

Abstract
Histone deacetylase inhibitors have emerged as a new class of anticancer therapeutic drugs. Their clinical utility in oncology stems from their intrinsic cytotoxic properties and combinatorial effects with other conventional cancer therapies. To date, the histone deacetylase inhibitors suberoylanilide hydroxamic acid (Vorinostat, Zolinza®) and depsipeptide (Romidepsin, Istodax®) have been approved by the US Food and Drug Administration for the treatment of refractory cutaneous T-cell lymphoma. Further, there are currently over 100 clinical trials involving the use of histone deacetylase inhibitors in a wide range of solid and hematological malignancies. The therapeutic potential of histone deacetylase inhibitors has also been investigated for numerous other diseases. For example, the cytotoxic properties of histone deacetylase inhibitors are currently being harnessed as a potential treatment for malaria, whereas the efficacy of these compounds for HIV relies on de-silencing latent virus. The anti-inflammatory properties of histone deacetylase inhibitors are the predominant mechanisms for other diseases, such as hepatitis, systemic lupus erythematosus and a wide range of neurodegenerative conditions. Additionally, histone deacetylase inhibitors have been shown to be efficacious in animal models of cardiac hypertrophy and asthma. Broad-spectrum histone deacetylase inhibitors are clinically available and have been used almost exclusively in preclinical systems to date. However, it is emerging that class- or isoform-specific compounds, which are becoming more readily available, may be more efficacious particularly for non-oncological applications. The aim of this review is to provide an overview of the effects and clinical potential of histone deacetylase inhibitors in various diseases. Apart from applications in oncology, the discussion is focused on the potential efficacy of histone deacetylase inhibitors for the treatment of neurodegenerative diseases, cardiac hypertrophy and asthma.
Keywords: Chromatin modifications, histone acetylation, histone deacetylase inhibitor, Trichostatin A, neurodegeneration, cardiac hypertrophy, asthma
Introduction
Chromatin is a dynamic structure that undergoes remodeling to facilitate metabolic processes including transcription, replication and repair [1]. These structural changes are mediated largely by DNA methylation and post-translational modifications of histones. Of the various post-translational modifications, histone acetylation is relatively well-characterized, with the first reports highlighting the importance of this modification in RNA synthesis dating to 1964 [2, 3]. Histone acetylation status is regulated by the opposing actions of histone acetyl-transferases (HATs) and histone deacetylases (HDACs) [4]. HATs transfer the acetyl moiety of acetyl-coA resulting in acetylation of the ε-amino tails of lysine residues in histones [5]. This neutralizes the positive charge on histone tails, weakening the interaction between histones and negatively DNA, yielding a more open, transcriptionally permissive chromatin conformation [4, 5]. Conversely, HDACs remove acetyl groups from histones resulting in a more condensed, transcriptionally repressive chromatin conformation [6]. In addition to the core H2A, H2B, H3 and H4 histones, numerous non-histones proteins are targets for acetylation / deacetylation. These include key cell motility proteins (e.g. α-tubulin, cortactin), chaperones (e.g. HSP90, HSP70), DNA repair proteins (e.g. Ku70, Ku86) and transcription factors and co-regulators (e.g. p53, MyoD, c-Myc) [7-10].
The 18 mammalian HDAC enzymes identified to date are classified into two distinct families – the metal dependent enzymes which are represented by class I, II and IV HDACs and the class III sirtuins [7, 11-13]. Class III HDACs include sirtuins 1-7 which are homologous to the yeast silent information regulator 2 [14]. Deacetylation of lysine residues by sirtuins requires consumption nicotinamide adenine dinucleotide (NAD+) [14]. The metal-dependent enzymes are typically referred to as the classical HDACs and require co-ordination of divalent metal ion for catalytic activity [15]. The 11 classical HDACs are categorized into three classes based on their homology to yeast proteins (Figure 1) [15-18]. Briefly, class I enzymes include HDAC1, 2, 3 and 8 and share homology with the yeast transcriptional regulator RDP3 [16, 17]. They are expressed ubiquitously, localized predominantly in the nucleus and HDACs 1-3 are part of multi-protein nuclear repressor proteins [12, 13]. Overall, it is thought that class I enzymes have a critical role in cell survival and proliferation [12, 13, 19]. Class II enzymes are related to yeast HDA1 and are further subdivided into IIa (HDACs 4, 5, 7 and 9) and IIb (HDACs 6 and 10) [20, 21]. They shuttle between the nucleus and cytoplasm and have more tissue-specific expression patterns and functions [8, 10, 12, 13]. HDAC6 is a major cytoplasmic protein with key substrates such as α-tubulin, HSP90 and the important redox regulatory proteins peroxiredoxin I and II [22, 23]. It has diverse roles in modulating cell growth, migration and cellular survival [24-26].
Figure 1.

Schematic representation of the classical class I, II and IV mammalian histone deacetylases (HDACs). Class I enzymes are homologous with the yeast, reduced potassium dependency-3 (Rpd3) and consist of HDAC1, 2, 3 and 8. The class II HDACs are homologous to the yeast, histone deacetylase-1 (Hda1) and enzymes in this class are further subdivided into two sub-classes. Class IIa is comprised of HDAC4, 5, 7 and 9; class IIb consists of HDAC6 and 10. The HDACs have a conserved deacetylase (DAC) domain depicted as a blue cylinder with the C- and N-terminal tails represented as black lines. Green cylinders represent the myocyte enhancer factor-2 (MEF2)-binding domains and short grey cylinders depict the 14-3-3 binding motifs with Ser phosphorylation sites. The number of amino acid residues of the longest isoform of each HDAC is shown on the right and the chromosomal site of each HDAC is shown in brackets. H. sapiens, Homo sapiens; S. cerevisiae, Saccharomyces cerevisiae; SE14, Ser-Glu containing tetradecapeptide repeats; ZnF, ubiquitin-binding zinc finger domain. Adapted from [18].
Histone deacetylase inhibitors: anticancer effects
HDAC enzymes have important roles in modulating proliferation, apoptosis, migration and differentiation [12, 13]. Further, aberrant HDAC expression and activity has been observed in numerous malignancies [27-33]. These provided the basis for the development of HDAC inhibitors as anticancer therapies. Indeed, the HDAC inhibitors suberoylanilide hydroxamic acid (SAHA, Vorinostat, Zolinza®) and depsipeptide (Romidepsin, Istodax®) have been approved by the US Food and Drug Administration (FDA) for the treatment of refractory cutaneous T-cell lymphoma, in 2006 and 2009, respectively [34, 35].
Briefly, the main structural groups of HDAC inhibitors include the hydroxamic acids such as Trichostatin A, SAHA and Panobinostat (LBH5890) and the cyclic peptides which include depsipeptide and trapoxin [7, 12, 13, 17, 36]. These are relatively potent HDAC inhibitors with activity in the nanomolar to low micromolar range. The benzamides (e.g. Entinostat) and electrophilic ketones (e.g. α-ketomide) are also potent HDAC inhibitors [7, 36]. The short-chain fatty acids which include phenylbutyrate, sodium butyrate and valproic acid are less potent with HDAC inhibition activity in the millimolar range [7, 36]. Although these HDAC inhibitors show some selectivity in cell-free assay systems, they are typically referred to as broad-spectrum compounds as they inhibit multiple class I, II and IV HDAC enzymes. Although it is still controversial as to whether they would provide a therapeutic advantage, particularly in oncology, more class-selective and isoform-selective analogues are increasingly becoming available. Prime examples include tubacin and PC-34501 which selectively inhibit HDACs 6 and 8, respectively [23, 37-39].
