

Abstract
Certain aminoglycosides are capable of inducing “translational readthrough” of premature termination codons (PTCs). However, toxicity and relative lack of efficacy deter treatment with clinically available aminoglycosides for genetic diseases caused by PTCs, including cystic fibrosis (CF). Using a structure-based approach, the novel aminoglycoside NB54 was developed that exhibits reduced toxicity and enhanced suppression of PTCs in cell-based reporter assays relative to gentamicin. We examined whether NB54 administration rescued CFTR protein and function in clinically relevant CF models. In a fluorescence-based halide efflux assay, NB54 partially restored halide efflux in a CF bronchial epithelial cell line (CFTR genotype W1282X/F508del), but not in a CF epithelial cell line lacking a PTC (F508del/F508del). In polarized airway epithelial cells expressing either a CFTR-W1282X or -G542X cDNA, treatment with NB54 increased stimulated short-circuit current (ISC) with greater efficiency than gentamicin. NB54 and gentamicin induced comparable increases in forskolin-stimulated ISC in primary airway epithelial cells derived from a G542X/F508del CF donor. Systemic administration of NB54 to Cftr−/− mice expressing a human CFTR-G542X transgene restored 15–17% of the average stimulated transepithelial chloride currents observed in wild-type (Cftr+/+) mice, comparable to gentamicin. NB54 exhibited reduced cellular toxicity in vitro and was tolerated at higher concentrations than gentamicin in vivo. These results provide evidence that synthetic aminoglycosides are capable of PTC suppression in relevant human CF cells and a CF animal model and support further development of these compounds as a treatment modality for genetic diseases caused by PTCs.
Keywords: Cystic fibrosis transmembrane conductance regulator, Premature termination codons, Aminoglycosides, Short-circuit current, Primary human bronchial epithelial cells
Introduction
Cystic fibrosis (CF) is caused by mutations in the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) and results in significant morbidity and early mortality in affected individuals. Over 1,500 disease-causing mutations in CFTR have been identified and can be divided into six major classes [1]. Nonsense mutations (a class I mutation type) are caused by a single nucleotide change that introduces an in-frame premature termination codon (PTC or nonsense mutation), resulting in absent functional protein and typically a severe CF phenotype [2, 3]. In-frame PTCs account for ∼11% of all described gene lesions causing human genetic diseases and 5–10% of disease-causing CF mutations in Caucasian populations. PTCs within CFTR are especially common in individuals of Ashkenazi Jewish decent, where they account for nearly 64% of CFTR mutations [4].
A potential treatment for conditions caused by nonsense mutations includes the use of aminoglycosides or other agents to suppress the normal proofreading function of the ribosome at in-frame PTCs. This leads to insertion of a near-cognate amino acid, which then allows continued translation of the remainder of the open reading frame. This “translational readthrough” of nonsense mutations has been shown to restore protein function in a number of preclinical settings [5–11]. In several studies, aminoglycosides have been shown to partially rescue CFTR activity in human subjects with CF due to nonsense mutations, including following systemic administration of gentamicin [12, 13] and topical gentamicin delivered to the nasal airway [14, 15]. However, the approach has not been successful in all CF trials [16], suggesting more effective compounds may be needed. Furthermore, significant toxicity is associated with long-term administration of aminoglycoside antibiotics. While one alternative is to use novel non-aminoglycoside small molecules such as ataluren (PTC124) [17, 18], it may also be possible to rationally synthesize aminoglycosides and/or aminoglycoside mimetics optimized for maximal PTC suppression and minimal toxicity. Using a systematic structure-based approach, two synthetic aminoglycosides (termed NB30 and NB54) have been shown to enhance suppression of PTCs using dual luciferase readthrough reporters while also exhibiting reduced toxicity in cellular assays (Fig. 1) [19–21]. Whereas the first-generation lead, NB30, exhibited significantly reduced cytotoxicity in comparison to gentamicin [20] and promoted dose-dependent suppression of nonsense mutations of the PCDH15 gene [21], one of the underlying causes of type 1 Usher syndrome, suppression efficacy was significantly lower relative to that of gentamicin. In attempts to further improve suppression efficiency and reduce the toxicity of NB30, NB54 was developed as a second-generation lead structure [19]. NB54 exhibited significantly reduced cell, cochlear and acute toxicities, and substantially higher PTC readthrough potency than gentamicin in both in vitro and ex vivo studies [19].
Fig. 1.
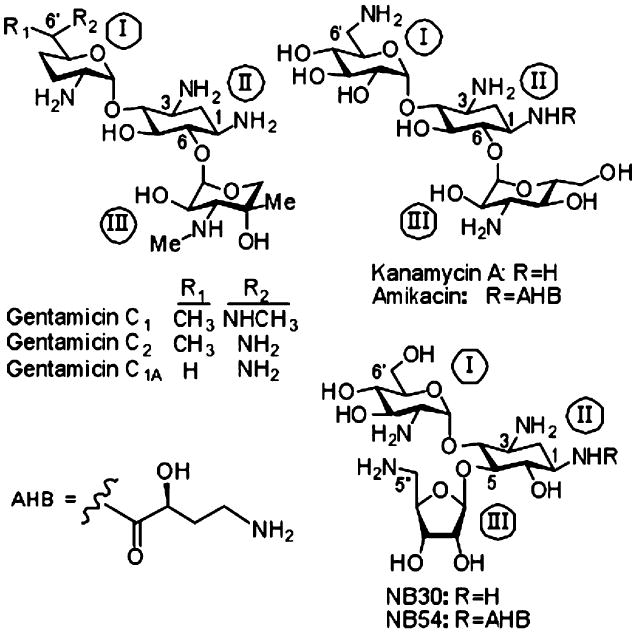
Shown are chemical structures of a series of 2-deoxystreptamine-derived (ring II) aminoglycosides relevant to this study, including the gentamicin congeners C1, C1A, and C2; kanamycin A; amikacin; NB30; and NB54. Ring numbers are designated by roman numerals. AHB (S)-4-amino-2 hydroxybutanoyl group
In this study, we provide evidence in several human CF cell lines and in primary human airway cells that NB30 and NB54 induce readthrough of CFTR PTCs and partially restore CFTR function as well as (and in some cases better than) gentamicin. We also show that NB54 restores a comparable level of CFTR expression and function as gentamicin in transgenic mice expressing a human CFTR-G542X cDNA. This provides the first evidence that a synthetic aminoglycoside is capable of PTC suppression in cell-based or animal models of a human disease.
Materials and methods
Materials used in this study
Gentamicin sulfate injection, USP (10 mg/ml), was purchased from the UAB Hospital pharmacy (manufactured by Hospira, Inc., Lake Forest, IL). The synthetic compounds NB30 and NB54 were prepared and purified for biological assays as reported previously by us [19, 20]. All other chemicals and biochemicals, unless otherwise stated, were obtained from commercial sources. In all biological tests, synthetic aminoglycosides were in their sulfate salt forms [MW (in grams per mole) of the sulfate salts were as follows: NB30—563.0, NB54— 652.8]. Purity of NB30 and NB54 were determined by using HPLC-ESI-MS analysis, which indicated 98.69% and 95.55% purity, respectively.
Growth of stable cell lines expressing recombinant CFTR CFTR-W1282X
cDNA were stably transfected into HeLa and CFBE41o- cells using the TranzVector™ (Tranzyme, Inc., Birmingham, AL) as described [22]. Expression of the CFTR gene was coupled to the puromycin-N-acetyltransferase gene (puro) gene, allowing selection of a stable pool of cells expressing CFTR by growth with puromycin (4 mg/ml). Once selected, puromycin was removed and cells were maintained in DMEM supplemented with 10% FBS, penicillin, and streptomycin for all experiments, as previously described [23].
