

Summary
Type 2 diabetes (T2D) has become an epidemic in our modern lifestyle, likely due to calorie-rich diets overwhelming our adaptive metabolic pathways. One such pathway is mediated by nicotinamide phosphoribosyltransferase (NAMPT), the rate-limiting enzyme in mammalian NAD+ biosynthesis, and the NAD+-dependent protein deacetylase SIRT1. Here we show that NAMPT-mediated NAD+ biosynthesis is severely compromised in metabolic organs by high-fat diet (HFD). Strikingly, nicotinamide mononucleotide (NMN), a product of the NAMPT reaction and a key NAD+ intermediate, ameliorates glucose intolerance by restoring NAD+ levels in HFD-induced T2D mice. NMN also enhances hepatic insulin sensitivity and restores gene expression related to oxidative stress, inflammatory response, and circadian rhythm, partly through SIRT1 activation. Furthermore, NAD+ and NAMPT levels show significant decreases in multiple organs during aging, and NMN improves glucose intolerance and lipid profiles in age-induced T2D mice. These findings provide critical insights into a potential nutriceutical intervention against diet- and age-induced T2D.
Introduction
Recent studies have raised an interesting possibility that various physiological mechanisms that mediate metabolic adaptation have evolved in response to nutritionally scarce conditions such as famine and drought (Lazar, 2005). In our modern, sedentary lifestyle with calorie-rich diets, such adaptive mechanisms could be seriously overwhelmed, causing an epidemic of obesity and T2D worldwide (Yach et al., 2006). In mammals, one such mechanism comprises NAMPT-mediated NAD+ biosynthesis and the NAD+-dependent protein deacetylase SIRT1 (Haigis and Sinclair, 2010; Imai, 2010; Imai and Guarente, 2010). NAMPT-mediated NAD+ biosynthesis and SIRT1 together play critical roles in regulating a variety of biological processes that include metabolism, stress response, cellular differentiation, and circadian rhythm, and also mediating adaptive responses to limited energy intake, such as fasting and diet restriction (Imai, 2010). For example, in skeletal muscle, both nutritional deprivation and exercise increase Nampt expression through the activation of AMP-activated protein kinase (AMPK), enhancing NAD+ biosynthesis and SIRT1 activity (Canto et al., 2010; Fulco et al., 2008). In pancreatic β cells, both NAMPT-mediated NAD+ biosynthesis and SIRT1 regulate glucose-stimulated insulin secretion (GSIS) in response to glucose availability (Moynihan et al., 2005; Revollo et al., 2007). Additionally, in the liver and white adipose tissue (WAT), NAMPT and SIRT1 comprise a novel transcriptional-enzymatic feedback loop for the regulation of circadian rhythm, a powerful effecter for metabolism (Imai, 2010).
How nutritional and environmental perturbations affect the system dynamics of this NAMPT/NAD+/SIRT1-driven adaptive, systemic regulatory network, named the “NAD World” (Imai, 2010), still remains unclear. Here we show that HFD and aging compromise NAMPT-mediated NAD+ biosynthesis, contributing to the pathogenesis of T2D. Importantly, we also provide evidence that promoting NAD+ biosynthesis by using nicotinamide mononucleotide (NMN), a product of the NAMPT reaction and a key NAD+ intermediate, could be an effective intervention against diet- and age-induced T2D.
Results
NAMPT-mediated NAD+ biosynthesis is compromised by HFD
To examine the connection between NAMPT-mediated NAD+ biosynthesis and T2D, wild-type B6 male and female mice at 3–6 months of age were fed a HFD containing 42% calories from fat. Both males and females developed overt diabetes after 3.5 and 6 months, respectively. In these mice, we found that NAMPT protein levels were significantly reduced in the liver and WAT, but not in skeletal muscle, compared to regular chow (RC)-fed control mice (Figures 1A, S1A, and S1B). Consistent with these decreases in NAMPT levels, NAD+ levels were also significantly reduced in the liver and WAT, but not in skeletal muscle (Figures 1B and S1C), indicating that there is an underlying defect in NAD+ biosynthesis in the liver and WAT of HFD-induced diabetic mice.