In summary, HDAC inhibitors decrease proliferation, induce cell-death and apoptosis, cause differentiation and cell cycle arrest and alter migration in malignant and transformed cell lines (Figure 2) [12, 13, 40]. The cytotoxic effects of HDAC inhibitors are much more pronounced, in malignant and transformed cells compared to normal cells. This provides the advantageous therapeutic window in oncology. In addition to their intrinsic cytotoxic properties HDAC inhibitors have been shown to induce at least additive cytotoxic effects with other anti-cancer modalities (Figure 2). Effective combinations with HDAC inhibitors include those with conventional cancer modalities such as chemotherapy (anthracyclines and retinoic acid) and radiotherapy [8, 12, 13, 41-45]. Further, HDAC inhibitors have been shown to augment the effects ultraviolet radiation and range other biological agents such as tumor-necrosis-factor apoptosis-inducing ligand [46, 47]. The clinical potential of HDAC inhibitors in oncology is highlighted by the fact that there are currently there are over 100 clinical trials involving these compounds for a wide range of malignancies.
Figure 2.
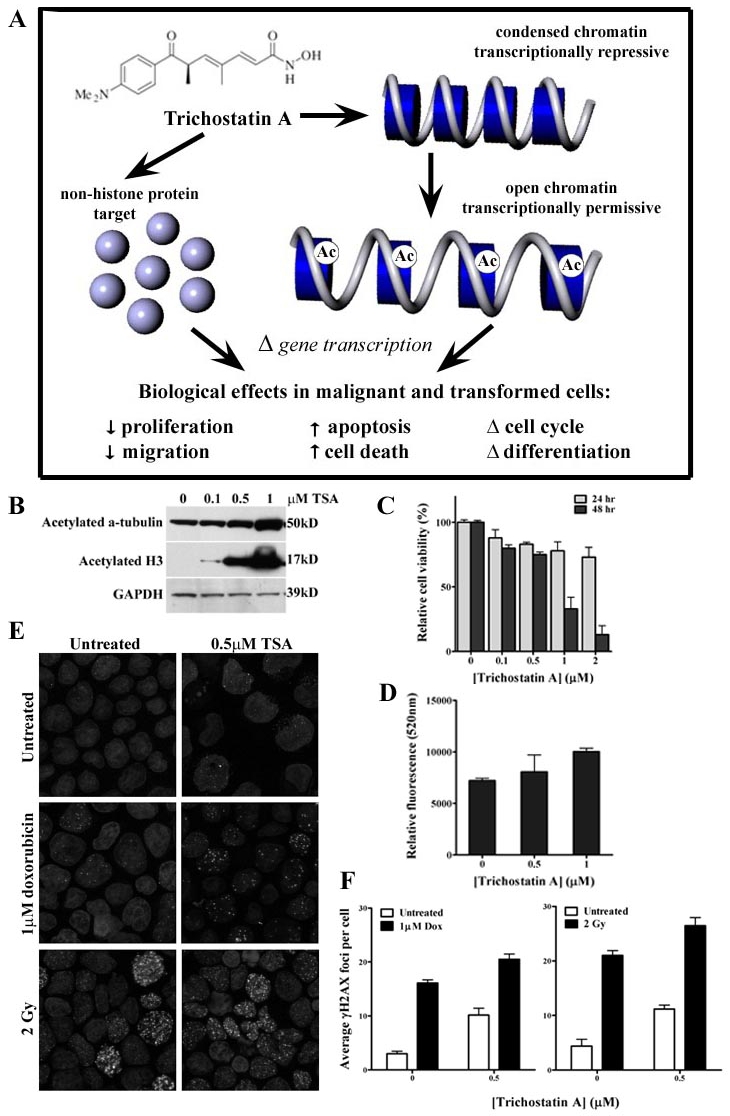
Summary of the biological effects of the prototypical broad-spectrum HDAC inhibitor (HDAC inhibitors) Trichostatin A (TSA). (A) TSA alters gene transcription by inhibiting histone deacetylase activity and remodelling chromatin architecture. Hyperacetylation of the core histones results in a more open, transcriptionally permissive chromatin structure. TSA also results in the deacetylation of numerous non-histone protein targets. The overall effect of TSA in malignant and transformed cells is decreased proliferation, increased cell-death, induction of apoptosis, decreased migration, cell cycle arrest (predominantly G1 arrest and G1 and G2/M arrest at higher concentrations) and differentiation. (B) TSA results in the accumulation of hyperacetylated histone H3 and α-tubulin in T-cell leukemic CEM-CCRF cells. Cells were treated with the indicated concentrations of TSA for 24 hours prior to extraction of whole cells lysates. Immunoblots of acetylated histone H3 and α-tubulin with a GAPDH loading control are shown. (C) TSA decreases the relative cell viability of CEM-CCRF cells. Cells were treated with the indicated concentrations of TSA for 24 and 48 hours and relative cell viability was measured using the Cell Titre (Promega) assay kit. (D) TSA induces apoptosis in CEM-CCRF cells. Cells were treated with the indicated concentrations of TSA for 24 hours and caspase 3/7 activity was measured using the Apo-One (Promega) assay kit. (E) TSA augments doxorubicin- and radiation-induced DNA double-strand breaks in CEM-CCRF cells. Micrographs of CEM-CCRF cells immunostained forγH2AX (depicted as white foci) are shown. (F) Cells were treated with 0.5μM TSA for 24 hours prior to one hour incubation with 1μM doxorubicin at 37°C. Cells were washed and incubated for a further 24 hours prior to staining for γH2AX. In separate experiments cells were treated with 0.5μM TSA for 24 hours prior to irradiation with 2 Gy (137Cs). Cells were stained for γH2AX foci one hour following irradiation.
Non-oncological applications of histone deacetylase inhibitors
Apart from applications in oncology, the therapeutic potential of HDAC inhibitors has also been investigated for a wide range of other diseases. Examples include HIV infection where the potential of HDAC inhibitors in de-silencing latent virus was evaluated and ulcerative colitis where inhibition of NF-κB activation by butyrate was proposed to be beneficial [48, 49]. Other examples include hepatitis for which HDAC inhibitors were shown to function by inhibiting TNF-α and INFγ and similarly, downregulation of pro-inflammatory cytokines is thought to provide the basis for the potential use of HDAC inhibitors in systemic lupus erythematosus [50, 51]. The potential of HDAC inhibitors as antimalarial agents has also been investigated. It has been shown that the mammalian HDAC inhibitors Trichostatin A and apicidin cause histone acetylation and inhibit the growth of Plasmodium falciparum, the main malarial parasite in humans [52-54]. Similarly, the mammalian HDAC inhibitors azelaic bishydroxamic acid and suberohydroxamic acid exhibit antimalarial activity against P. falciparum [55]. This prompted the development of analogues based on the structures of L-cysteine and 2-aminosuberic acid yielding compounds with greater selectivity for the protozoan HDAC enzymes [56, 57]. In addition to diseases described above, a considerable research effort has been aimed at evaluating the potential of HDAC inhibitors as therapeutics for neurodegenerative disorders, cardiac hypertrophy and more recently asthma, which will be the focus of discussion of the remaining of this review.
Histone deacetylase inhibitors for the treatment of neurodegenerative disorders
Aberrant acetylation has been associated with numerous neurodegenerative diseases; a primary example of which is Rubinstein-Taybi syndrome. Rubinstein-Taybi syndrome is a developmental disorder characterized by mental retardation and is caused by mutations in cyclic AMP response element binding protein (CREB) and p300 genes with HAT function [58]. Relevant animal models which exhibit defects in chromatin acetylation and impairment of long term-memory, such as late phase of hippocampal long-term potentiation have been used to evaluate the effects of restoration of acetylation defects [59]. It has been shown that enhancing CREB-dependent genes improves the condition of the mice. Similarly, pharmacological intervention with the broad-spectrum HDAC inhibitor Trichostatin A, partially restored long-term memory loss providing the basis for the possible use of HDAC inhibition for the treatment of Rubinstein-Taybi syndrome [60].