Procurement and growth of primary human bronchial epithelial cells
The UAB institutional review board approved use of human cells and tissues. Primary human airway epithelial cells were derived from lung explants of CF subjects who provided written informed consent and were confirmed to harbor two severe CFTR mutations (including the G542X premature termination codon) by methods described previously [24, 25]. Only first or second passage cells were seeded on permeable supports for studies. Once cells were 80–90% confluent, they were seeded on Snapwell 1.13 cm2 permeable supports (1×106 cells/filter; Bayer, Pittsburgh, PN) or Costar 0.4-μm permeable supports (5×105 cells/filter; Bethesda, MD) after coating with NIH 3T3 fibroblast conditioned media, and grown in differentiating media containing DMEM/F12 (Invitrogen, Carlsbad, CA), 2% Ultroser-G (Pall, New York, NY), 2% Fetal Clone II (Hyclone, Logan, UT), 2.5 μg/ml insulin (Sigma), 0.25% bovine brain extract (LONZA), 20 nM hydrocortisone (Sigma-Aldrich), 500 nM triodothyronine (Sigma), 2.5 μg/ml transferrin (Invitrogen), 250 nM ethanolamine (Sigma-Aldrich), 1.5 μM epinephrine (Sigma-Aldrich), 250 nM phosphoethanolamine (Sigma-Aldrich), and 10 nM retinoic acid (Sigma-Aldrich) until terminally differentiated, as previously described [24, 25].
SPQ studies of halide efflux in heterologous cells
HeLa cells stably transfected with a lentiviral system carrying a CFTR-W1282X cDNA were studied with the halide quenched dye 6-methoxy-N-(3-sulfopropyl)-quinolinium (SPQ, Molecular Probes Inc., Eugene, OR) as described by the manufacturer and previously published [23, 26, 27]. Methods were similar for the functional screen in IB3 and CFNPE monolayers using SPQ fluorescence, with only minor differences. Cells were grown in a 96-well (full area) microtiter plates according to a microtiter plate map design. Cells were loaded with SPQ (2 mg/ml in DMEM) overnight (18 h), concurrent with administration of test drugs. SPQ fluorescence following addition of NaNO3 buffer was read across the microtiter plate within a Turner Designs Multi-mode Plate Reader at a single time point. NaCl Ringer was used instead of NaI Ringer to quench SPQ fluorescence in the initial and recovery phase of the assay. For each assay, results were expressed as the change in fluorescence over basal, as previously published [23, 28, 29]. Cell toxicity of aminoglycosides was monitored using the CellTiterGLO luminescent cell viability assay (Promega, Madison, WI).
Voltage clamp studies in Ussing chambers
Inserts were mounted in Ussing chambers, and short-circuit current (ISC) was measured under voltage clamp conditions using MC8 voltage clamps and P2300 Ussing chambers (Physiologic Instruments, San Diego, CA) as previously described [24]. Monolayers were initially bathed on both sides with identical Ringers solutions containing (in millimolars) 115 NaCl, 25 NaHCO3, 2.4 KH2PO4, 1.24 K2HPO4, 1.2 CaCl2, 1.2 MgCl2, and 10 D-glucose (pH 7.4). Bath solutions were vigorously stirred and gassed with 95% O2 to 5% CO2. Short-circuit current measurements were obtained using an epithelial voltage clamp (Physiologic Instruments). A 3-mV pulse of 1-s duration was imposed every 3 s to monitor resistance, which was calculated using Ohm's law. Where indicated, the mucosal bathing solution was changed to a low Cl− solution containing (in millimolars) 1.2 NaCl, 115 Na gluconate, and all other components as above. Amiloride (100 μM) was added to block residual ENaC current, followed by the CFTR agonists forskolin and genistein, as indicated (minimum 5 min observation at each concentration). CFTRInh-172 (10 μM) was added to the mucosal bathing solution at the end of experiments to block CFTR-dependent ISC. Agonist stimulation was initiated within 30 min of placement into the chambers. Results were expressed as the change with agonist stimulation and normalized for the particular cell type where noted.
Mouse lines and treatment protocols
The CFTR-G542X mice used in this study contained the Cftrtm1Cam knockout [30] and expressed a human CFTR transgene with the G542X premature stop mutation [17, 31, 32] (referred to as Cftr−/− hCFTR-G542X mice). Treatments consisted of subcutaneous injections of gentamicin or NB54 delivered in the hind limb once daily for 14 days. Additional detail is available in the online supplement. The animal protocols used in this work were reviewed and approved by the UAB Institutional Animal Care and Use Committee.
Short-circuit current measurements in mouse intestines
Transepithelial ISC measurements and immunohistochemical staining of murine intestinal tissues were carried out as previously described [17, 31, 32]. In the setting of a serosal to mucosal chloride gradient, forskolin (10 μM) was added to both the mucosal and serosal solutions and ISC was continuously monitored for ≥10 min to ensure that a sustained response was obtained. Carbachol (100 μM) was then added to the serosal solution to further augment the chloride secretory gradient, and incubation was continued for an additional 5 min. The chloride gradient is imposed to improve sensitivity to detect CFTR activity, measured as the change in ISC from baseline (see dotted line, Fig. 6). While the baseline can frequently exhibit a gradual but consistent positive slope, based on previous experience [17, 31–33], the stimulated ISC induced by the forskolin/IBMX is consistent with a cAMP-stimulated chloride current induced by the CFTR channel and is also inhibited by the CFTR inhibitor glybenclamide (the ISC returns to the extrapolated baseline).
Fig. 6.
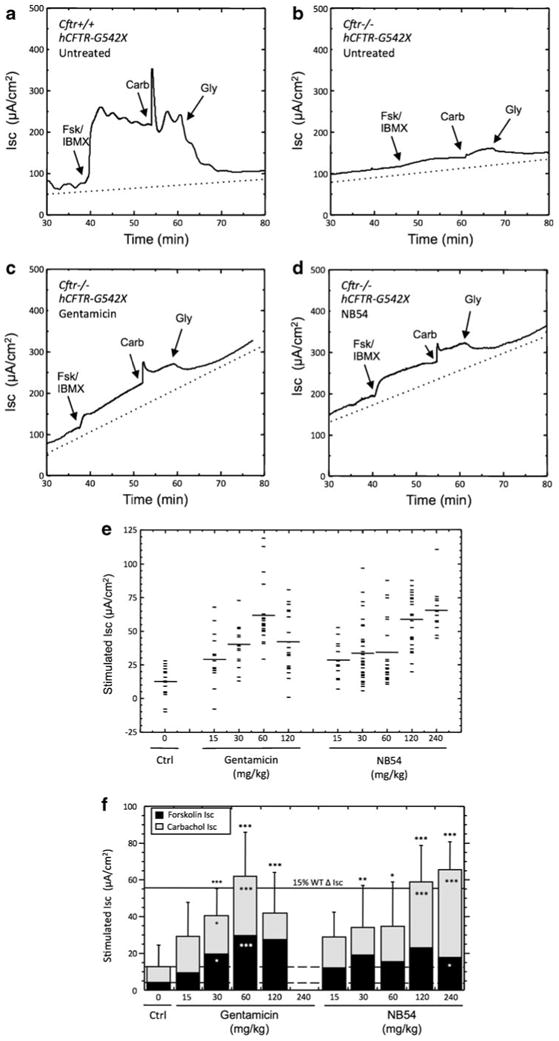
cAMP-stimulated transepithelial chloride currents in ileum sections from mice treated with gentamicin or NB54. Representative short-circuit current tracings are shown from a untreated Cftr+/+ hCFTR-G542X mice (WT control), b untreated Cftr−/− hCFTR-G542X (negative control), c gentamicin-treated Cftr−/− hCFTR-G542X (60 mg/kg), and d NB54-treated Cftr−/− hCFTR-G542X (120 mg/kg). e Scatter plot of the total ISC data from Cftr−/− hCFTR-G542X mice (untreated, gentamicin treated, and NB54 treated) that combines the forskolin/IBMX and carbachol responses. f Mean transepithelial cAMP-stimulated short-circuit chloride currents in intestinal tissues (ileum) from mice treated with the indicated doses of gentamicin or NB54. Treatment consisted of a subcutaneous injection once daily with the indicated dose of compound for 2 weeks. Values are expressed as mean±standard deviation. *P<0.05, **P<0.01, ***P<0.001, n≥14/condition, ±SEM
Immunohistochemical staining of murine intestinal tissues
Immunofluorescence experiments were carried out as previously described [17, 31, 32]. The human CFTR-specific antiserum #4,562 (rabbit) was raised against a TrpE fusion protein that included hCFTR NBD1 and a portion of the R domain (hCFTR amino acids 521–828).
Statistical analysis
For cell-based and mouse intestinal short-circuit current measurements and fluorescence-based halide efflux, descriptive statistics (mean, SD, and SEM) and paired and unpaired t tests or ANOVA were performed using SigmaStat software (Jandel, CA), Graphpad Prism software (La Jolla, CA), and Microsoft Excel (Seattle, WA), as appropriate. All statistical tests were two sided and were performed at a 5% significance level (i.e., α=0.05).