Figure 1. NMN ameliorates defects in NAMPT-mediated NAD+ biosynthesis in HFD-induced diabetic mice.
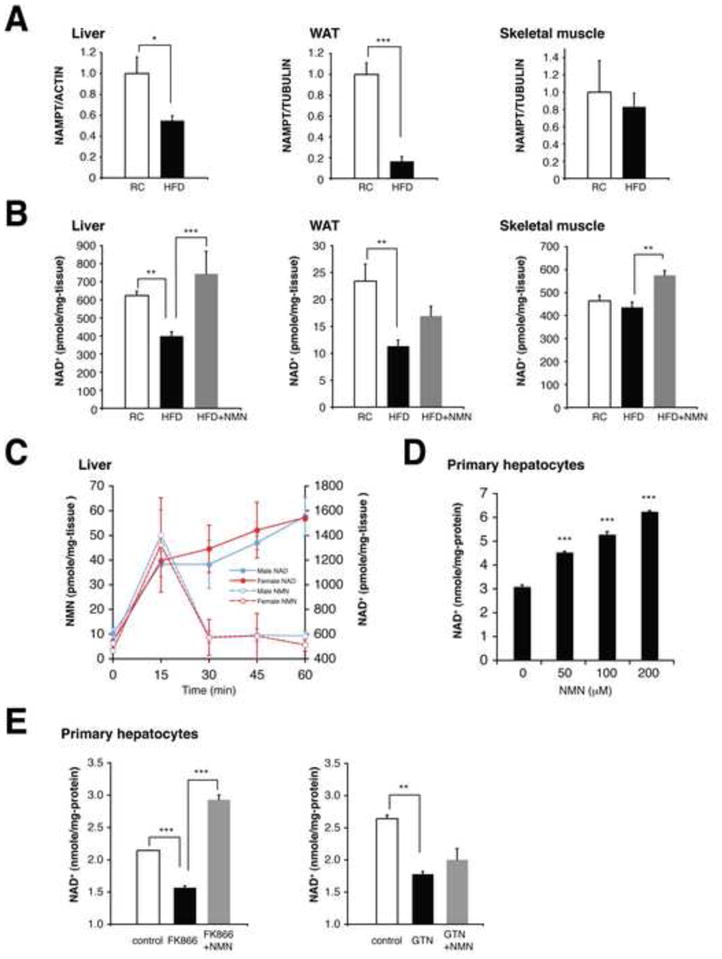
(A) NAMPT protein levels in the liver, WAT, and skeletal muscle. Female mice were fed a RC or a HFD for 6–8 months. NAMPT levels were normalized to ACTIN (liver) or TUBULIN (WAT and skeletal muscle) (n=4 to 5 mice per group). (B) Tissue NAD+ levels in the liver, WAT, and skeletal muscle from RC, HFD, and NMN-treated HFD mice (n=5 to 13 mice per group). NMN (500mg/kg body weight/day) was given intraperitoneally to HFD-fed female mice for 7 consecutive days. (C) Changes in NMN and NAD+ levels in the liver after administering a single dose of NMN to B6 mice (n=3 to 5 mice for each time point). (D and E) Intracellular NAD+ levels in mouse primary hepatocytes. Cells were treated with NMN at the indicated concentrations (D), or with enzyme inhibitors [500 nM FK866 or 100 μM gallotannin (GTN)] in the presence or absence of 100 μM NMN (E), for 4 hrs (n=3 per group). Data were analyzed by Student’s unpaired t test (A) and one-way ANOVA with the Fisher’s PLSD post-hoc test (B, D, E). All values are presented as mean ± SEM. *P < 0.05; **P <0.01; ***P < 0.001.