The potential efficacy of broad-spectrum HDAC inhibitors particularly, Trichostatin A, butyrates and valproic acid, have been investigated in a wide range of other neurodegenerative conditions [61, 62]. For example, valproic acid, Trichostatin A and phenylbutyrate have been investigated for their potential in ameliorating the effects of stroke [63-68]. Overall, the findings indicate the HDAC inhibitors may restore histone acetylation status, enhance neurogenesis and decrease neuroinflammatory responses. Similarly, restoration of acetylation status – both histone and the non-histone substrate α-tubulin–anti-inflammatory effects and decreased dopaminergic neuronal death have been shown to be responsible for the beneficial effects of HDAC inhibitors in models of Parkinson's disease [69-72]. Further, HDAC inhibition has been shown to be effective in models of Huntington's disease due to restored histone acetylation status and normalization of striatal atrophy [62, 73-76]. Suppression of motor neuron degeneration and muscle atrophy as well as increased expression of SMN2 provides the basis for the potential use of HDAC inhibitors in spinal muscular atrophy [77-84]. Valproic acid and phenylbutyrate, either alone in combination with antioxidants or lithium have also shown beneficial effects in models of amyotrophic lateral sclerosis [85-90]. Like for other neurodegenerative conditions, the beneficial effects of the HDAC inhibitors involve restored histone acetylation status, suppression of motor neuronal death and improved motor function and survival.
Alzheimer's disease is a very common neurodegenerative condition and represents a leading cause of death in industrialized countries [91]. It is characterized by progressive memory loss and ultimately dementia [91]. The main pathobiological features of Alzheimer's disease include the accumulation of insoluble β-amyloid due to aberrant cleavage of amyloid precursor protein by secretases and accumulation of neurofibrillary tangles due to hyperphosphorylation of Tau protein [92, 93]. Numerous relevant in vivo models of Alzheimer's disease have been developed and these have assisted in the understanding of the molecular basis of disease and also allowed for the evaluation of pharmacological interventions. The broad-spectrum histone deacetylase inhibitors valproic acid, sodium butyrate, phenylbutyrate and Trichostatin A have shown beneficial effects in models of Alzheimer's disease [94-96]. In general, HDAC inhibitors have been shown to restore histone acetylation status and improve synaptic plasticity. Further, HDAC inhibitors have been found to improve learning and memory and to reverse spatial memory defects [94-96]. Additionally, findings indicate that HDAC inhibitors decrease expression of β-amyloid and phosphorylation of Tau in relevant models, providing a basis for their potential efficacy in Alzheimer's disease [94-96].
Although broad-spectrum HDAC inhibitors generally display beneficial effects in various models of neurodegenerative diseases, they do exhibit toxicity in various cell types of the central nervous system. This is not unexpected given the biological effects of HDAC inhibitors as anti-cancer agents. Therefore, class or isoform-selective inhibitors may provide a therapeutic advantage and studies with SAHA which predominantly inhibited class I HDAC enzymes in a model of Alzheimer's disease is a step in this direction [97]. Although HDAC2 has been shown to regulate memory formation and neuronal plasticity, the function of the different HDAC isoforms in neurodegenerative diseases is still poorly understood [98]. Therefore, it is expected that further research will be aimed at identifying the role of the different HDAC enzymes and evaluating isoform-specific HDAC inhibitors in models of neurodegenerative diseases. In this context, the detailed map of HDAC1-11 enzymes in >50 regions of the rat brain will provide a starting point for examination of the roles of specific HDAC enzymes in the brain [99]. Finally, an important study in this direction utilized pharmacological and genetic approaches to specifically inhibit HDAC6, highlighting the potential for selective inhibition as a nontoxic therapeutic strategy for protection and regeneration following injury in the central nervous system [100].
Potential of histone deacetylase inhibitors for the treatment of cardiac hypertrophy
The therapeutic potential of HDAC inhibitors in models of heart disease, including cardiac hypertrophy and myocardial ischemia-reperfusion injury, has emerged [101-106]. For example, a recent study indicates that Trichostatin A protects from load- and agonist-induced hypertrophy in vivo by suppressing autophagy [107]. Similarly, a recent study indicates that valproic acid prevents pulmonary artery banding-induced right ventricular hypertrophy in rats [108]. However, the clinical potential of HDAC inhibitors for the treatment of heart disease remains controversial. This is highlighted from another recent study which indicates, in contrast to the findings with valproic acid, that Trichostatin A worsens pulmonary artery banding-induced right ventricular dysfunction in rats [109]. It was proposed that HDAC inhibitor induced-apoptosis and antiangiogenic effects are likely to be responsible for the disadvantageous effects of Trichostatin A on the pressure-overloaded right ventricle [109]. In vitro studies from our laboratory, using doxorubicin-induced hypertrophy in rat cardiac myocytes as a model system, also suggest detrimental effects of broad-spectrum HDAC inhibitors (Figure 3). Firstly, our findings indicate that Trichostatin A augments doxorubicin-induced hypertrophy, at least in part, by modulating the expression of the hypertrophy-associated genes, ventricular myosin light chain-2, the alpha isoform of myosin heavy chain and atrial natriuretic peptide [110]. Further, our findings indicate that pre-treatment, but not post-treatment, of cells with the broad-spectrum HDAC inhibitors, Trichostatin A, valproic acid and sodium butyrate enhance doxorubicin-induced DNA damage as monitored with γH2AX – a molecular marker of DNA double-strand breaks [110, 111].
Figure 3.

Histone acetylation status is regulated by the opposing actions of histone acetyltransferase (HATs) and histone deacetylases (HDACs). The precise function of HDAC enzymes in regulating cardiac hypertrophy remains unknown. Similarly, the potential of HDAC inhibitors in the treatment of cardiac hypertrophy remains controversial. A number of reports have indicated a beneficial effect of broad-spectrum HDAC inhibitors in animal models of cardiac hypertrophy. However, others have indicated the opposite effect. (A) We have shown that broad-spectrum HDAC inhibitors augment doxorubicin induced hypertrophy in vitro. H9c2 myoblasts were differentiated into cardiac myocytes after 7 days culture in the presence of 10 nM all-trans retinoic acid. (i) Cells were then treated with 1 μM Trichostatin A (TSA), 10 mM sodium butyrate (NaB) and 10 mM valproic acid (VPA) for 24 hours prior to one hour incubation with 1 μM doxorubicin. Cells were washed and incubated for a further 24 hours in fresh media prior to immunostainingfor γH2AX. Analysis of γH2AX foci indicates that broad-spectrum HDAC inhibitors augment doxorubicin-induced DNA double strand breaks. (ii) Cells treated with HDAC inhibitors also induced a hypertrophic response with an increase in the mean of the longest diameter of H9c2 cells. (iii) Photomicrographs of differentiated H9c2 cardiac myocytes treated with 1 μM TSA and immunostained for γH2AX (depicted as white foci) are shown. (B) Recent evidence indicates that class I HDACs promote cardiac hypertrophy whereas class II enzymes inhibit pro-hypertrophic pathways which may be regulated by the myocyte enhancer factor-2 (MEF2) transcription factor. Therefore, it is anticipated that class I-specific HDAC inhibitors may be more beneficial for the potential treatment of cardiac hypertrophy.
Evidence indicates that the uncertainty surrounding the clinical potential of HDAC inhibitors in cardiac hypertrophy stems from the disparate actions of class I and class II HDACs in this disease [112-116]. Overall, class I HDACs potentiate cardiac hypertrophy and class II HDACs are thought to suppress pro-hypertrophic responses (Figure 3). The role of different HDAC enzymes in regulating cardiac hypertrophy has been reviewed extensively [113-115]. Briefly, class II HDACs prevent hypertrophic responses, largely, by inactivating myocyte enhancer 2 (MEF2), a transcription factor which drives cardiac hypertrophy in response to stress [115, 117]. In addition, the activities of numerous other transcription factors involved in myocardial growth including, serum response factor, myocardin and calmodulin binding transcription activator 2, are modulated by class II HDAC enzymes [114, 118]. Although the precise mechanisms are yet to be fully elucidated, direct evidence for the function of class II HDACs in suppressing stress-induced hypertrophy comes from in vivo experiments involving mice lacking either HDAC5 or HDAC9 [115, 119]. Mice lacking HDAC5 and HDAC9, which are typically highly expressed in the heart, develop extremely enlarged hearts following cardiac stress [115, 119]. Given the differential functions of HDAC enzymes in cardiac hypertrophy it is widely anticipated that class I HDAC inhibitors may be more efficacious in this disease. In this context, Spiruchostatin A, a potent inhibitor with selectivity towards class I HDACs in cardiac myocytes, has been shown to abrogate the pro-hypertrophic effects of phenylephrine and urocortin [120]. Further research in this direction is expected.