Results
Translational readthrough screen of synthetic aminoglycosides in human epithelial cell lines
To determine whether the synthetic aminoglycosides NB30 and NB54 induce enough translational readthrough of CFTR PTCs to partially restore CFTR function using a stimulated halide transport assay, we first tested the compounds using HeLa cells stably transduced with a lentiviral system driving CFTR-W1282X expression. Aminoglycoside-induced translational read-through of CFTR PTCs was previously achieved in this cell line [6, 23], which allows individual cells to be monitored for stimulated halide efflux using the halide-sensitive fluorescent dye SPQ. Following incubation of cells with NB30, NB54, or gentamicin (all at a dose of 500 μg/ml for 24 h), we found that forskolin (20 μM) and genistein (50 μM) stimulated halide transport to a similar extent under all three treatment conditions (Fig. 2a). At baseline, these cells also exhibited a small but detectable increase in stimulated halide transport over baseline, as previously reported [23], probably due to basal activation of CFTR channels following treatment with forskolin and genistein.
Fig. 2.
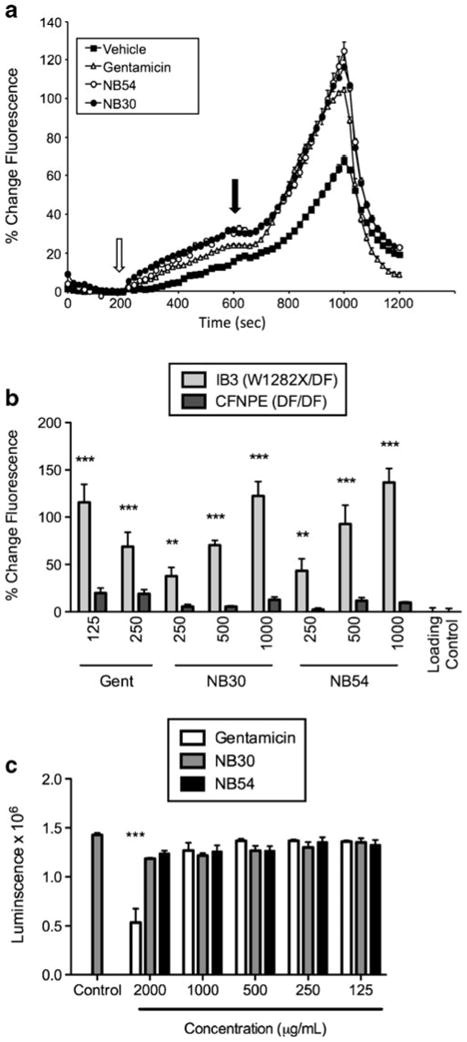
Fluorescence-based halide efflux assay of cells expressing CFTR-W1282X following treatment with gentamicin, NB30, or NB54. a HeLa cells expressing CFTR-W1282X were grown on glass coverslips were treated with 500 μg/ml of the indicated aminoglycoside (gentamicin, NB30, or NB54) for 24 h, or mock treated with vehicle, and studied by fluorescence-based halide efflux (SPQ). Following perfusion of quenching buffer (NaI), dequench buffer was added (NaNO3, open arrow) followed by NaNO3 with the CFTR agonists (closed arrow) forskolin (20 μM) and genistein (50 μM). Increased slope following addition of agonist represents activation of halide transport (P<0.001 for each aminoglycoside tested compared to vehicle control). Cell fluorescence was quenched with NaI at the end of the experiment for confirmation. Fluorescence was measured for 30–45 cells/condition. b IB3-1 cells (endogenous CFTR genotype W1282X/F508Del) were incubated with the aminoglycosides NB30, NB54, or gentamicin at the concentrations indicated for 24 h, and halide efflux was quantified by percentage change in fluorescence over basal compared to control following activation with forskolin (20 μM) and genistein (50 μM). Loading control cells were treated with vehicle (1% sterile water in media) in the same fashion. As a genotype control, CFNPE cells (F508Del/F508Del) were treated in the same manner. c Number of viable IB3-1 cells was quantified by luminescence following treatment with the exact same conditions shown in b. *P<0.05, **P<0.005, ***P<0.0005; n=8/condition, ±SEM
To confirm the effects of aminoglycosides on read-through in a more physiologically relevant cell type with endogenous CFTR expression, we next examined the IB3-1 bronchial epithelial cell line (CFTR genotype W1282X/F508del) in comparison to a CF epithelial cell line derived from nasal polyps from a homozygous CFTR-F508del donor (CFNPE cells). Here, halide efflux monitored by the fluorescent dye SPQ was used to monitor CFTR function using a higher throughput, microtiter plate method in order to monitor halide efflux at different doses of each compound. Following incubation of cells for 24 h at the indicated concentrations of NB30, NB54, or gentamicin, we found that addition of the CFTR agonists forskolin (20 μM) and genistein (50 μM) stimulated halide efflux in IB3-1 cells, but had no detectable effect in CFNPE cells (Fig. 2b). This indicated specificity for PTC suppression. In addition, NB30 and NB54 were each associated with dose-dependent increases in stimulated halide efflux. Cells treated with NB30 and NB54 at a dose of 1,000 μg/ml exhibited peak stimulated fluorescence that was 122% and 136% higher, respectively, than cells treated with vehicle alone. In contrast, cells treated with 125 μg/ml gentamicin showed a 115% increase in peak fluorescence as compared to cells treated with vehicle alone. Notably, we observed a reduced stimulation of halide transport in cells treated with 250 μg/ml gentamicin, although cell viability was not altered as determined using a luminescence cell viability assay (Fig. 2c). These results suggested that an attenuation of readthrough occurs in the presence of higher concentrations of gentamicin. Cells tolerated NB30 and NB54 treatment at a concentration of 2,000 μg/ml, whereas this concentration of gentamicin reduced cell number indicative of toxicity. Based on these results, and the greater efficacy of NB54 in comparison to NB30 in ex vivo studies [20], we prioritized this compound for additional testing in biologically relevant models of CF caused by PTCs.
PTC readthrough by synthetic aminoglycosides in polarized epithelial monolayers
Previous studies have shown that differences in cell type and cell polarization are known to affect the efficiency of CFTR processing [34]. To further test whether synthetic aminoglycosides can suppress PTCs and restore CFTR function in vitro, we next used ISC measurements to monitor stimulated chloride transport in polarized airway epithelial monolayers. In CFBE41o- cells stably transduced with a CFTR-W1282X cDNA, NB54 treatment (500 μg/ml) increased ISC 0.38 μA/cm2 more than in cells treated with vehicle alone. This increase was also 0.31 μA/cm2 greater than the stimulated ISC observed following gentamicin treatment (Fig. 3). To test the more common CFTR-G542X allele and examine dose dependence, CFBE41o- human airway cells transduced with an adenovirus vector expressing a CFTR-G542X cDNA were tested following treatment with gentamicin or NB54. We observed a dose-dependent increase in stimulated chloride transport upon the sequential addition of forskolin (20 μM) and genistein (50 μM) following a 24-h incubation with gentamicin or NB54 but not in control cells treated with vehicle alone (Fig. 4). For each concentration tested (500, 1,000, and 2,000 μg/ml), NB54 elicited a larger increase in stimulated ISC than gentamicin in this cell line. Stimulated currents were sensitive to the CFTR-specific inhibitor CFTRInh-172 (Fig. 4a), confirming the specificity of the effect on CFTR-mediated anion transport. The most effective NB54 concentration tested (2,000 μg/ml) induced a total CFTR-dependent ISC following forskolin and genistein stimulation that was 17% of the activity seen in cells transduced with the corresponding wild-type CFTR construct (18.8 μA/cm2, Fig. 4b). Notably, this concentration of NB54 resulted in 10.4% of the activity observed in cells expressing wild-type CFTR following stimulation with forskolin alone (17.8 μA/cm2). Transepithelial resistance was similar for treated and untreated cells, indicating that significant cell toxicity did not occur under the conditions tested (Fig. 4c).
Fig. 3.
