are remote from the active site have been described (5–9) and have provided novel insights into clinically important phenotypes associated with specific mutations, viz. pyridoxine responsiveness. A subset of pathogenic mutations, clustered in the CBS1 domain of the regulatory module are associated with an interesting phenotype, i.e. they show robust basal activity but are unresponsive to allosteric activation by AdoMet (6, 9, 14). Biophysical characterization of one such mutant, D444N, revealed that it is conformationally locked in an activated state that is observed with wild-type enzyme only in the presence of AdoMet (21). These studies reveal that AdoMet binding to the regulatory domain shifts the conformation of cystathionine β-synthase from a kinetically distinguishable “basal” state to an “activated” state, which in turn, increases the activity of the enzyme. In this study, we have examined the penalties associated with a pair of linked mutations P78R/K102N, described in homocystinuric patients and compared them to the deficits associated with the single mutations.
Wild-type human cystathionine β-synthase exists as a mixture of oligomeric states with higher-order oligomers being the predominant species. Analysis of wild-type cystathionine β-synthase by blue native gel electrophoresis reveals the presence of oligomers ranging from a dimer to ~16-mers (10). However, the kinetic properties associated with the full-length dimer have not been reported because of the difficulty in separating the various oligomeric states in pure form. P78 is located at the periphery of the dimer interface in the crystal structure of truncated cystathionine β-synthase (22, 26) (Figure 2). Unlike wild-type enzyme, chromatography of P78R results in partial separation of two oligomeric states monitored by native gel chromatography, which correspond to a predominantly full-length dimeric form (P78R-II) and a mixture of higher oligomeric forms (P78R-I). The P78R-I form is unstable and is converted to the P78R-II state over 24 h at 4°C, permitting limited kinetic characterization of the full-length dimeric form of cystathionine β-synthase (Table 1). The principal findings for P78R-II, which is enriched in the full-length dimer, is that it is active, albeit ~3-fold less so than wild-type enzyme, and is responsive to allosteric activation by AdoMet. The KM values for both serine and homocysteine for P78R-II are similar to those for wild-type enzyme. The P78R-I form, which is a heterogeneous mixture of oligomeric states is 3-fold less active than P78R-II and exhibits KM values for the two substrates that are similar to wild-type enzyme. Extrapolating from these results to wild-type enzyme, suggests that a dimeric form of cystathionine β-synthase would be active and AdoMet responsive. The physiological relevance of the oligomeric heterogeneity associated with purified human cystathionine β-synthase is not known, nor what ligands or metabolic state might favor one versus other structures. Since the full-length dimer of P78R is associated with higher activity compared to the mixed higher-order oligomeric state, P78R-I, it raises the possibility that stabilization of the full-length dimer may be used as a mechanism for upregulation of wild-type cystathionine β-synthase under certain conditions.
The instability of the P78R-I form together with the absence of the homogenous dimeric form in the P78R-II pool, limited our ability to further characterize their oligomeric states by biophysical methods viz. analytical ultracentrifugation, which yielded variable results. It also raises questions about whether the kinetic properties of the P78R-II species that we have determined, truly reflect those for the full-length dimer. In fact, we estimate that ~30% of the P78R-II pool represents higher-order oligomers. Unfortunately, we have been unable to purify the dimeric form any further using various chromatographic columns.2 Nonetheless, the observed differences in the kinetic properties between P78R-I and -II, particularly with respect to specific activity, lends confidence at least to the qualitative differences between the dimer and the mixed higher oligomeric states that are reported in this study.
The principal in vitro biochemical phenotypes associated with the linked patient mutation, P78R/K102N, are high basal activity comparable to wild-type enzyme and AdoMet unresponsiveness. The high basal activity exhibited by the P78R/K102N enzyme indicates that the mutant retains a full complement of PLP. This is curious in light of the B6-responsive phenotype associated with patients carrying this mutation (23). It is possible that the B6-responsiveness is associated with the second mutation in these patients, E239K. We also note the seeming paradox between pyridoxine responsiveness in patients carrying the I278T mutation in cystathionine β-synthase, which is not reflected in an altered affinity for PLP in the expressed protein (27). This observation led the authors of the study to suggest that the clinical response to pyridoxine may involve multiple mechanisms (27).
Since neither single mutant exhibits this property, we conclude that their interaction in the double mutant is responsible for this phenotype. The insensitivity of the double mutant to AdoMet could arise in principle from its inability to bind the allosteric activator and therefore be locked in the “basal” conformation or to it being locked in the “activated” conformation that is seen with wild-type enzyme in the presence of AdoMet (Figure 4A). The AdoMet responsive peptide identified by hydrogen exchange MS reveals that in the catalytic domain mutations, P78R/K102N, as in the regulatory mutation, D444N, the exchange kinetics for this peptide in the absence of AdoMet mimic that for wild type enzyme, in the presence of AdoMet (Figure 4B). Thus, the double mutant exhibits an identical conformational signature in a regulatory domain peptide that is seen with wild type enzyme in the presence of AdoMet or the regulatory domain mutant, D444N (Figure 4). To our knowledge, this is the first demonstration of mutations in the catalytic core that mimic the AdoMet insensitivity of a subclass of CBS1-domain mutations (6, 9).