Effects of histone deacetylase inhibitors in models of asthma
Like for cardiac hypertrophy, the potential clinical utility of HDAC inhibitors in asthma remains controversial. Studies indicate that aberrant HAT and HDAC expression and activity may be implicated in asthma pathogenesis. Analysis of bronchial biopsies has indicated reduced HDAC activity and reduced HDAC1 and HDAC2 protein expression is asthmatic compared to normal subjects [121]. The same study HAT activity is increased in subjects with asthma [121]. Further, it was shown that treatment with inhaled steroids reduces HAT and increases HDAC activity, providing evidence for a mechanism accounting for the increased expression of multiple inflammatory genes in asthma [121]. Similarly, it has been shown that conditional deletion of HDAC1 results in increased Th2 cytokine production and enhanced airway inflammation in an in vivo allergic airway inflammation model [122]. These findings suggest that HDAC inhibition would be contraindicated in asthma. However, paradoxical findings have shown that HDAC inhibition attenuates airway hyperresponsiveness (AHR) and inflammation in animals of inflammation and asthma [123, 124]. Of particular interest are the findings that the broad-spectrum HDAC inhibitor, Trichostatin A, improved AHR and reduced inflammation by decreasing the expression of Th2 cytokines using a mouse model of allergic airways disease [124]. More recent findings have indicated that Trichostatin A inhibits AHR but not inflammation in a mouse asthma model [125]. Incidentally, published findings from our laboratory indicate the beneficial effects of the dietary class III HDAC agonist, resveratrol, in inhibiting AHR and inflammation in a chronic ovalbumin-challenge and sensitization model of allergic airways disease [126]. In similar experiments (unpublished observations) we have found beneficial effect of both Trichostatin A and valproic acid in the chronic mouse model of allergic airways disease. Overall, the recent findings are indicating the potential for HDAC inhibition as a therapeutic modality in asthma. In this context, it will be important to systematically investigate the function of specific HDAC isoforms in asthma pathobiology. Similarly, the effects of selective HDAC inhibitors may clarify the clinical utility of this class of compounds in asthma.
Conclusions
The potential versatility of HDAC inhibitors extends beyond their current application in oncology. The efficacy of HDAC inhibitors in a wide range of conditions including neurodegenerative disorders, cardiac hypertrophy and asthma has been investigated in relevant systems. While broad-spectrum HDAC inhibitors have been evaluated almost exclusively to date, it is emerging that class- or isoform-specific compounds, which are increasingly becoming available, may be more beneficial. However, the molecular pathways regulated by different HDAC-isoforms in disease processes remain poorly understood. Given the clinical potential of HDAC inhibitors, this issue will undoubtedly be clarified by further research. Already, evidence is pointing to the importance of investigating class - or isoform-selective HDAC inhibitors as potential therapeutics for cardiac diseases, with the expectation that class I HDACs will be more efficacious.
Acknowledgments
The support of the Australian Institute of Nuclear Science and Engineering is acknowledged. TCK was the recipient of AINSE awards. Epigenomic Medicine Laboratory is supported by the National Health and Medical Research Council of Australia. KV is supported by a Baker IDI postgraduate scholarship. Supported in part by the Victorian Government's Operational Infrastructure Support Program. The authors would like to acknowledge the use of the facilities provided by Monash Micro Imaging @ AMREP and particularly, the expert assistance from Drs Stephen Cody and Iśka Carmichael.
References
- 1.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 2.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and Methylation of Histones and Their Possible Role in the Regulation of Rna Synthesis. Proc Natl Acad Sci USA. 1964;51:786–794. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allfrey VG, Mirsky AE. Structural Modifications of Histones and their Possible Role in the Regulation of RNA Synthesis. Science. 1964;144:559. doi: 10.1126/science.144.3618.559. [DOI] [PubMed] [Google Scholar]
- 4.Smith BC, Denu JM. Chemical mechanisms of histone lysine and arginine modifications. Biochim Biophys Acta. 2009;1789:45–57. doi: 10.1016/j.bbagrm.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roth SY, Denu JM, Allis CD. Histone acetyl-transferases. Annu Rev Biochem. 2001;70:81–120. doi: 10.1146/annurev.biochem.70.1.81. [DOI] [PubMed] [Google Scholar]
- 6.Kuo MH, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays. 1998;20:615–626. doi: 10.1002/(SICI)1521-1878(199808)20:8<615::AID-BIES4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 7.Dokmanovic M, Marks PA. Prospects: histone deacetylase inhibitors. J Cell Biochem. 2005;96:293–304. doi: 10.1002/jcb.20532. [DOI] [PubMed] [Google Scholar]
- 8.Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat Rev Cancer. 2006;6:38–51. doi: 10.1038/nrc1779. [DOI] [PubMed] [Google Scholar]
- 9.Rosato RR, Grant S. Histone deacetylase inhibitors: insights into mechanisms of lethality. Expert Opin Ther Targets. 2005;9:809–824. doi: 10.1517/14728222.9.4.809. [DOI] [PubMed] [Google Scholar]
- 10.Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541–5552. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- 11.de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marks PA. Histone deacetylase inhibitors: a chemical genetics approach to understanding cellular functions. Biochim Biophys Acta. 2010;1799:717–725. doi: 10.1016/j.bbagrm.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marks PA, Xu WS. Histone deacetylase inhibitors: Potential in cancer therapy. J Cell Biochem. 2009;107:600–608. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tanner KG, Landry J, Sternglanz R, Denu JM. Silent information regulator 2 family of NAD- dependent histone/protein deacetylases generates a unique product, 1-O-acetyl-ADPribose. Proc Natl Acad Sci USA. 2000;97:14178–14182. doi: 10.1073/pnas.250422697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 16.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 17.Marks P, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2001;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 18.Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008;9:206–218. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yang XJ, Seto E. Collaborative spirit of histone deacetylases in regulating chromatin structure and gene expression. Curr Opin Genet Dev. 2003;13:143–153. doi: 10.1016/s0959-437x(03)00015-7. [DOI] [PubMed] [Google Scholar]
- 20.Martin M, Kettmann R, Dequiedt F. Class IIa histone deacetylases: regulating the regulators. Oncogene. 2007;26:5450–5467. doi: 10.1038/sj.onc.1210613. [DOI] [PubMed] [Google Scholar]