NMN ameliorates defects in NAD+ biosynthesis and glucose metabolism in T2D mice
Based on these findings, we hypothesized that the defect in NAMPT-mediated NAD+ biosynthesis could be ameliorated by administering NMN to diabetic mice. To test this idea, we initially examined how NMN administration influences NAD+ biosynthesis in the liver, pancreas, and WAT. NMN was immediately utilized and converted to NAD+ within 15 min, resulting in significant increases in NAD+ levels over 60 min (Figures 1C and S1D). We also observed a mild increase (~5-fold) in nicotinamide riboside (NR) levels compared to the ~15-fold increase of NMN, implying that a part of NMN might be converted to NR to get into the liver (Figure S1E). We further confirmed that NMN enhanced NAD+ biosynthesis in mouse primary hepatocytes in a dose-dependent manner (Figure 1D). NMN was also able to overcome NAD+ deficits caused by a potent NAMPT inhibitor FK866 (Hasmann and Schemainda, 2003) but unable to alleviate the NAD+ reduction due to the inhibition of nicotinamide/nicotinic acid mononucleotide adenylyltransferases (NMNATs) by their potent inhibitor gallotannin (Berger et al., 2005) (Figures 1E and S1F). These results demonstrate the effectiveness and specificity of NMN at stimulating NAD+ biosynthesis in vivo and in vitro.
Given these findings, we administered NMN at a dose of 500 mg/kg body weight/day intraperitoneally to HFD-fed male and female diabetic mice for 10 and 7 consecutive days, respectively. No overt abnormalities or changes in body weight were detected during this time (data not shown). NMN administration successfully restored NAD+ levels in the liver and WAT of diabetic mice, and even in diabetic skeletal muscle, a moderate but significant increase in NAD+ was detected (Figure 1B). These results demonstrate the efficacy of NMN treatment in ameliorating the underlying defect in NAD+ biosynthesis in HFD-induced diabetes.
Strikingly, NMN administration completely normalized impaired glucose tolerance in diabetic female mice (Figure 2A). Whereas plasma insulin levels during intraperitoneal glucose tolerance tests (IPGTTs) did not differ before and after NMN treatment, insulin tolerance was significantly improved in these females (Figures 2B and 2C). In diabetic males, NMN did improve impaired glucose tolerance, but the effect was milder compared to the females (Figure 2D). Different from females, GSIS in males was enhanced at 15- and 30-min time points in IPGTTs after NMN administration (Figure 2E). Using primary pancreatic islets isolated from diabetic males, we also confirmed that both NAD+ levels and GSIS were enhanced by NMN (Figure S2). On the other hand, insulin tolerance remained unchanged in males (Figures 2F), suggesting that NMN exerts its major effects on different target tissues between males and females. Although the reason for this sex difference is currently unclear, these results demonstrate that NMN treatment can ameliorate impaired glucose tolerance by improving either insulin sensitivity or insulin secretion in HFD-induced diabetic mice.
Figure 2. NMN administration improves impaired glucose tolerance in HFD-induced diabetic mice.
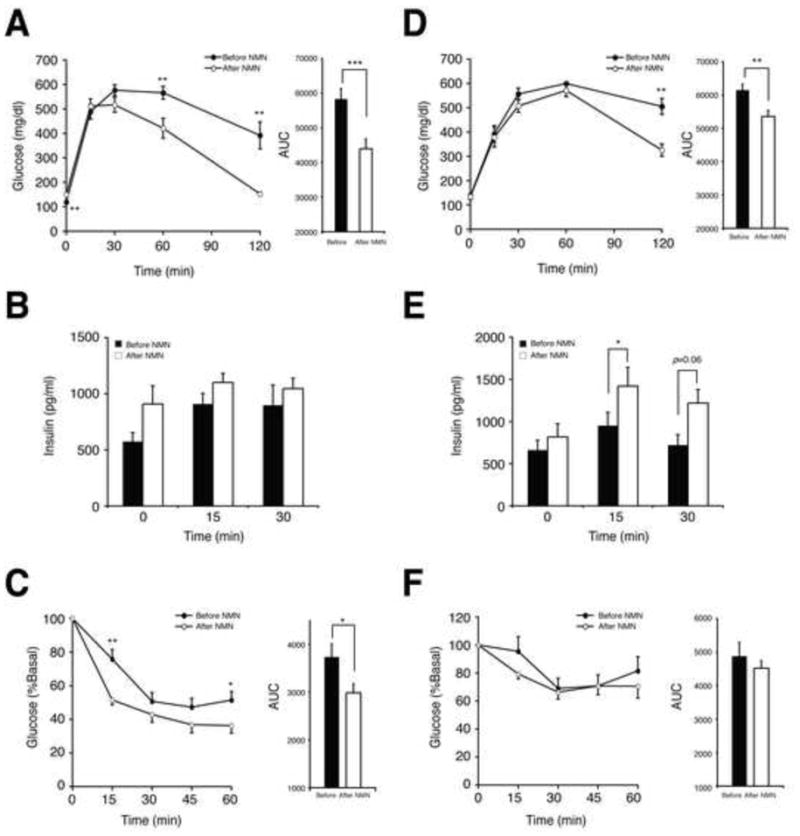