The K102N mutation exhibits only mild deficits being ~2- to 3-fold less active than wild type enzyme in the absence or presence of AdoMet (Table 1). The distinguishing kinetic feature of this mutant is that it exhibits non-Michaelis Menten kinetics when homocysteine is the variable substrate and the sigmoidal dependence is associated with a Hill coefficient of 3. This feature is also observed in the double mutant, which exhibits a KM for homocysteine that is comparable in value to that of wild-type enzyme but exhibits positive cooperativity with a Hill coefficient of 3. Thus, the change in interaction between subunits in the double mutant with respect to homocysteine binding can be ascribed to the missense mutation at the K102 locus. Interestingly, the individual single mutants are each less active than the double mutant indicating favorable compensatory changes in the linked pair that restores basal activity to wild-type levels.
An earlier study had reported preliminary characterization of the single and double mutants in E. coli cell extracts (23). Both the P78R and K102N single mutants exhibited fairly variable activity that were 2- to 10-fold lower than wild type enzyme, depending on the clone. The authors reported seeing no activity with the double mutant. These results are inconsistent with those reported in this study for the P78R/K102N mutant and also with the absence of a classical homocystinuric clinical phenotype in one of the siblings in whom this mutation has been described, especially since the other mutation in this patient, E239K, has also been reported by the same group to be associated with no activity in cell extracts (23). The basis of the discrepancy between the two studies is not known. However, it is interesting to note that other patients with nonclassical homocystinuria have been described who also have mutations that map to the regulatory domain of cystathionine β-synthase and result in AdoMet-unresponsiveness (15). These patients exhibit high plasma homocysteine levels in the absence of the neurological or connective tissue defects classically associated with homocystinuria due to cystathionine β-synthase deficiency. This clinical presentation is reminiscent of the male patient with the AdoMet-unresponsive P78R/K102N double mutation, associated with nonclassical homocystinuria (23).
Computational studies have revealed that insensitivity to allosteric activation by AdoMet can result in hyperhomocysteinemia particularly under conditions of high methionine intake when the transsulfuration pathway becomes especially important in disposing off excess methionine and homocysteine (16). While the AdoMet-unresponsiveness of the P78R/K102N mutation could similarly predispose patients to hyperhomocysteinemia, the interaction with subunits containing the second mutation found in these patients, E239K, may significantly alter the phenotype of the encoded protein. Alternatively, the nucleotide changes associated with the double mutation could alter gene expression.
Finally, although known for some time in laboratories that work on this protein, the oligomeric heterogeneity associated with wild-type cystathionine β-synthase has not been characterized primarily due to the difficulty in stably resolving the different enzyme forms. The P78R mutation appears to afford some stabilization of the dimer, which has permitted the limited characterization reported in this study. It is not known if some or all of the multiple oligomeric states of cystathionine β-synthase are constructed from monomers (or dimers) that have slightly different conformations, and therefore qualify as “morpheeins”, a model for allostery in which the monomer conformation dictates the resulting oligomeric state (28). The analysis of an uncommon allele of human porphobilinogen synthase allowed isolation and crystallization of a hexameric form, distinct from the octamer seen with wild-type enzyme and led to the morpheein concept (29). Elegant studies have since shown that it is the quaternary structure change rather than the amino acid change that underlies the differences in the kinetic properties of the two variants of porphobilinogen synthase (29, 30). We currently have insufficient data to evaluate whether or not a dynamic morpheein equilibrium is responsible for the multiple quaternary states of cystathionine β-synthase but note that some of the kinetic attributes of morpheeins viz. dependence of activity on order of substrate addition and hysteresis, have been observed with cystathionine β-synthase (13).
Acknowledgments
We thank Dr. Jiong Yu in the Mass Spectrometry Center of the Redox Biology Center (University of Nebraska, Lincoln) for help with data accumulation.
Footnotes
This work was supported in part by a grant from the National Institutes of Health (HL58984 to RB). The Mass Spectrometry Core Facility of the Redox Biology Center was supported by an NIH grant (P20RR17675).
Abbreviations Used: PLP: pyridoxal 5′-phosphate, AdoMet: S-adenosylmethionine, MS: mass spectrometry
A gel filtration column, Superdex-200 (Amerhsam) was used in addition to the anion exchange and GST-columns described under Methods to further purify dimeric P78R-II from the higher oligomeric forms.