- 21.Witt O, Deubzer HE, Milde T, Oehme I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009;277:8–21. doi: 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 22.Kovacs JJ, Murphy PJ, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18:601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]
- 23.Parmigiani RB, Xu WS, Venta-Perez G, Erdjument-Bromage H, Yaneva M, Tempst P, Marks PA. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proc Natl Acad Sci USA. 2008;105:9633–9638. doi: 10.1073/pnas.0803749105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gao YS, Hubbert CC, Yao TP. The microtubule-associated histone deacetylase 6 (HDAC6) regulates epidermal growth factor receptor (EGFR) endocytic trafficking and degradation. J Biol Chem. 2010;285:11219–11226. doi: 10.1074/jbc.M109.042754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, Yoshida M, Wang XF, Yao TP. HDAC6 is a microtubule-associated deacetylase. Nature. 2002;417:455–458. doi: 10.1038/417455a. [DOI] [PubMed] [Google Scholar]
- 26.Kawaguchi Y, Kovacs JJ, McLaurin A, Vance JM, Ito A, Yao TP. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell. 2003;115:727–738. doi: 10.1016/s0092-8674(03)00939-5. [DOI] [PubMed] [Google Scholar]
- 27.Chang HH, Chiang CP, Hung HC, Lin CY, Deng YT, Kuo MY. Histone deacetylase 2 expression predicts poorer prognosis in oral cancer patients. Oral Oncol. 2009;45:610–614. doi: 10.1016/j.oraloncology.2008.08.011. [DOI] [PubMed] [Google Scholar]
- 28.Gloghini A, Buglio D, Khaskhely NM, Georgakis G, Orlowski RZ, Neelapu SS, Carbone A, Younes A. Expression of histone deacetylases in lymphoma: implication for the development of selective inhibitors. Br J Haematol. 2009;147:515–525. doi: 10.1111/j.1365-2141.2009.07887.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakagawa M, Oda Y, Eguchi T, Aishima S, Yao T, Hosoi F, Basaki Y, Ono M, Kuwano M, Tanaka M, Tsuneyoshi M. Expression profile of class I histone deacetylases in human cancer tissues. Oncol Rep. 2007;18:769–774. [PubMed] [Google Scholar]
- 30.Ropero S, Fraga MF, Ballestar E, Hamelin R, Yamamoto H, Boix-Chornet M, Caballero R, Alaminos M, Setien F, Paz MF, Herranz M, Palacios J, Arango D, Orntoft TF, Aaltonen LA, Schwartz S, Jr, Esteller M. A truncating mutation of HDAC2 in human cancers confers resistance to histone deacetylase inhibition. Nat Genet. 2006;38:566–569. doi: 10.1038/ng1773. [DOI] [PubMed] [Google Scholar]
- 31.Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, Meeh P, Markowitz SD, Willis J, Dawson D, Willson JK, Gazdar AF, Hartigan J, Wu L, Liu C, Parmigiani G, Park BH, Bachman KE, Papadopoulos N, Vogelstein B, Kinzler KW, Velculescu VE. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 32.Weichert W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009;280:168–176. doi: 10.1016/j.canlet.2008.10.047. [DOI] [PubMed] [Google Scholar]
- 33.Zhang Z, Yamashita H, Toyama T, Sugiura H, Ando Y, Mita K, Hamaguchi M, Hara Y, Kobayashi S, Iwase H. Quantitation of HDAC1 mRNA expression in invasive carcinoma of the breast*. Breast Cancer Res Treat. 2005;94:11–16. doi: 10.1007/s10549-005-6001-1. [DOI] [PubMed] [Google Scholar]
- 34.Marks PA, Breslow R. Dimethyl sulfoxide to vorinostat: development of this histone deacetylase inhibitor as an anticancer drug. Nat Biotechnol. 2007;25:84–90. doi: 10.1038/nbt1272. [DOI] [PubMed] [Google Scholar]
- 35.Campas-Moya C. Romidepsin for the treatment of cutaneous T-cell lymphoma. Drugs Today (Barc) 2009;45:787–795. doi: 10.1358/dot.2009.45.11.1437052. [DOI] [PubMed] [Google Scholar]
- 36.Dokmanovic M, Clarke C, Marks PA. Histone deacetylase inhibitors: overview and perspectives. Mol Cancer Res. 2007;5:981–989. doi: 10.1158/1541-7786.MCR-07-0324. [DOI] [PubMed] [Google Scholar]
- 37.Haggarty SJ, Koeller KM, Wong JC, Grozinger CM, Schreiber SL. Domain-selective smallmolecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc Natl Acad Sci USA. 2003;100:4389–4394. doi: 10.1073/pnas.0430973100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Namdar M, Perez G, Ngo L, Marks PA. Selective inhibition of histone deacetylase 6 (HDAC6) induces DNA damage and sensitizes transformed cells to anticancer agents. Proc Natl Acad Sci USA. 2010;107:20003–20008. doi: 10.1073/pnas.1013754107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang W, Luo T, Greenberg EF, Bradner JE, Schreiber SL. Discovery of histone deacetylase 8 selective inhibitors. Bioorg Med Chem Lett. 2011;21:2601–2605. doi: 10.1016/j.bmcl.2011.01.134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kwa FA, Balcerczyk A, Licciardi P, El-Osta A, Karagiannis TC. Chromatin modifying agents - the cutting edge of anticancer therapy. Drug Discov Today. 2011;16:543–547. doi: 10.1016/j.drudis.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 41.De los Santos M, Zambrano A, Aranda A. Combined effects of retinoic acid and histone deacetylase inhibitors on human neuroblastoma SH-SY5Y cells. Mol Cancer Ther. 2007;6:1425–1432. doi: 10.1158/1535-7163.MCT-06-0623. [DOI] [PubMed] [Google Scholar]
- 42.Kim HJ, Bae SC. Histone deacetylase inhibitors: molecular mechanisms of action and clinical trials as anti-cancer drugs. Am J Transl Res. 2011;3:166–179. [PMC free article] [PubMed] [Google Scholar]
- 43.Karagiannis TC, Harikrishnan KN, El-Osta A. The histone deacetylase inhibitor, Trichostatin A, enhances radiation sensitivity and accumulation of gammaH2A.X. Cancer Biol Ther. 2005;4:787–793. doi: 10.4161/cbt.4.7.1922. [DOI] [PubMed] [Google Scholar]
- 44.Karagiannis TC, Harikrishnan KN, El-Osta A. Disparity of histone deacetylase inhibition on repair of radiation-induced DNA damage on euchromatin and constitutive heterochromatin compartments. Oncogene. 2007;26:3963–3971. doi: 10.1038/sj.onc.1210174. [DOI] [PubMed] [Google Scholar]
- 45.Sanchez-Gonzalez B, Yang H, Bueso-Ramos C, Hoshino K, Quintas-Cardama A, Richon VM, Garcia-Manero G. Antileukemia activity of the combination of an anthracycline with a histone deacetylase inhibitor. Blood. 2006;108:1174–1182. doi: 10.1182/blood-2005-09-008086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Briggs B, Ververis K, Rodd AL, Foong LJ, Silva FM, Karagiannis TC. Photosensitization by iodinated DNA minor groove binding ligands: Evaluation of DNA double-strand break induction and repair. J Photochem Photobiol B. 2011;103:145–152. doi: 10.1016/j.jphotobiol.2011.02.022. [DOI] [PubMed] [Google Scholar]
- 47.Singh TR, Shankar S, Srivastava RK. HDAC inhibitors enhance the apoptosis-inducing potential of TRAIL in breast carcinoma. Oncogene. 2005;24:4609–4623. doi: 10.1038/sj.onc.1208585. [DOI] [PubMed] [Google Scholar]
- 48.Lehrman G, Hogue IB, Palmer S, Jennings C, Spina CA, Wiegand A, Landay AL, Coombs RW, Richman DD, Mellors JW, Coffin JM, Bosch RJ, Margolis DM. Depletion of latent HIV-1 infection in vivo: a proof-of-concept study. Lancet. 2005;366:549–555. doi: 10.1016/S0140-6736(05)67098-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Luhrs H, Gerke T, Muller JG, Melcher R, Schauber J, Boxberge F, Scheppach W, Menzel T. Butyrate inhibits NF-kappaB activation in lamina propria macrophages of patients with ulcerative colitis. Scand J Gastroenterol. 2002;37:458–466. doi: 10.1080/003655202317316105. [DOI] [PubMed] [Google Scholar]