(A and D) Glucose tolerance in HFD female (A) and male mice (D) before and after NMN treatment (n=10 for females, and n=6 for males). IPGTTs were conducted with the same individuals before (closed circles) and after (open circles) NMN. NMN (500mg/kg body weight/day) was administered to female and male mice for 7 and 10 consecutive days, respectively. The areas under each glucose tolerance curve are presented next to the glucose tolerance curves. (B and E) Plasma insulin levels in female (B) and male (E) mice during IPGTTs before and after NMN treatment (n=10 for females, and n=6 for males). (C and F) Insulin tolerance in HFD female (C) and male (F) mice before and after NMN treatment (n=10 for females, and n=6 for males). ITTs were performed before (closed circles) and after (open circles) NMN. ITTs were conducted several days before or after IPGTTs. The areas under each insulin tolerance curve are presented next to the insulin tolerance curves. Data were analyzed by Student’s paired t test. All values are presented as mean ± SEM. *P < 0.05; **P <0.01; ***P < 0.001.
NMN enhances hepatic insulin sensitivity by reversing gene expression caused by HFD
Given that NMN administration restored normal NAD+ levels in diabetic livers (Figure 1B) and also that the liver is known to have a major effect on insulin sensitivity in mice, we examined whether NMN improves hepatic insulin sensitivity in diabetic females. We first assessed phosphorylation status of AKT, a downstream kinase in insulin signaling, in diabetic livers with or without NMN treatment. Clear increases in AKT phosphorylation were detected in NMN-treated diabetic mice, indicating that hepatic insulin sensitivity is improved by NMN treatment (Figure 3A). We next compared gene expression profiles between RC-fed, HFD-fed, and NMN-treated HFD-fed livers. We also performed the parametric analysis of gene set enrichment (PAGE) (Kim and Volsky, 2005) using these gene expression data and GO gene sets. Interestingly, the biological pathways and genes related to oxidative stress, inflammatory response, immune response, and lipid metabolism, all of which are known to contribute to hepatic insulin resistance (Mattson, 2009; Shoelson et al., 2006), were affected by HFD and reversed by NMN (Figures 3B and S3A). For example, pathways related to glutathione S-transferases, which play an important role in the protection from lipid peroxidation products and thereby the maintenance of hepatic insulin sensitivity (Mattson, 2009), were suppressed by HFD and restored by NMN (Figure 3B, GO:0004364 and GO: 0016765). Indeed, expression levels of the glutathione S-transferase alpha 2 gene (Gsta2) were significantly reduced by HFD and recovered by NMN (Figures 3C). Gsta1 and Gsta4 also showed the same directions of changes (12.3 and 6.18 for the sum of Z ratio, respectively), although their false discovery rates did not reach statistical significance (data not shown). Pathways related to damage, inflammatory, and immune responses were induced by HFD and suppressed by NMN (Figure 3B, GO:0006952, GO:0006954, GO:0009611, GO:0006955, and GO:0009605). Consistent with these pathway alterations, expression levels of the interleukin 1β gene (Il1b) and the S100 calcium binding protein A8 and A9 genes (S100a8 and S100a9), which are all direct NF-κB target genes and play important roles in hepatic insulin resistance (Nemeth et al., 2009; Nov et al., 2010), were significantly up-regulated by HFD and down-regulated by NMN (Figures 3C and S3A). Other genes that showed significant changes by both HFD and NMN, such as Lipin1 (Croce et al., 2007) and pyruvate dehydrogenase kinase 4 (Pdk4) (Jeoung and Harris, 2008), have also been connected to insulin resistance. It should also be noted that expression of genes related to circadian rhythm (Dbp, Dec1, and Rev-erb-α) were altered by HFD and reversed by NMN (Figures 3C). The genes regulated by DBP, such as Cyp2a5 and Rgs16, also showed similar expression profiles (Figure 3C and S3A). These findings provide strong support for the importance of NAMPT-mediated NAD+ biosynthesis in the connection between circadian rhythm and metabolic disorders (Imai, 2010).