References
- 1.Banerjee R, Evande R, Kabil O, Ojha S, Taoka S. Reaction mechanism and regulation of cystathionine beta-synthase. Biochim Biophys Acta. 2003;1647:30–5. doi: 10.1016/s1570-9639(03)00044-x. [DOI] [PubMed] [Google Scholar]
- 2.Banerjee R, Zou CG. Redox regulation and reaction mechanism of human cystathionine-beta-synthase: a PLP-dependent hemesensor protein. Arch Biochem Biophys. 2005;433:144–56. doi: 10.1016/j.abb.2004.08.037. [DOI] [PubMed] [Google Scholar]
- 3.Miles EW, Kraus JP. Cystathionine {beta}-Synthase: Structure, Function, Regulation, and Location of Homocystinuria-causing Mutations. J Biol Chem. 2004;279:29871–29874. doi: 10.1074/jbc.R400005200. [DOI] [PubMed] [Google Scholar]
- 4.Kraus JP, Janosik M, Kozich V, Mandell R, Shih V, Sperandeo MP, Sebastio G, de Franchis R, Andria G, Kluijtmans LA, Blom H, Boers GH, Gordon RB, Kamoun P, Tsai MY, Kruger WD, Koch HG, Ohura T, Gaustadnes M. Cystathionine beta-synthase mutations in homocystinuria. Hum Mutat. 1999;13:362–75. doi: 10.1002/(SICI)1098-1004(1999)13:5<362::AID-HUMU4>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 5.Kabil O, Banerjee R. Deletion of the regulatory domain in the pyridoxal phosphate-dependent heme protein cystathionine beta-synthase alleviates the defect observed in a catalytic site mutant. J Biol Chem. 1999;274:31256–60. doi: 10.1074/jbc.274.44.31256. [DOI] [PubMed] [Google Scholar]
- 6.Janosik M, Kery V, Gaustadnes M, Maclean KN, Kraus JP. Regulation of human cystathionine beta-synthase by S-adenosyl-L- methionine: evidence for two catalytically active conformations involving an autoinhibitory domain in the C-terminal region. Biochemistry. 2001;40:10625–33. doi: 10.1021/bi010711p. [DOI] [PubMed] [Google Scholar]
- 7.Janosik M, Oliveriusova J, Janosikova B, Sokolova J, Kraus E, Kraus JP, Kozich V. Impaired heme binding and aggregation of mutant cystathionine beta- synthase subunits in homocystinuria. Am J Hum Genet. 2001;68:1506–13. doi: 10.1086/320597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ojha S, Wu J, LoBrutto R, Banerjee R. Effects of heme ligand mutations including a pathogenic variant, H65R, on the properties of human cystathionine beta syntase. Biochemistry. 2002;41:4649–4654. doi: 10.1021/bi011827o. [DOI] [PubMed] [Google Scholar]
- 9.Evande R, Boers GHJ, Blom HJ, Banerjee R. Alleviation of Intrasteric Inhibition by the Pathogenic Activation Domain Mutation, D444N, in Human Cystathionine beta-synthase. Biochemistry. 2002;41:11832–11837. doi: 10.1021/bi026248d. [DOI] [PubMed] [Google Scholar]
- 10.Frank N, Kery V, Maclean KN, Kraus JP. Solvent-accessible cysteines in human cystathionine beta-synthase:Crucial role of cysteine 431 in S-adenosyl L-methionine binding. Biochemistry. 2006;45:11021–11029. doi: 10.1021/bi060737m. [DOI] [PubMed] [Google Scholar]
- 11.Kery V, Poneleit L, Kraus J. Trypsin cleavage of human cystathionine beta-synthase into an evolutionarily conserved active core: Structural and functional consequences. Arch Biochem Biophys. 1998;355:222–232. doi: 10.1006/abbi.1998.0723. [DOI] [PubMed] [Google Scholar]
- 12.Shan X, Kruger WD. Correction of disease causing CBS mutations in yeast. Nature Genetics. 1998;19:91–93. doi: 10.1038/ng0598-91. [DOI] [PubMed] [Google Scholar]
- 13.Taoka S, Widjaja L, Banerjee R. Assignment of enzymatic functions to specific regions of the PLP-dependent hemeprotein cystathionine β-synthase. Biochemistry. 1999;38:13155–13161. doi: 10.1021/bi990865t. [DOI] [PubMed] [Google Scholar]
- 14.Kluitjmans LAJ, Boers GHJ, Stevens EMB, Renie WO, Kraus JP, Trijbels FJM, Heuvel LPWJvd, Blom HJ. Defective cystathionine beta-synthase regulation by S-adenosylmethionine in a partially pyridoxine responsive homocystinuria. J Clin Invest. 1996;98:285–289. doi: 10.1172/JCI118791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maclean KN, Gaustadnes M, Oliveriusova J, Janosik M, Kraus E, Kozich V, Kery V, Skovby F, Rudiger N, Ingerslev J, Stabler SP, Allen RH, Kraus JP. High homocysteine and thrombosis without connective tissue disorders are associated with a novel class of cystathionine beta-synthase (CBS) mutations. Hum Mutat. 2002;19:641–55. doi: 10.1002/humu.10089. [DOI] [PubMed] [Google Scholar]