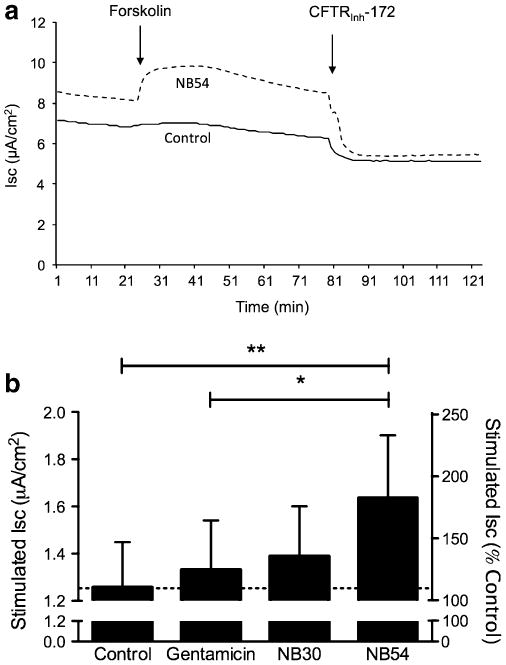
Increased W1282X-CFTR dependent short-circuit current following incubation of synthetic aminoglycosides. a Representative short-circuit current tracings obtained from CFBE41o- expressing W1282X CFTR mounted in modified Ussing chambers and studied under voltage clamp conditions. In the setting of a chloride secretory gradient and amiloride (100 μM), CFTR dependent ISC was stimulated with forskolin (20 μM) followed by block with CFTRInh-172 (10 μM). Cells were grown under polarized conditions at air– liquid interface and treated with 500 μg/ml of gentamicin, NB30, NB54, or control conditions for 24 h prior to assay. Representative tracings using NB54 versus vehicle control are shown. b Summary data indicating forskolin-stimulated ISC. *P<0.05; **P<0.005; ±SEM, n=16
Fig. 4.
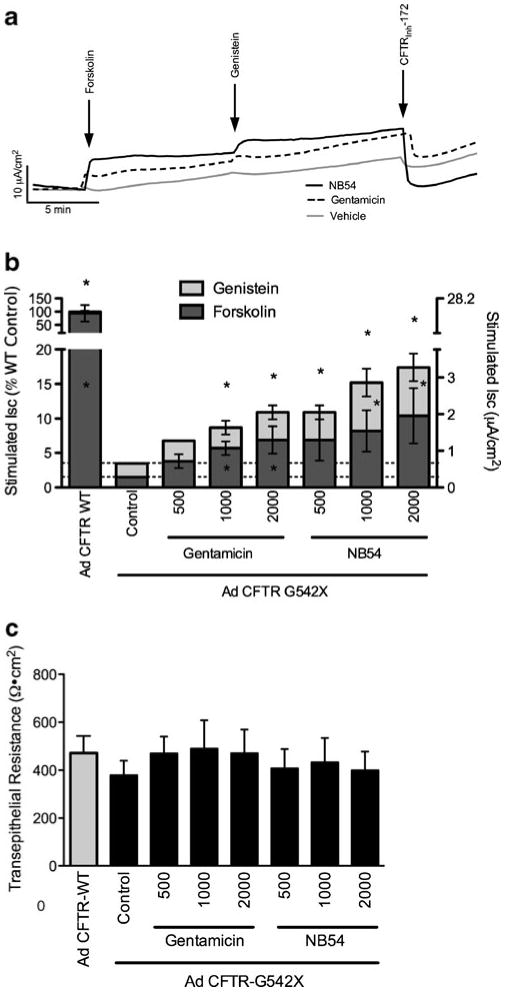
Short-circuit current assay of CFBE41o- cells transduced with an adenovirus expressing CFTR-G542X following treatment with gentamicin, NB30, or NB54. a Representative short-circuit current tracings of CFBE41o- cells were grown on air–liquid interface, transduced with adenovirus encoding CFTR-G542X, allowed to recover 48 h, and exposed to aminoglycosides (500 μg/ml) for 24 h. Monolayers were then mounted in modified Ussing chambers and examined under voltage clamp conditions. Cells were exposed to a chloride secretory gradient in the presence of amiloride (100 μM) then stimulated with sequential administration of the CFTR agonists forskolin (20 μM) and genistein (50 μM). CFTRInh-172 (10 μM) was added to confirm CFTR dependence. b Mean stimulated short-circuit currents measured in CFBE41o- cells transduced with adenovirus CFTR-G542X following treatment with the indicated doses of gentamicin or NB54. Short-circuit currents stimulated by forskolin and genistein are shown. *P<0.05, n=4 monolayers per condition, ±SEM. c Baseline transepithelial resistance for the experiments shown in b was measured under voltage clamp conditions prior to addition of ion transport agonists. No statistically significant differences were observed ±SEM
Rescue of primary human airway epithelial monolayers
Fully differentiated primary human bronchial epithelial mono-layers (HBE cells) have been shown to be highly predictive of in vivo results with the CFTR modulator VX-770, a CFTR potentiator recently shown to increase CFTR channel open probability and enhance CFTR-dependent chloride secretion [25]. These cells also represent an excellent model to determine the ability of synthetic aminoglycosides to partially restore CFTR function by suppressing PTCs expressed by the endogenous promoter. We obtained primary HBE cells from a compound heterozygote subject (G542X/F508del) following a lung transplant and examined the effects of NB54 on CFTR activity. These cells were cultured until fully differentiated monolayers formed [24, 35] and then were treated with either NB54 or gentamicin (250 μg/ml for 48 h). Concentrations were chosen based on previous experience with HBE cells and the limited number of cells available for study. We found that NB54 partially restored cAMP-stimulated ISC under these conditions, while gentamicin did not (Fig. 5). However, a comparable rescue of cAMP-stimulated ISC induced by gentamicin was observed after a more extended treatment period (96 h). Due to limited availability of these primary HBE cells, the effect of NB54 treatment for 96 h could not be determined. However, the cAMP-stimulated ISC observed following NB54 treatment for 48 h (0.8 μA/cm2) was significantly greater (P<0.05) than the same primary HBE cells treated with vehicle alone (0.4 μA/cm2). The increase in current obtained was equivalent to achieving ∼9.4% of CFTR activity based on the differences in current observed in a panel of CF and non-CF HBE cells. Importantly, this level of CFTR function was previously shown to correlate with improved clinical outcomes in CF subjects based on genotype–phenotype correlations in the disease [36]. There was no significant difference in the transepithelial resistance of cells following NB54 (202.6±51.6 Ω cm2) or gentamicin (141±4.9 Ω cm2) treatment compared to vehicle alone (144±30.7 Ω cm2), indicating that the treatment conditions used were not cytotoxic. From these experiments, we conclude that NB54 can induce a cAMP-stimulated ISC comparable to gentamicin in primary HBE cells and does so after a shorter treatment period.
Fig. 5.
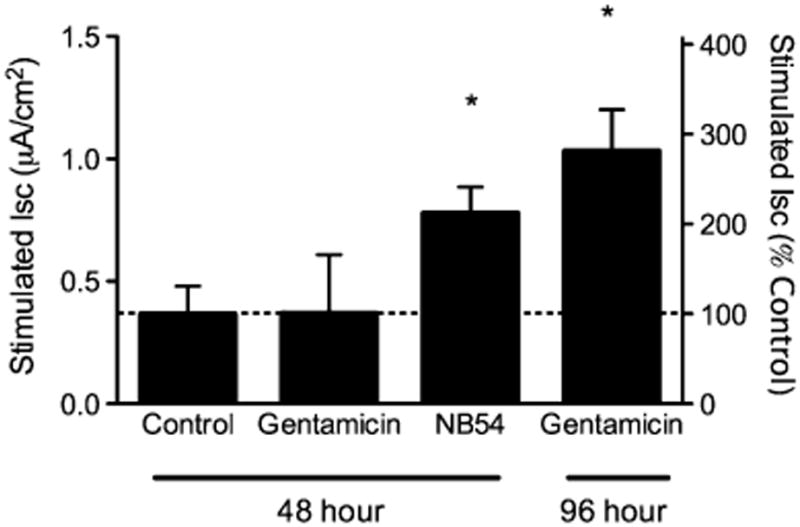
Stimulated short-circuit currents in CF primary human airway cells grown in polarizing conditions treated with gentamicin or NB54. Fully differentiated primary airway cells derived from a CF (G542X/F508Del) donor were grown at air–liquid interface until terminally differentiated (e.g., 90% ciliated), and then treated with NB54, gentamicin, or vehicle (500 μg/ml) for the times indicated, then mounted in modified Ussing chambers under voltage clamp conditions. Short-circuit current was stimulated with forskolin (20 μM) in the presence of amiloride and a chloride secretory gradient, followed by block with CFTRInh-172 (10 μM) to confirm CFTR dependence. Forskolin-dependent ISC is shown. *P<0.05, n=4–5 monolayers/condition, ±SEM
Readthrough of CFTR-G542X in vivo induced by the synthetic aminoglycoside NB54
To examine whether NB54 could suppress a CFTR PTC in vivo, we next utilized a transgenic human CFTR-G542X (hCFTR-G542X) mouse line [17, 31–33]. In this mouse model, the hCFTR-G542X transgene is expressed from the rat intestinal FABP promoter in a mouse Cftr−/− background. Samples from the ileum of untreated Cftr+/+ littermates carrying the transgene were used as positive controls (Fig. 6a), while corresponding samples from untreated Cftr−/− mice carrying the hCFTR-G542X transgene were used as negative controls (Fig. 6b). Representative ISC measurements from the ileum of mice treated with 60 mg/kg gentamicin (Fig. 6c) or 120 mg/kg NB54 are shown (Fig. 6d).