Figure 3. NMN ameliorates hepatic insulin resistance and restores gene expression related to oxidative stress, inflammatory response, and circadian rhythm.
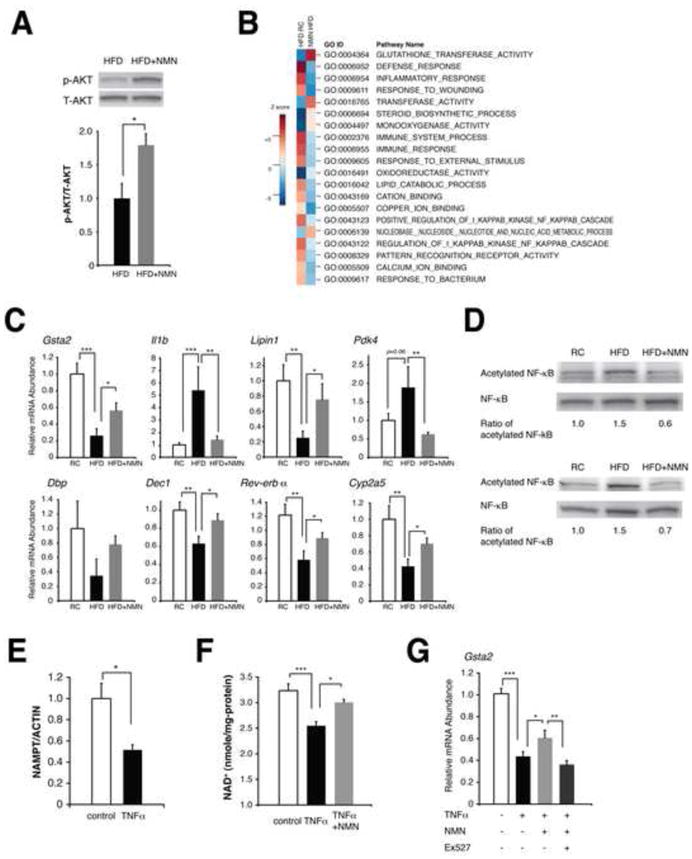
(A) The phosphorylation status of AKT in HFD and NMN-treated HFD female livers (n=3 mice per group). Signal levels of phosphorylated AKT were normalized to total AKT protein levels. (B) Biological pathways that were altered by HFD and reversed by NMN in female livers. Parametric analysis of gene-set enrichment (PAGE) was performed to identify pathways that were significantly up-regulated (red) or down-regulated (blue) by either HFD or NMN using our microarray data (n=4 mice for each condition). Twenty top pathways are listed following the sum of the absolute values of Z scores between two comparisons. (C) Quantitative RT-PCR results for representative genes related to oxidative stress, inflammatory response, circadian rhythm, and metabolism (n=4 to 5 mice per group). Gsta2, glutathione S-transferase alpha 2; Il1b, interleukin 1 beta; Pdk4, pyruvate dehydrogenase kinase isozyme 4; Dbp, D site of albumin promoter (albumin D-box) binding protein; Dec1, deleted in esophageal cancer 1. (D) Acetylation status of NF-κB p65 in RC, HFD, and NMN-treated HFD livers. Two independent sets of mice were used for this analysis, and numbers below each panel represent normalized ratios of acetylated to total p65 levels. (E–G) The effects of TNF-α on NAMPT-mediated NAD+ biosynthesis and gene expression in mouse primary hepatocytes. Cells were treated with 50 ng/ml TNF-α for 72 hrs and given indicated reagents for 6 hrs prior to harvesting for measurements. (E) NAMPT protein levels were normalized to ACTIN (n=3 per group). (F) TNF-α-treated cells were given 100 μM NMN prior to NAD+ measurements (n=3–6 per group). (G) TNF-α-treated cells were cultured with NMN or NMN plus 40 μM EX527 and examined for Gsta2 expression (n=6–9 per group). Data were analyzed by Student’s unpaired t test (A, E). Differences in Ct values or NAD+ levels were analyzed with one-way ANOVA with the Fisher’s PLSD post-hoc test (C, F, G). All values are presented as mean ± SEM. *P < 0.05; **P <0.01; ***P < 0.001.