- 50.Leoni F, Fossati G, Lewis EC, Lee JK, Porro G, Pagani P, Modena D, Moras ML, Pozzi P, Reznikov LL, Siegmund B, Fantuzzi G, Dinarello CA, Mascagni P. The histone deacetylase inhibitor ITF2357 reduces production of pro-inflammatory cytokines in vitro and systemic inflammation in vivo. Mol Med. 2005;11:1–15. doi: 10.2119/2006-00005.Dinarello. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mishra N, Reilly CM, Brown DR, Ruiz P, Gilkeson GS. Histone deacetylase inhibitors modulate renal disease in the MRL-lpr/lpr mouse. J Clin Invest. 2003;111:539–552. doi: 10.1172/JCI16153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Colletti SL, Myers RW, Darkin-Rattray SJ, Gurnett AM, Dulski PM, Galuska S, Allocco JJ, Ayer MB, Li C, Lim J, Crumley TM, Cannova C, Schmatz DM, Wyvratt MJ, Fisher MH, Meinke PT. Broad spectrum antiprotozoal agents that inhibit histone deacetylase: structure-activity relationships of apicidin. Part 2. Bioorg Med Chem Lett. 2001;11:113–117. doi: 10.1016/s0960-894x(00)00605-3. [DOI] [PubMed] [Google Scholar]
- 53.Colletti SL, Myers RW, Darkin-Rattray SJ, Gurnett AM, Dulski PM, Galuska S, Allocco JJ, Ayer MB, Li C, Lim J, Crumley TM, Cannova C, Schmatz DM, Wyvratt MJ, Fisher MH, Meinke PT. Broad spectrum antiprotozoal agents that inhibit histone deacetylase: structure-activity relationships of apicidin. Part 1. Bioorg Med Chem Lett. 2001;11:107–111. doi: 10.1016/s0960-894x(00)00604-1. [DOI] [PubMed] [Google Scholar]
- 54.Darkin-Rattray SJ, Gurnett AM, Myers RW, Dulski PM, Crumley TM, Allocco JJ, Cannova C, Meinke PT, Colletti SL, Bednarek MA, Singh SB, Goetz MA, Dombrowski AW, Polishook JD, Schmatz DM. Apicida novel antiprotozoal agent that inhibits parasite histone deacetylase. Proc Natl Acad Sci USA. 1996;93:13143–13147. doi: 10.1073/pnas.93.23.13143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andrews KT, Walduck A, Kelso MJ, Fairlie DP, Saul A, Parsons PG. Anti-malarial effect of histone deacetylation inhibitors and mammalian tumour cytodifferentiating agents. Int J Parasitol. 2000;30:761–768. doi: 10.1016/s0020-7519(00)00043-6. [DOI] [PubMed] [Google Scholar]
- 56.Andrews KT, Tran TN, Lucke AJ, Kahnberg P, Le GT, Boyle GM, Gardiner DL, Skinner-Adams TS, Fairlie DP. Potent antimalarial activity of histone deacetylase inhibitor analogues. Antimicrob Agents Chemother. 2008;52:1454–1461. doi: 10.1128/AAC.00757-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Andrews KT, Tran TN, Wheatley NC, Fairlie DP. Targeting histone deacetylase inhibitors for anti-malarial therapy. Curr Top Med Chem. 2009;9:292–308. doi: 10.2174/156802609788085313. [DOI] [PubMed] [Google Scholar]
- 58.Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC, Masuno M, Tommerup N, van Ommen GJ, Goodman RH, Peters DJ, Breuning MH. Rubinstein-Taybi syndrome caused by mutations in the transcriptional co-activator CBP. Nature. 1995;376:348–351. doi: 10.1038/376348a0. [DOI] [PubMed] [Google Scholar]
- 59.Barco A. The Rubinstein-Taybi syndrome: modeling mental impairment in the mouse. Genes Brain Behav. 2007;6(Suppl 1):32–39. doi: 10.1111/j.1601-183X.2007.00320.x. [DOI] [PubMed] [Google Scholar]
- 60.Vecsey CG, Hawk JD, Lattal KM, Stein JM, Fabian SA, Attner MA, Cabrera SM, McDonough CB, Brindle PK, Abel T, Wood MA. Histone deacetylase inhibitors enhance memory and synaptic plasticity via CREB:CBPdependent transcriptional activation. J Neurosci. 2007;27:6128–6140. doi: 10.1523/JNEUROSCI.0296-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008;7:854–868. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- 62.Chuang DM, Leng Y, Marinova Z, Kim HJ, Chiu CT. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009;32:591–601. doi: 10.1016/j.tins.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhong Y, Zhou LJ, Ren WJ, Xin WJ, Li YY, Zhang T, Liu XG. The direction of synaptic plasticity mediated by C-fibers in spinal dorsal horn is decided by Src-family kinases in microglia: the role of tumor necrosis factor-alpha. Brain Behav Immun. 2010;24:874–880. doi: 10.1016/j.bbi.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 64.Kim HJ, Rowe M, Ren M, Hong JS, Chen PS, Chuang DM. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: multiple mechanisms of action. J Pharmacol Exp Ther. 2007;321:892–901. doi: 10.1124/jpet.107.120188. [DOI] [PubMed] [Google Scholar]
- 65.Faraco G, Pancani T, Formentini L, Mascagni P, Fossati G, Leoni F, Moroni F, Chiarugi A. Pharmacological inhibition of histone deacetylases by suberoylanilide hydroxamic acid specifically alters gene expression and reduces ischemic injury in the mouse brain. Mol Pharmacol. 2006;70:1876–1884. doi: 10.1124/mol.106.027912. [DOI] [PubMed] [Google Scholar]
- 66.Kim HJ, Leeds P, Chuang DM. The HDAC inhibitor, sodium butyrate, stimulates neurogenesis in the ischemic brain. J Neurochem. 2009;110:1226–1240. doi: 10.1111/j.1471-4159.2009.06212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qi X, Hosoi T, Okuma Y, Kaneko M, Nomura Y. Sodium 4-phenylbutyrate protects against cerebral ischemic injury. Mol Pharmacol. 2004;66:899–908. doi: 10.1124/mol.104.001339. [DOI] [PubMed] [Google Scholar]
- 68.Sinn DI, Kim SJ, Chu K, Jung KH, Lee ST, Song EC, Kim JM, Park DK, Kun Lee S, Kim M, Roh JK. Valproic acid-mediated neuroprotection in intracerebral hemorrhage via histone deacetylase inhibition and transcriptional activation. Neurobiol Dis. 2007;26:464–472. doi: 10.1016/j.nbd.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 69.Kontopoulos E, Parvin JD, Feany MB. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum Mol Genet. 2006;15:3012–3023. doi: 10.1093/hmg/ddl243. [DOI] [PubMed] [Google Scholar]
- 70.Chen PS, Peng GS, Li G, Yang S, Wu X, Wang CC, Wilson B, Lu RB, Gean PW, Chuang DM, Hong JS. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol Psychiatry. 2006;11:1116–1125. doi: 10.1038/sj.mp.4001893. [DOI] [PubMed] [Google Scholar]
- 71.Wu X, Chen PS, Dallas S, Wilson B, Block ML, Wang CC, Kinyamu H, Lu N, Gao X, Leng Y, Chuang DM, Zhang W, Lu RB, Hong JS. Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int J Neuropsychopharmacol. 2008;11:1123–1134. doi: 10.1017/S1461145708009024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gardian G, Yang L, Cleren C, Calingasan NY, Klivenyi P, Beal MF. Neuroprotective effects of phenylbutyrate against MPTP neurotoxicity. Neuromolecular Med. 2004;5:235–241. doi: 10.1385/NMM:5:3:235. [DOI] [PubMed] [Google Scholar]
- 73.Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PA, Steffan JS, Marsh JL, Thompson LM, Lewis CM, Marks PA, Bates GP. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc Natl Acad Sci USA. 2003;100:2041–2046. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pallos J, Bodai L, Lukacsovich T, Purcell JM, Steffan JS, Thompson LM, Marsh JL. Inhibition of specific HDACs and sirtuins suppresses pathogenesis in a Drosophila model of Huntington's disease. Hum Mol Genet. 2008;17:3767–3775. doi: 10.1093/hmg/ddn273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gardian G, Browne SE, Choi DK, Klivenyi P, Gregorio J, Kubilus JK, Ryu H, Langley B, Ratan RR, Ferrante RJ, Beal MF. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington's disease. J Biol Chem. 2005;280:556–563. doi: 10.1074/jbc.M410210200. [DOI] [PubMed] [Google Scholar]