A scatter plot of the total ISC (ΔISC following forskolin/IBMX + carbachol) from all Cftr−/− hCFTR-G542X mice (untreated, gentamicin treated, and NB54 treated) is presented in Fig. 6e, while an analysis of that data is represented in Fig. 6f. In Cftr+/+ hCFTR-G542X mice, ileal tissues yielded a mean cAMP-stimulated ISC of 161 μA/cm2 10 min after forskolin/IBMX addition, and a further increase in ISC of 209 μA/cm2 following the addition of carbachol (a muscarinic agonist that transiently enhances the cAMP-dependent chloride secretory response in intestinal tissues by activating Ca2+-dependent potassium channels in the basolateral plasma membrane [37]). This yielded a total stimulated ISC (the sum of the two responses) of 370 μA/cm2. In contrast, stimulated responses were effectively absent in untreated Cftr−/− hCFTR-G542X control mice. We observed a mean ISC of 4.3 μA/cm2 after forskolin/IBMX addition and a mean ISC of 8.6 μA/cm2 following carbachol addition. This yielded a total stimulated ISC of only 12.8 μA/cm2 (3.5% of WT).
We initially compared the relative efficacy of gentamicin and NB54 to restore activated ISC, a measure of full-length CFTR protein function resulting from suppression of the hCFTR-G542X mutation. Each compound was administered once daily for 14 days by subcutaneous injection at doses ranging from 15 to 240 mg/kg (Fig. 6e, f). These doses were chosen based on our prior experience with aminoglycoside dosing for PTC suppression in vivo, with the goal of comparing both the toxicity and PTC suppression potential of NB54 under conditions that met or exceeded the optimal doses previously established for gentamicin [31–33]. We observed significant increases in the total stimulated short-circuit currents at 30, 60, and 120 mg/kg gentamicin. For the 30 mg/kg dose, the mean total ISC measured was 40.4 μA/cm2. This increased to 61.8 μA/cm2 in mice administered with 60 mg/kg gentamicin, the highest level of short-circuit current obtained with this compound. Mice treated with 120 mg/kg gentamicin showed a lower total ISC (41.8 μA/cm2), suggesting that a narrow window existed for optimal PTC suppression by gentamicin in vivo (similar to our observations in the IB3-1 cell line). We were unable to administer 240 mg/kg gentamicin for the 2-week treatment period due to toxicity, as indicated by significant weight loss that necessitated termination of the treatment protocol.
Next, we examined the ability of NB54 to induce read-through and restore CFTR activity in the Cftr−/− hCFTR-G542X transgenic mouse model. As before, NB54 was administered once daily by subcutaneous injection at doses ranging from 15 to 240 mg/kg (Fig. 6e, f). We observed significant increases in the total stimulated short-circuit currents at 30, 60, and 120 mg/kg (33.8, 34.4, and 58.5 μA/cm2, respectively). In contrast to gentamicin, we did not observe an attenuation of total stimulated short-circuit current in mice administered with 120 mg/kg NB54. Furthermore, we were able to successfully administer NB54 to mice at a dose of 240 mg/kg, which resulted in a further increase in the total stimulated ISC to 65.6 μA/cm2. These data indicate that NB54 induced a level of PTC suppression in vivo that is comparable to the peak level obtained with gentamicin (although at a twofold higher dose), that NB54 exhibited peak PTC suppression over a broader range of doses than gentamicin, and confirmed the previous finding that NB54 is less toxic than gentamicin in vivo [19].
To confirm that the stimulated ISC we observed in mouse intestine was due to partial restoration of human CFTR expression, we also carried out immunocytochemistry on samples from the mouse duodenum using a polyclonal antiserum that detects human CFTR, but not mouse CFTR [17, 31–33]. Only background staining was observed in untreated Cftr−/− hCFTR-G542X mice (Fig. 7a). In contrast, hCFTR-specific fluorescence concentrated primarily to the apical region of columnar epithelial cells lining the intestinal glands was observed in mice treated with either 60 mg/kg gentamicin (Fig. 7b) or 120 mg/kg NB54 (Fig. 7c). These results confirmed that the stimulated ISC observed correlated with the appearance of human CFTR protein at the apical surface of columnar cells in the intestinal glands following treatment with gentamicin or NB54.
Fig. 7.
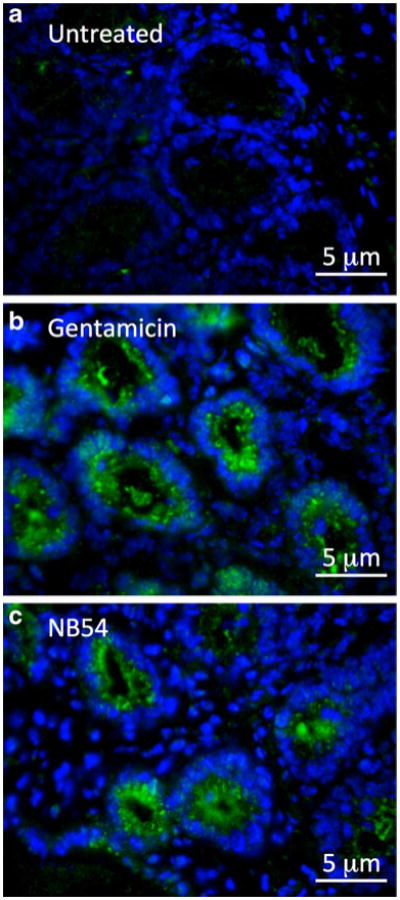
Human CFTR immunofluorescence in intestinal tissues from mice treated with gentamicin or NB54. Fixed tissue sections from the duodenum were incubated with CFTR-NBD1 antiserum that recognizes human, but not murine, CFTR. The human CFTR-specific primary antibody (rabbit IgG) was detected with anti-rabbit IgG (goat), Alexa Fluor 488-labeled secondary antibody (green). Nuclei were stained with DAPI (blue). Treatment consisted of a subcutaneous injection once daily with the indicated dose of compound for 2 weeks. a Samples from untreated Cftr−/− hCFTR-G542X, b samples from Cftr−/− hCFTR-G542X mice treated with 60 mg/kg gentamicin, and c samples from Cftr−/− hCFTR-G542X mice treated with 120 mg/kg NB54. A total of seven sections were examined for each strain and treatment condition with similar results
Discussion
Previous studies have shown that aminoglycosides can suppress translation termination at disease-causing PTCs and partially restore the expression of functional proteins in mammalian cells (for reviews, see [38–40]). In particular, gentamicin has been shown to suppress PTCs and partially restore protein expression in mouse models of Duchenne muscular dystrophy [10, 41] and CF [31, 32]. However, the use of aminoglycosides is commonly associated with serious side effects that limit long-term administration, including nephrotoxicity and ototoxicity [42, 43].