SIRT1 is one of the mediators for the effect of NMN
To elucidate what transcription factors mediate expression changes by HFD and NMN, biological network analysis was performed using the shortest path and the transcription regulation (TR) algorithms (Figure S3B). We noticed that transcription factors that formed relatively large hubs in the deduced network, such as c-Myc, NF-κB, PPARγ, and p53, are all reported targets of SIRT1 (Figure S3C), implying that SIRT1 might be one of the mediators for the observed gene expression changes. Given that NF-κB is regulated by SIRT1-mediated deacetylation (Yeung et al., 2004) and plays a critical role in hepatic insulin resistance (Shoelson et al., 2006), we analyzed acetylation status of NF-κB in livers from each experimental condition (Figure 3D). Consistent with changes in NAD+ levels and NF-κB target gene expression, levels of acetylated p65, a component of NF-κB, increased by HFD and decreased considerably by NMN, suggesting that SIRT1 activity is suppressed by HFD and restored by NMN.
Because both inflammatory cytokines and oxidative stress are linked to hepatic insulin resistance (Evans et al., 2005; Peraldi and Spiegelman, 1998), we also tested whether TNF-α and menadione can affect NAMPT-mediated NAD+ biosynthesis and cause similar gene expression changes in vitro. Interestingly, whereas both reagents were able to significantly reduce NAMPT and NAD+ levels in primary hepatocytes, only TNF-α showed gene expression patterns similar to those in HFD-fed diabetic livers (Figure 3E, 3F, S3D, and data not shown). This TNF-α-mediated decrease in NAD+ levels was ameliorated by NMN (Figure 3F). In TNF-α/NMN-treated primary hepatocytes, NMN was able to restore the Gsta2 expression to a similar level observed in HFD-fed, NMN-treated livers, and its effect was abrogated by EX527, a potent SIRT1-specific inhibitor, further confirming that SIRT1 is at least one of the mediators for the effect of NMN (Figure 3G).
NMN ameliorates metabolic complications in age-induced T2D mice
In addition to calorie-rich diets, aging is one of the greatest risk factors for developing T2D (Moller et al., 2003). We found that NAD+ levels showed significant decreases in the pancreas, WAT, and skeletal muscle and also the same trend with a much larger variation in the liver in old mice, compared to those in young mice (Figure 4A). NAMPT protein levels also decreased in those tissues (Figures S4A and S4B). Based on these findings, we speculated that NMN treatment might also be effective in age-induced T2D models. We screened RC wild-type B6 males and females at 15–26 months of age and found ~15% of males diabetic (>200 mg/dl at 2-hr time point in IPGTTs). To these aged, naturally occurring diabetic male mice, we intraperitoneally administered the same dose of NMN. Surprisingly, just one dose of NMN normalized impaired glucose tolerance (Figure 4B). In these mice, GSIS at 15- and 30-min time points during IPGTTs were higher after NMN treatment, although the difference did not reach statistical significance (Figure 4C). This is consistent with the results in HFD-induced diabetic male mice (Figure 2E). Importantly, NMN did not convey any significant effects to aged non-diabetic male mice (Figures S4C and S4D), indicating that NMN treatment is effective for age-induced diabetic individuals and does not negatively affect glucose homeostasis in non-diabetic individuals. Because females proved difficult to naturally develop T2D, we fed aged female mice a HFD for 7 weeks. Unlike younger females, aged females were very susceptible to HFD and developed severe diabetes within this time frame. In these aged, diabetic females, 11 consecutive injections of NMN completely normalized severely impaired glucose tolerance (Figure 4D). Qualitatively consistent with this improvement, their respiratory quotient (RQ) showed a significant increase after NMN treatment, implicating better glucose utilization, although their oxygen consumption did not change (Figure S4E). Rectal body temperature also decreased, which might be explained by a significant shift in energy utilization from fat to glucose (Figure S4F). Furthermore, hyperlipidemia induced by HFD was also corrected by NMN (Figure 4E). Thus, these results support the efficacy of NMN to significantly improve impaired glucose tolerance in age-induced T2D, as well as in HFD-induced T2D.