- 76.Dompierre JP, Godin JD, Charrin BC, Cordelieres FP, King SJ, Humbert S, Saudou F. Histone deacetylase 6 inhibition compensates for the transport deficit in Huntington's disease by increasing tubulin acetylation. J Neurosci. 2007;27:3571–3583. doi: 10.1523/JNEUROSCI.0037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chang JG, Hsieh-Li HM, Jong YJ, Wang NM, Tsai CH, Li H. Treatment of spinal muscular atrophy by sodium butyrate. Proc Natl Acad Sci USA. 2001;98:9808–9813. doi: 10.1073/pnas.171105098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Andreassi C, Angelozzi C, Tiziano FD, Vitali T, De Vincenzi E, Boninsegna A, Villanova M, Bertini E, Pini A, Neri G, Brahe C. Phenylbutyrate increases SMN expression in vitro: relevance for treatment of spinal muscular atrophy. Eur J Hum Genet. 2004;12:59–65. doi: 10.1038/sj.ejhg.5201102. [DOI] [PubMed] [Google Scholar]
- 79.Brichta L, Hofmann Y, Hahnen E, Siebzehnrubl FA, Raschke H, Blumcke I, Eyupoglu IY, Wirth B. Valproic acid increases the SMN2 protein level: a well-known drug as a potential therapy for spinal muscular atrophy. Hum Mol Genet. 2003;12:2481–2489. doi: 10.1093/hmg/ddg256. [DOI] [PubMed] [Google Scholar]
- 80.Hauke J, Riessland M, Lunke S, Eyupoglu IY, Blumcke I, El-Osta A, Wirth B, Hahnen E. Survival motor neuron gene 2 silencing by DNA methylation correlates with spinal muscular atrophy disease severity and can be bypassed by histone deacetylase inhibition. Hum Mol Genet. 2009;18:304–317. doi: 10.1093/hmg/ddn357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Avila AM, Burnett BG, Taye AA, Gabanella F, Knight MA, Hartenstein P, Cizman Z, Di Prospero NA, Pellizzoni L, Fischbeck KH, Sumner CJ. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J Clin Invest. 2007;117:659–671. doi: 10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tsai LK, Tsai MS, Ting CH, Li H. Multiple therapeutic effects of valproic acid in spinal muscular atrophy model mice. J Mol Med (Berl) 2008;86:1243–1254. doi: 10.1007/s00109-008-0388-1. [DOI] [PubMed] [Google Scholar]
- 83.Mercuri E, Bertini E, Messina S, Pelliccioni M, D'Amico A, Colitto F, Mirabella M, Tiziano FD, Vitali T, Angelozzi C, Kinali M, Main M, Brahe C. Pilot trial of phenylbutyrate in spinal muscular atrophy. Neuromuscul Disord. 2004;14:130–135. doi: 10.1016/j.nmd.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 84.Brahe C, Vitali T, Tiziano FD, Angelozzi C, Pinto AM, Borgo F, Moscato U, Bertini E, Mercuri E, Neri G. Phenylbutyrate increases SMN gene expression in spinal muscular atrophy patients. Eur J Hum Genet. 2005;13:256–259. doi: 10.1038/sj.ejhg.5201320. [DOI] [PubMed] [Google Scholar]
- 85.Ryu H, Smith K, Camelo SI, Carreras I, Lee J, Iglesias AH, Dangond F, Cormier KA, Cudkowicz ME, Brown RH, Jr, Ferrante RJ. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J Neurochem. 2005;93:1087–1098. doi: 10.1111/j.1471-4159.2005.03077.x. [DOI] [PubMed] [Google Scholar]
- 86.Del Signore SJ, Amante DJ, Kim J, Stack EC, Goodrich S, Cormier K, Smith K, Cudkowicz ME, Ferrante RJ. Combined riluzole and sodium phenylbutyrate therapy in transgenic amyotrophic lateral sclerosis mice. Amyotroph Lateral Scler. 2009;10:85–94. doi: 10.1080/17482960802226148. [DOI] [PubMed] [Google Scholar]
- 87.Petri S, Kiaei M, Kipiani K, Chen J, Calingasan NY, Crow JP, Beal MF. Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2006;22:40–49. doi: 10.1016/j.nbd.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 88.Sugai F, Yamamoto Y, Miyaguchi K, Zhou Z, Sumi H, Hamasaki T, Goto M, Sakoda S. Benefit of valproic acid in suppressing disease progression of ALS model mice. Eur J Neurosci. 2004;20:3179–3183. doi: 10.1111/j.1460-9568.2004.03765.x. [DOI] [PubMed] [Google Scholar]
- 89.Rouaux C, Panteleeva I, Rene F, Gonzalez de Aguilar JL, Echaniz-Laguna A, Dupuis L, Menger Y, Boutillier AL, Loeffler JP. Sodium valproate exerts neuroprotective effects in vivo through CREB-binding proteindependent mechanisms but does not improve survival in an amyotrophic lateral sclerosis mouse model. J Neurosci. 2007;27:5535–5545. doi: 10.1523/JNEUROSCI.1139-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Feng HL, Leng Y, Ma CH, Zhang J, Ren M, Chuang DM. Combined lithium and valproate treatment delays disease onset, reduces neurological deficits and prolongs survival in an amyotrophic lateral sclerosis mouse model. Neuroscience. 2008;155:567–572. doi: 10.1016/j.neuroscience.2008.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cummings JL. Alzheimer's disease. N Engl J Med. 2004;351:56–67. doi: 10.1056/NEJMra040223. [DOI] [PubMed] [Google Scholar]
- 92.Alonso AC, Grundke-Iqbal I, Iqbal K. Alzheimer's disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med. 1996;2:783–787. doi: 10.1038/nm0796-783. [DOI] [PubMed] [Google Scholar]
- 93.Karran E, Mercken M, Strooper BD. The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10:698–712. doi: 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- 94.Ricobaraza A, Cuadrado-Tejedor M, Perez-Mediavilla A, Frechilla D, Del Rio J, Garcia-Osta A. Phenylbutyrate ameliorates cognitive deficit and reduces tau pathology in an Alzheimer's disease mouse model. Neuropsychopharmacology. 2009;34:1721–1732. doi: 10.1038/npp.2008.229. [DOI] [PubMed] [Google Scholar]
- 95.Qing H, He G, Ly PT, Fox CJ, Staufenbiel M, Cai F, Zhang Z, Wei S, Sun X, Chen CH, Zhou W, Wang K, Song W. Valproic acid inhibits Abeta production, neuritic plaque formation, and behavioral deficits in Alzheimer's disease mouse models. J Exp Med. 2008;205:2781–2789. doi: 10.1084/jem.20081588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Francis YI, Fa M, Ashraf H, Zhang H, Staniszewski A, Latchman DS, Arancio O. Dysregulation of histone acetylation in the APP/PS1 mouse model of Alzheimer's disease. J Alzheimers Dis. 2009;18:131–139. doi: 10.3233/JAD-2009-1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kilgore M, Miller CA, Fass DM, Hennig KM, Haggarty SJ, Sweatt JD, Rumbaugh G. Inhibitors of class 1 histone deacetylases reverse contextual memory deficits in a mouse model of Alzheimer's disease. Neuropsychopharmacology. 2010;35:870–880. doi: 10.1038/npp.2009.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature. 2009;459:55–60. doi: 10.1038/nature07925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Broide RS, Redwine JM, Aftahi N, Young W, Bloom FE, Winrow CJ. Distribution of histone deacetylases 1-11 in the rat brain. J Mol Neurosci. 2007;31:47–58. doi: 10.1007/BF02686117. [DOI] [PubMed] [Google Scholar]