Aminoglycosides currently used in the clinic were developed based on their antibacterial properties, rather than the ability to suppress PTCs in mammalian genes. These antibacterial properties correlate with some features of the toxicity associated with this class of compounds. Accordingly, we pursued strategies to design derivatives that maintain (or enhance) readthrough of mammalian PTCs, while simultaneously reducing antibacterial activity and mammalian cytotoxicity. Initial attempts led to the synthesis of the novel synthetic aminoglycosides NB30 [20] and NB54 [19]. NB30, a pseudosaccharide derivative of the clinical aminoglycoside paromomycin, showed reduced toxicity but only limited readthrough potential in cell-based reporter assays [21]. NB54 improved on NB30 by the addition of an (S)-4-amino-2 hydroxybutanoyl (AHB) group at the N-1 position, which is predicted to increase the binding affinity to helix 44 in the ribosomal decoding site (Fig. 1). The AHB group is found on the aminoglycoside amikacin, which has been shown to induce PTC readthrough in CF models [32]. The importance of the AHB moiety is clearly demonstrated by the superior readthrough associated with amikacin compared to kanamycin A, which differs from amikacin only by the absence of the AHB moiety. Kanamycin A does not show measurable readthrough activity in mammalian translation systems [11]. In a preliminary study, the AHB modification on NB54 was shown to enhance readthrough in cell-based reporter assays while exhibiting a significantly lower toxicity profile than gentamicin [19].
Here, we extended the characterization of NB30 and NB54 using a series of cell-based and in vivo CF model systems, and show that these agents maintain (and in some cases, enhance) readthrough efficacy while reducing toxicity. We tested the ability of these compounds to suppress two CFTR PTCs (G542X and W1282X) in a variety of CF cellular models, including heterologous CFTR expression systems, immortalized CF cell lines, and primary HBE cells isolated from a CF lung. In addition, we examined the ability of these compounds to suppress the human CFTR-G542X mutation in a transgenic mouse model. The broad range of CF models examined in this study provides a unique and comprehensive analysis of the efficacy and toxicity of these compounds for the treatment of CFTR PTCs. This study demonstrates for the first time that synthetic aminoglycosides rationally designed to augment readthrough have successfully suppressed PTCs in clinically relevant human cells and an animal model of this (or any) genetic disease.
We found that NB30 and NB54 treatment restored a level of cAMP-stimulated halide transport that was comparable to gentamicin in both HeLa cells stably transduced with a lentiviral system expressing CFTR-W1282X and in the IB3-1 bronchial epithelial cell line (W1282X/F508del, Fig. 2). While the doses of NB30 and NB54 required for maximal responses were generally higher than those required for gentamicin, the toxicity observed was also significantly less. The maximal ISC measured in CFBE41o-airway epithelial monolayers stably transduced with a lentivirus vector expressing CFTR-W1282X or an adenovirus vector expressing CFTR-G542X was consistently higher with NB54 than gentamicin, and NB54 was also nontoxic at these doses (Figs. 3 and 4). NB54 also induced a cAMP-stimulated ISC that was comparable to gentamicin in primary HBE cells carrying the CFTR-G542X mutation, and the induction of that response occurred sooner than we observed with gentamicin treatment (Fig. 5). Notably, primary HBE cells were shown to correlate well with in vivo results in a recent study using the CFTR modulator VX-770 [25, 44]. When taken together, the results of these studies indicate that NB54 is capable of suppressing these CFTR PTC mutations at least as efficiently as gentamicin and is tolerated at higher concentrations in vitro. The differences in the effective doses of these compounds observed in different cell lines are most likely due to differences in their cellular uptake or the effects of baseline levels of expression, and highlight the importance of using a variety of complementary cell types to evaluate new compounds for PTC suppression. Other factors known to influence readthrough efficiency include nonsense-mediated decay, the identity of the PTC to be suppressed, and the sequence context surrounding the PTC (particularly the first 3′ nucleotide). Our studies did not examine the relative effects of synthetic aminoglycosides on these factors, but they should be examined in future studies.
As a further test of the ability of NB54 to suppress CF-related PTCs, we examined the propensity of NB54 to restore human CFTR expression in Cftr−/− hCFTR-G542X transgenic mice (Fig. 6). We found that NB54 induced significant and dose-dependent increases in stimulated ISC in Cftr−/− hCFTR-G542X transgenic mice, resulting in 15.8% of the current measured in WT mice at a dose of 120 mg/kg. This level of stimulated ISC was comparable to the maximal response obtained with gentamicin (60 mg/kg). While a higher dose of NB54 was required to restore a comparable level of CFTR activity, we found that NB54 was also tolerated at higher doses in vivo. When taken together, these results indicate that NB54 can induce comparable readthrough of CFTR PTCs in vitro and in vivo and is also significantly less toxic than gentamicin. Since it was demonstrated in a previous study that gentamicin and PTC124 (ataluren) restored a comparable level of CFTR activity in the same hCFTR-G542X Cftr−/− mouse model [17], the level of CFTR function restored by NB54 should be similar to PTC124 as well.
The overall concordance of the relative levels of PTC suppression obtained following NB54 treatment in most of the CF models tested was reassuring, given that major differences in exposure and/or drug uptake by cultured cells relative to murine tissues are likely. The uptake of aminoglycosides by cultured cells is probably primarily driven by constant high concentrations of the compound in the culture medium, resulting in nonspecific uptake playing a prominent role. In contrast, once daily administration used in the animal model results in a transient peak serum level minutes after administration, followed by re-equilibration to a trough level shortly thereafter [32]. This inherently short serum half-life relegates a majority of the uptake to different (and potentially more specific) aminoglycoside transport mechanisms, such as the cationic endocytic receptor megalin [45]. While the differences between the results in vivo and in the cell lines were generally small, they also point to the continued need to determine the model systems most predictive of response to readthrough agents in the clinic.
The results described in this study have important implications. First, they provide the first demonstration that a synthetic aminoglycoside specifically designed to maintain PTC suppression with significantly reduced toxicity can partially restore protein function in an animal model of a genetic disease [19]. They also support testing the synthetic aminoglycosides NB30 and NB54 in models of other genetic disorders caused by nonsense mutations. Finally, the current results support continuing systematic research efforts towards the development of synthetic aminoglycosides to maximize drug-induced suppression while minimizing toxicity. Further progress in this regard could provide additional advantages in efficacy and long-term tolerability that may be necessary to support the widespread use of this novel therapeutic approach to treat genetic diseases caused by nonsense mutations.
Supplementary Material
Acknowledgments
This research was funded by the US National Institute of Health grants 1K23DK075788-01 and 1R03DK084110-01 (to SMR), 1P30DK072482-01A1 and Cystic Fibrosis Foundation grant R464-CR02 (to EJS), US-Israel Binational Science Foundation grant 2006/301 and Mitchel Fund 2012386 (to TB), and Horowitz Fund (grant no. 2010826 to TB and DMB). VB acknowledges the financial support by the Center of Absorption in Science, the Ministry of Immigration Absorption, and the Ministry of Science and Technology, Israel (Kamea Program). We are grateful to Cheryl Owens for assistance with preparation of the manuscript.
The authors thank Kim Keeling for critically reading the manuscript. This study was supported by US National Institute of Health grants 1K23DK075788-01 and 1R03DK084110-01 (to SMR), 1P30DK072482-01A1 and Cystic Fibrosis Foundation grant R464-CR02 (to EJS), US-Israel Binational Science Foundation grant 2006/301 and Mitchel Fund 2012386 (to TB), and Horowitz Fund (grant no. 2010826 to TB and DMB). VB acknowledges the financial support by the Center of Absorption in Science, the Ministry of Immigration Absorption, and the Ministry of Science and Technology, Israel (Kamea Program).
Abbreviations
- AHB
(S)-4-Amino-2 hydroxybutanoyl
- CF
Cystic fibrosis
- CFTR
Cystic fibrosis transmembrane conductance regulator
- ISC
Short-circuit current
- PTC
Premature termination codons
- SPQ
6-Methoxy-N-(3-sulfopropyl)-quinolinium
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00109-011-0787-6) contains supplementary material, which is available to authorized users.
Disclosure statement SMR serves on the advisory board for PTC Therapeutics, Inc. DMB serves as a consultant for PTC Therapeutics, Inc.
Contributor Information
Steven M. Rowe, Email: smrowe@uab.edu, Department of Medicine, University of Alabama at Birmingham, Birmingham, AL, USA; Department of Pediatrics, University of Alabama at Birmingham, Birmingham, AL, USA; Department of Physiology and Biophysics, University of Alabama at Birmingham, Birmingham, AL, USA; Gregory Fleming James Cystic Fibrosis Research Center, University of Alabama at Birmingham, Birmingham, AL, USA; University of Alabama at Birmingham, MCLM 768, 1500 3rd Ave South, Birmingham, AL 35294-0005, USA.