- 16.Prudova A, Martinov MV, Vitvitsky V, Ataullakhanov F, Banerjee R. Analysis of pathological defects in methionine metabolism using a simple mathematical model. Biochim Biophys Acta. 2005;1741:331–8. doi: 10.1016/j.bbadis.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 17.Meier M, Janosik M, Kery V, Kraus JP, Burkhard P. Structure of human cystathionine beta-synthase: a unique pyridoxal 5′-phosphate-dependent heme protein. EMBO J. 2001;20:3910–6. doi: 10.1093/emboj/20.15.3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taoka S, Lepore BW, Kabil Ö, Ojha S, Ringe D, Banerjee R. Human cystathionine beta-synthase is a heme sensor protein. Evidence that the redox sensor is heme and not the vicinal cysteines in the CXXC motif seen in the crystal structure of the truncated enzyme. Biochemistry. 2002;41:10454–61. doi: 10.1021/bi026052d. [DOI] [PubMed] [Google Scholar]
- 19.Bateman A. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Trends Biochem Sci. 1997;22:12–13. doi: 10.1016/s0968-0004(96)30046-7. [DOI] [PubMed] [Google Scholar]
- 20.Scott JW, Hawley SA, Green KA, Anis M, Stewart G, Scullion GA, Norman DG, Hardie DG. CBS domains form energy-sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J Clin Invest. 2004;113:274–84. doi: 10.1172/JCI19874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sen S, Yu J, Yamanishi M, Schellhorn D, Banerjee R. Mapping Peptides Correlated with Transmission of Intrasteric Inhibition and Allosteric Activation in Human Cystathionine beta-Synthase. Biochemistry. 2005;44:14210–6. doi: 10.1021/bi051046d. [DOI] [PubMed] [Google Scholar]
- 22.Yamanishi M, Kabil Ö, Sen S, Banerjee R. Structural Insights into Pathogenic Mutations in Heme-dependent Cystathionine-β-synthase. J Inorg Bioc. 2006 doi: 10.1016/j.jinorgbio.2006.08.020. accepted for publication. [DOI] [PubMed] [Google Scholar]
- 23.de Franchis R, Kozich V, McInnes RR, Kraus JP. Identical genotypes in siblings with different homocystinuric phenotypes: identification of three mutations in cystathionine beta-synthase using an improved bacterial expression system. Hum Mol Genet. 1994;3:1103–8. doi: 10.1093/hmg/3.7.1103. [DOI] [PubMed] [Google Scholar]
- 24.Taoka S, Ohja S, Shan X, Kruger WD, Banerjee R. Evidence for heme-mediated redox regulation of human cystathionine β-synthase activity. J Biol Chem. 1998;273:25179–25184. doi: 10.1074/jbc.273.39.25179. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Z, Smith DL. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 1993;2:522–31. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meier M, Oliveriusova J, Kraus JP, Burkhard P. Structural insights into mutations of cystathionine beta-synthase. Biochim Biophys Acta. 2003;1647:206–13. doi: 10.1016/s1570-9639(03)00048-7. [DOI] [PubMed] [Google Scholar]
- 27.Chen X, Wang L, Fazlieva R, Kruger WD. Contrasting behaviors of mutant cystathionine beta-synthase enzymes associated with pyridoxine response. Hum Mutat. 2006;27:474–82. doi: 10.1002/humu.20320. [DOI] [PubMed] [Google Scholar]
- 28.Jaffe EK. Morpheeins--a new structural paradigm for allosteric regulation. Trends Biochem Sci. 2005;30:490–7. doi: 10.1016/j.tibs.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 29.Breinig S, Kervinen J, Stith L, Wasson AS, Fairman R, Wlodawer A, Zdanov A, Jaffe EK. Control of tetrapyrrole biosynthesis by alternate quaternary forms of porphobilinogen synthase. Nat Struct Biol. 2003;10:757–63. doi: 10.1038/nsb963. [DOI] [PubMed] [Google Scholar]
- 30.Tang L, Stith L, Jaffe EK. Substrate-induced interconversion of protein quaternary structure isoforms. J Biol Chem. 2005;280:15786–93. doi: 10.1074/jbc.M500218200. [DOI] [PubMed] [Google Scholar]