- 100.Rivieccio MA, Brochier C, Willis DE, Walker BA, D'Annibale MA, McLaughlin K, Siddiq A, Kozikowski AP, Jaffrey SR, Twiss JL, Ratan RR, Langley B. HDAC6 is a target for protection and regeneration following injury in the nervous system. Proc Natl Acad Sci USA. 2009;106:19599–19604. doi: 10.1073/pnas.0907935106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kook H, Lepore JJ, Gitler AD, Lu MM, Wing-Man Yung W, Mackay J, Zhou R, Ferrari V, Gruber P, Epstein JA. Cardiac hypertrophy and histone deacetylase-dependent transcriptional repression mediated by the atypical homeodomain protein Hop. J Clin Invest. 2003;112:863–871. doi: 10.1172/JCI19137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kong Y, Tannous P, Lu G, Berenji K, Rothermel BA, Olson EN, Hill JA. Suppression of class I and II histone deacetylases blunts pressureoverload cardiac hypertrophy. Circulation. 2006;113:2579–2588. doi: 10.1161/CIRCULATIONAHA.106.625467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kee HJ, Sohn IS, Nam KI, Park JE, Qian YR, Yin Z, Ahn Y, Jeong MH, Bang YJ, Kim N, Kim JK, Kim KK, Epstein JA, Kook H. Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation. 2006;113:51–59. doi: 10.1161/CIRCULATIONAHA.105.559724. [DOI] [PubMed] [Google Scholar]
- 104.Berry JM, Cao DJ, Rothermel BA, Hill JA. Histone deacetylase inhibition in the treatment of heart disease. Expert Opin Drug Saf. 2008;7:53–67. doi: 10.1517/14740338.7.1.53. [DOI] [PubMed] [Google Scholar]
- 105.Granger A, Abdullah I, Huebner F, Stout A, Wang T, Huebner T, Epstein JA, Gruber PJ. Histone deacetylase inhibition reduces myocardial ischemia-reperfusion injury in mice. FASEB J. 2008;22:3549–3560. doi: 10.1096/fj.08-108548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Antos CL, McKinsey TA, Dreitz M, Hollingsworth LM, Zhang CL, Schreiber K, Rindt H, Gorczynski RJ, Olson EN. Dosedependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors. J Biol Chem. 2003;278:28930–28937. doi: 10.1074/jbc.M303113200. [DOI] [PubMed] [Google Scholar]
- 107.Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, Kong Y, Rothermel BA, Gillette TG, Hill JA. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci USA. 2011;108:4123–4128. doi: 10.1073/pnas.1015081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cho YK, Eom GH, Kee HJ, Kim HS, Choi WY, Nam KI, Ma JS, Kook H. Sodium valproate, a histone deacetylase inhibitor, but not captopril, prevents right ventricular hypertrophy in rats. Circ J. 2010;74:760–770. doi: 10.1253/circj.cj-09-0580. [DOI] [PubMed] [Google Scholar]
- 109.Bogaard HJ, Mizuno S, Hussaini AA, Toldo S, Abbate A, Kraskauskas D, Kasper M, Natarajan R, Voelkel NF. Suppression of histone deacetylases worsens right ventricular dysfunction after pulmonary artery banding in rats. Am J Respir Crit Care Med. 2011;183:1402–1410. doi: 10.1164/rccm.201007-1106OC. [DOI] [PubMed] [Google Scholar]
- 110.Karagiannis TC, Lin AJ, Ververis K, Chang L, Tang MM, Okabe J, El-Osta A. Trichostatin A accentuates doxorubicin-induced hypertrophy in cardiac myocytes. Aging (Albany NY) 2010;2:659–668. doi: 10.18632/aging.100203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ververis K, Rodd AL, Tang MM, El-Osta A, Karagiannis TC. Histone deacetylase inhibitors augment doxorubicin-induced DNA damage in cardiomyocytes. Cell Mol Life Sci. 2011 doi: 10.1007/s00018-011-0727-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.McKinsey TA, Olson EN. Dual roles of histone deacetylases in the control of cardiac growth. Novartis Found Symp. 2004;259:132–141. discussion 141-135, 163-139. [PubMed] [Google Scholar]
- 113.Backs J, Olson EN. Control of cardiac growth by histone acetylation/deacetylation. Circ Res. 2006;98:15–24. doi: 10.1161/01.RES.0000197782.21444.8f. [DOI] [PubMed] [Google Scholar]
- 114.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110:479–488. doi: 10.1016/s0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Olson EN, Backs J, McKinsey TA. Control of cardiac hypertrophy and heart failure by histone acetylation/deacetylation. Novartis Found Symp. 2006;274:3–12. discussion 13-19, 152-155, 272-156. [PubMed] [Google Scholar]
- 117.Kolodziejczyk SM, Wang L, Balazsi K, DeRepentigny Y, Kothary R, Megeney LA. MEF2 is upregulated during cardiac hypertrophy and is required for normal post-natal growth of the myocardium. Curr Biol. 1999;9:1203–1206. doi: 10.1016/S0960-9822(00)80027-5. [DOI] [PubMed] [Google Scholar]
- 118.Song K, Backs J, McAnally J, Qi X, Gerard RD, Richardson JA, Hill JA, Bassel-Duby R, Olson EN. The transcriptional coactivator CAMTA2 stimulates cardiac growth by opposing class II histone deacetylases. Cell. 2006;125:453–466. doi: 10.1016/j.cell.2006.02.048. [DOI] [PubMed] [Google Scholar]
- 119.Chang S, McKinsey TA, Zhang CL, Richardson JA, Hill JA, Olson EN. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol Cell Biol. 2004;24:8467–8476. doi: 10.1128/MCB.24.19.8467-8476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Davidson SM, Townsend PA, Carroll C, Yurek-George A, Balasubramanyam K, Kundu TK, Stephanou A, Packham G, Ganesan A, Latchman DS. The transcriptional coactivato p300 plays a critical role in the hypertrophic and protective pathways induced by phenylephrine in cardiac cells but is specific to the hypertrophic effect of urocortin. Chembiochem. 2005;6:162–170. doi: 10.1002/cbic.200400246. [DOI] [PubMed] [Google Scholar]
- 121.Ito K, Caramori G, Lim S, Oates T, Chung KF, Barnes PJ, Adcock IM. Expression and activity of histone deacetylases in human asthmatic airways. Am J Respir Crit Care Med. 2002;166:392–396. doi: 10.1164/rccm.2110060. [DOI] [PubMed] [Google Scholar]
- 122.Grausenburger R, Bilic I, Boucheron N, Zupkovitz G, El-Housseiny L, Tschismarov R, Zhang Y, Rembold M, Gaisberger M, Hartl A, Epstein MM, Matthias P, Seiser C, Ellmeier W. Conditional deletion of histone deacetylase 1 in T cells leads to enhanced airway inflammation and increased Th2 cytokine production. J Immunol. 2010;185:3489–3497. doi: 10.4049/jimmunol.0903610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Assem el SK, Peh KH, Wan BY, Middleton BJ, Dines J, Marson CM. Effects of a selection of histone deacetylase inhibitors on mast cell activation and airway and colonic smooth muscle contraction. Int Immunopharmacol. 2008;8:1793–1801. doi: 10.1016/j.intimp.2008.08.017. [DOI] [PubMed] [Google Scholar]
- 124.Choi JH, Oh SW, Kang MS, Kwon HJ, Oh GT, Kim DY. Trichostatin A attenuates airway inflammation in mouse asthma model. Clin Exp Allergy. 2005;35:89–96. doi: 10.1111/j.1365-2222.2004.02006.x. [DOI] [PubMed] [Google Scholar]
- 125.Banerjee A, Trivedi CM, Damera G, Jiang M, Jester W, Hoshi T, Epstein JA, Panettieri RA., Jr Trichostatin A Abrogates Airway Constriction, but not Inflammation in Mouse and Human Asthma Models. Am J Respir Cell Mol Biol. 2011 doi: 10.1165/rcmb.2010-0276OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Royce SG, Dang W, Gao Y, Tran J, El-Osta A, Karagiannis TC, Tang LKM. Resveratrol has protective effects against airway remodeling and airway inflammation in a murine model of allergic airways disease. Pathobiology of Aging & Age-related Diseases. 2011;1:7134. doi: 10.3402/pba.v1i0.7134. [DOI] [PMC free article] [PubMed] [Google Scholar]