Peter Sloane, Department of Medicine, University of Alabama at Birmingham, Birmingham, AL, USA; Gregory Fleming James Cystic Fibrosis Research Center, University of Alabama at Birmingham, Birmingham, AL, USA.
Li Ping Tang, Department of Medicine, University of Alabama at Birmingham, Birmingham, AL, USA; Gregory Fleming James Cystic Fibrosis Research Center, University of Alabama at Birmingham, Birmingham, AL, USA.
Kyle Backer, Department of Medicine, University of Alabama at Birmingham, Birmingham, AL, USA; Gregory Fleming James Cystic Fibrosis Research Center, University of Alabama at Birmingham, Birmingham, AL, USA.
Marina Mazur, Gregory Fleming James Cystic Fibrosis Research Center, University of Alabama at Birmingham, Birmingham, AL, USA.
Jessica Buckley-Lanier, Department of Microbiology, University of Alabama at Birmingham, Birmingham, AL, USA.
Igor Nudelman, The Edith and Joseph Fisher Enzyme Inhibitors Laboratory, Schulich Faculty of Chemistry, Technion-Israel Institute of Technology, Haifa 32000, Israel.
Valery Belakhov, The Edith and Joseph Fisher Enzyme Inhibitors Laboratory, Schulich Faculty of Chemistry, Technion-Israel Institute of Technology, Haifa 32000, Israel.
Zsuzsa Bebok, Gregory Fleming James Cystic Fibrosis Research Center, University of Alabama at Birmingham, Birmingham, AL, USA; Department of Cell Biology, University of Alabama at Birmingham, Birmingham, AL, USA.
Erik Schwiebert, DiscoveryBioMed, Inc., Innovation Depot, Birmingham, AL, USA.
Timor Baasov, The Edith and Joseph Fisher Enzyme Inhibitors Laboratory, Schulich Faculty of Chemistry, Technion-Israel Institute of Technology, Haifa 32000, Israel.
David M. Bedwell, Department of Physiology and Biophysics, University of Alabama at Birmingham, Birmingham, AL, USA; Department of Microbiology, University of Alabama at Birmingham, Birmingham, AL, USA; Gregory Fleming James Cystic Fibrosis Research Center, University of Alabama at Birmingham, Birmingham, AL, USA
References
- 1.Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352:1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- 2.Anonymous. Correlation between genotype and phenotype in patients with cystic fibrosis. The Cystic Fibrosis Genotype-Phenotype Consortium. N Engl J Med. 1993;329:1308–1313. doi: 10.1056/NEJM199310283291804. [DOI] [PubMed] [Google Scholar]
- 3.Shoshani T, Kerem E, Szeinberg A, Augarten A, Yahav Y, Cohen D, Rivlin J, Tal A, Kerem B. Similar levels of mRNA from the W1282X and the delta F508 cystic fibrosis alleles, in nasal epithelial cells. J Clin Invest. 1994;93:1502–1507. doi: 10.1172/JCI117128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kerem E. Pharmacologic therapy for stop mutations: how much CFTR activity is enough? Curr Opin Pulm Med. 2004;10:547–552. doi: 10.1097/01.mcp.0000141247.22078.46. [DOI] [PubMed] [Google Scholar]
- 5.Keeling KM, Brooks DA, Hopwood JJ, Li P, Thompson JN, Bedwell DM. Gentamicin-mediated suppression of Hurler syndrome stop mutations restores a low level of alpha-l-iduronidase activity and reduces lysosomal glycosaminoglycan accumulation. Hum Mol Genet. 2001;10:291–299. doi: 10.1093/hmg/10.3.291. [DOI] [PubMed] [Google Scholar]
- 6.Howard M, Frizzell RA, Bedwell DM. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat Med. 1996;2:467–469. doi: 10.1038/nm0496-467. [DOI] [PubMed] [Google Scholar]
- 7.Bedwell DM, Kaenjak A, Benos DJ, Bebok Z, Bubien JK, Hong J, Tousson A, Clancy JP, Sorscher EJ. Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nat Med. 1997;3:1280–1284. doi: 10.1038/nm1197-1280. [DOI] [PubMed] [Google Scholar]
- 8.Tok JB, Bi L. Aminoglycoside and its derivatives as ligands to target the ribosome. Curr Top Med Chem. 2003;3:1001–1019. doi: 10.2174/1568026033452131. [DOI] [PubMed] [Google Scholar]
- 9.Bonetti B, Fu L, Moon J, Bedwell DM. The efficiency of translation termination is determined by a synergistic interplay between upstream and downstream sequences in Saccharomyces cerevisiae. J Mol Biol. 1995;251:334–345. doi: 10.1006/jmbi.1995.0438. [DOI] [PubMed] [Google Scholar]
- 10.Barton-Davis ER, Cordier L, Shoturma DI, Leland SE, Sweeney HL. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Invest. 1999;104:375–381. doi: 10.1172/JCI7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manuvakhova M, Keeling K, Bedwell DM. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA. 2000;6:1044–1055. doi: 10.1017/s1355838200000716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clancy JP, Bebok Z, Ruiz F, King C, Jones J, Walker L, Greer H, Hong J, Wing L, Macaluso M, et al. Evidence that systemic gentamicin suppresses premature stop mutations in patients with cystic fibrosis. Am J Respir Crit Care Med. 2001;163:1683–1692. doi: 10.1164/ajrccm.163.7.2004001. [DOI] [PubMed] [Google Scholar]
- 13.Sermet-Gaudelus I, Renouil M, Fajac A, Bidou L, Parbaille B, Pierrot S, Davy N, Bismuth E, Reinert P, Lenoir G, et al. In vitro prediction of stop-codon suppression by intravenous gentamicin in patients with cystic fibrosis: a pilot study. BMC Med. 2007;5:5–14. doi: 10.1186/1741-7015-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilschanski M, Famini C, Blau H, Rivlin J, Augarten A, Avital A, Kerem B, Kerem E. A pilot study of the effect of gentamicin on nasal potential difference measurements in cystic fibrosis patients carrying stop mutations. Am J Respir Crit Care Med. 2000;161:860–865. doi: 10.1164/ajrccm.161.3.9904116. [DOI] [PubMed] [Google Scholar]
- 15.Wilschanski M, Yahav Y, Yaacov Y, Blau H, Bentur L, Rivlin J, Aviram M, Bdolah-Abram T, Bebok Z, Shushi L, et al. Gentamicin-induced correction of CFTR function in patients with cystic fibrosis and CFTR stop mutations. N Engl J Med. 2003;349:1433–1441. doi: 10.1056/NEJMoa022170. [DOI] [PubMed] [Google Scholar]
- 16.Clancy JP, Rowe SM, Bebok Z, Aitken ML, Gibson R, Zeitlin P, Berclaz P, Moss R, Knowles MR, Oster RA, et al. No detectable improvements in cystic fibrosis transmembrane conductance regulator by nasal aminoglycosides in patients with cystic fibrosis with stop mutations. Am J Respir Cell Mol Biol. 2007;37:57–66. doi: 10.1165/rcmb.2006-0173OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Du M, Liu X, Welch EM, Hirawat S, Peltz SW, Bedwell DM. PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model. Proc Natl Acad Sci U S A. 2008;105:2064–2069. doi: 10.1073/pnas.0711795105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kerem E, Hirawat S, Armoni S, Yaakov Y, Shoseyov D, Cohen M, Nissim-Rafinia M, Blau H, Rivlin J, Aviram M, et al. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: a prospective phase II trial. Lancet. 2008;372:719–727. doi: 10.1016/S0140-6736(08)61168-X. [DOI] [PubMed] [Google Scholar]
- 19.Nudelman I, Rebibo-Sabbah A, Cherniavsky M, Belakhov V, Hainrichson M, Chen F, Schacht J, Pilch DS, Ben-Yosef T, Baasov T. Development of novel aminoglycoside (NB54) with reduced toxicity and enhanced suppression of disease-causing premature stop mutations. J Med Chem. 2009;52:2836–2845. doi: 10.1021/jm801640k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nudelman I, Rebibo-Sabbah A, Shallom-Shezifi D, Hainrichson M, Stahl I, Ben-Yosef T, Baasov T. Redesign of aminoglycosides for treatment of human genetic diseases caused by premature stop mutations. Bioorg Med Chem Lett. 2006;16:6310–6315. doi: 10.1016/j.bmcl.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 21.Rebibo-Sabbah A, Nudelman I, Ahmed ZM, Baasov T, Ben-Yosef T. In vitro and ex vivo suppression by aminoglycosides of PCDH15 nonsense mutations underlying type 1 Usher syndrome. Hum Genet. 2007;122:373–381. doi: 10.1007/s00439-007-0410-7. [DOI] [PubMed] [Google Scholar]
- 22.Wu X, Wakefield JK, Liu H, Xiao H, Kralovics R, Prchal JT, Kappes JC. Development of a novel trans-lentiviral vector that affords predictable safety. Mol Ther. 2000;2:47–55. doi: 10.1006/mthe.2000.0095. [DOI] [PubMed] [Google Scholar]
- 23.Rowe SM, Varga K, Rab A, Bebok Z, Byram K, Li Y, Sorscher EJ, Clancy JP. Restoration of W1282X CFTR activity by enhanced expression. Am J Respir Cell Mol Biol. 2007;37:347–356. doi: 10.1165/rcmb.2006-0176OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rowe SM, Pyle LC, Jurkevante A, Varga K, Collawn J, Sloane PA, Woodworth B, Mazur M, Fulton J, Fan L, et al. DeltaF508 CFTR processing correction and activity in polarized airway and non-airway cell monolayers. Pulm Pharmacol Ther. 2010;23:268–278. doi: 10.1016/j.pupt.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, Turnbull A, Singh A, Joubran J, Hazlewood A, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A. 2009;106:18825–18830. doi: 10.1073/pnas.0904709106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clancy JP, Hong JS, Bebok Z, King SA, Demolombe S, Bedwell DM, Sorscher EJ. Cystic fibrosis transmembrane conductance regulator (CFTR) nucleotide-binding domain 1 (NBD-1) and CFTR truncated within NBD-1 target to the epithelial plasma membrane and increase anion permeability. Biochemistry. 1998;37:15222–15230. doi: 10.1021/bi980436f. [DOI] [PubMed] [Google Scholar]
- 27.Cobb BR, Ruiz F, King CM, Fortenberry J, Greer H, Kovacs T, Sorscher EJ, Clancy JP. A(2) adenosine receptors regulate CFTR through PKA and PLA(2) Am J Physiol Lung Cell Mol Physiol. 2002;282:L12–L25. doi: 10.1152/ajplung.2002.282.1.L12. [DOI] [PubMed] [Google Scholar]
- 28.Sermet-Gaudelus I, Vallee B, Urbin I, Torossi T, Marianovski R, Fajac A, Feuillet MN, Bresson JL, Lenoir G, Bernaudin JF, Edelman A. Normal function of the cystic fibrosis conductance regulator protein can be associated with homozygous (delta)F508 mutation. Pediatr Res. 2002;52:628–635. doi: 10.1203/00006450-200211000-00005. [DOI] [PubMed] [Google Scholar]
- 29.Jurkuvenaite A, Varga K, Nowotarski K, Kirk KL, Sorscher EJ, Li Y, Clancy JP, Bebok Z, Collawn JF. Mutations in the amino terminus of the cystic fibrosis transmembrane conductance regulator enhance endocytosis. J Biol Chem. 2006;281:3329–3334. doi: 10.1074/jbc.M508131200. [DOI] [PubMed] [Google Scholar]
- 30.Ratcliff R, Evans MJ, Cuthbert AW, MacVinish LJ, Foster D, Anderson JR, Colledge WH. Production of a severe cystic fibrosis mutation in mice by gene targeting. Nat Genet. 1993;4:35–41. doi: 10.1038/ng0593-35. [DOI] [PubMed] [Google Scholar]
- 31.Du M, Jones JR, Lanier J, Keeling KM, Lindsey RJ, Tousson A, Bebok Z, Whitsett JA, Dey CR, Colledge WH, et al. Aminoglycoside suppression of a premature stop mutation in a Cftr−/− mouse carrying a human CFTR-G542X transgene. J Mol Med. 2002;80:595–604. doi: 10.1007/s00109-002-0363-1. [DOI] [PubMed] [Google Scholar]
- 32.Du M, Keeling KM, Fan L, Liu X, Kovacs T, Sorscher E, Bedwell DM. Clinical doses of amikacin provide more effective suppression of the human CFTR-G542X stop mutation than gentamicin in a transgenic CF mouse model. J Mol Med. 2006;84:573–582. doi: 10.1007/s00109-006-0045-5. [DOI] [PubMed] [Google Scholar]
- 33.Du M, Keeling KM, Fan L, Liu X, Bedwell DM. Poly-l-aspartic acid enhances and prolongs gentamicin-mediated suppression of the CFTR-G542X mutation in a cystic fibrosis mouse model. J Biol Chem. 2009;284:6885–6892. doi: 10.1074/jbc.M806728200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Varga K, Jurkuvenaite A, Wakefield J, Hong JS, Guimbellot JS, Venglarik CJ, Niraj A, Mazur M, Sorscher EJ, Collawn JF, Bebok Z. Efficient intracellular processing of the endogenous cystic fibrosis transmembrane conductance regulator in epithelial cell lines. J Biol Chem. 2004;279:22578–22584. doi: 10.1074/jbc.M401522200. [DOI] [PubMed] [Google Scholar]
- 35.Pyle LC, Fulton JC, Sloane PA, Backer K, Mazur M, Prasain J, Barnes S, Clancy JP, Rowe SM. Activation of the cystic fibrosis transmembrane conductance regulator by the flavonoid quercetin: potential use as a biomarker of deltaF508 cystic fibrosis transmembrane conductance regulator rescue. Am J Respir Cell Mol Biol. 2009;43:607–616. doi: 10.1165/rcmb.2009-0281OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wilschanski M, Dupuis A, Ellis L, Jarvi K, Zielenski J, Tullis E, Martin S, Corey M, Tsui LC, Durie P. Mutations in the cystic fibrosis transmembrane regulator gene and in vivo transepithelial potentials. Am J Respir Crit Care Med. 2006;174:787–794. doi: 10.1164/rccm.200509-1377OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bijvelds MJ, Bot AG, Escher JC, De Jonge HR. Activation of intestinal Cl− secretion by lubiprostone requires the cystic fibrosis transmembrane conductance regulator. Gastroenterology. 2009;137:976–985. doi: 10.1053/j.gastro.2009.05.037. [DOI] [PubMed] [Google Scholar]
- 38.Rowe SM, Clancy JP. Pharmaceuticals targeting nonsense mutations in genetic diseases: progress in development. BioDrugs. 2009;23:165–174. doi: 10.2165/00063030-200923030-00003. [DOI] [PubMed] [Google Scholar]
- 39.Keeling K, Bedwell DM. Pharmacological suppression of premature stop mutations that cause genetic diseases. Current Pharmacogenomics. 2005;3:259–269. [Google Scholar]
- 40.Keeling K, Bedwell DM. Recoding therapies for genetic diseases. In: Gesteland R, Atkins J, editors. Recoding: expansion of decoding rules enriches gene expression. Springer; New York: 2010. pp. 123–146. [Google Scholar]
- 41.Malik V, Rodino-Klapac LR, Viollet L, Wall C, King W, Al-Dahhak R, Lewis S, Shilling CJ, Kota J, Serrano-Munuera C, et al. Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann Neurol. 2010;67:771–780. doi: 10.1002/ana.22024. [DOI] [PubMed] [Google Scholar]
- 42.Mingeot-Leclercq MP, Tulkens PM. Aminoglycosides: nephrotoxicity. Antimicrob Agents Chemother. 1999;43:1003–1012. doi: 10.1128/aac.43.5.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hutchin T, Cortopassi G. Proposed molecular and cellular mechanism for aminoglycoside ototoxicity. Antimicrob Agents Chemother. 1994;38:2517–2520. doi: 10.1128/aac.38.11.2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rowe SM, Clancy JP, Boyle M, Van Goor F, Ordonez C, Dong Q, Campbell P, Ashlock M, Accurso F. Parallel effects of VX-770 on transepithelial potential difference in vitro and in vivo. J Cyst Fibros. 2010;9:S20. [Google Scholar]
- 45.Moestrup SK, Cui S, Vorum H, Bregengard C, Bjorn SE, Norris K, Gliemann J, Christensen EI. Evidence that epithelial glycoprotein 330/megalin mediates uptake of polybasic drugs. J Clin Invest. 1995;96:1404–1413. doi: 10.1172/JCI118176. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.