Figure 4. NMN improves glucose and lipid homeostasis in age-induced T2D.
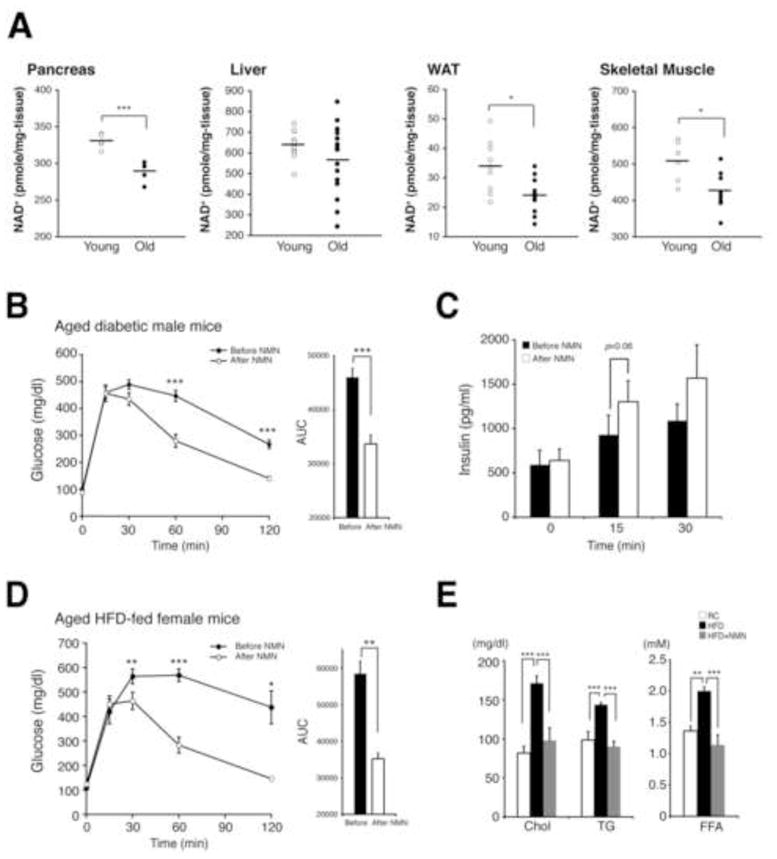
(A) NAD+ levels in metabolic tissues between young and old mice. Pancreas, liver, WAT, and skeletal muscle were collected from young (n=5–11) and old (n=5–15) mice at 3–6 and 25–31 months of age, respectively. (B) Glucose tolerance in aged, naturally occurring diabetic male mice before (closed circles) and after (open circles) a single dose of NMN (500 mg/kg body weight) (n=11). The areas under each glucose tolerance curve are presented next to the glucose tolerance curves. (C) Plasma insulin levels were measured during IPGTTs (n=5). (D and E) Glucose tolerance (D) and lipid levels (E) in aged HFD female mice before (closed circles) and after (open circles) NMN (n=5). Fasted plasma samples were collected from the same mice and subjected to the measurements of cholesterol (Chol), triglycerides (TG), and non-esterified free fatty acids (FFA). Data were analyzed by Student’s unpaired t test (A), paired t test (B–D), and one-way ANOVA with the Fisher’s PLSD post-hoc test (E). All values are presented as mean ± SEM. *P < 0.05; **P <0.01; ***P < 0.001.
Discussion
The results presented in this study demonstrate that NAMPT-mediated NAD+ biosynthesis is compromised by HFD and aging, contributing to the pathogenesis of T2D. We also provide proof of the concept that promoting NAD+ biosynthesis by administering NMN, a key NAD+ intermediate, can be an effective intervention to treat the pathophysiology of diet- and age-induced T2D (Figure S4G). In both diabetic models, NMN administration dramatically ameliorated impaired glucose tolerance by restoring normal NAD+ levels and enhancing either insulin sensitivity or insulin secretion, supporting our conclusion that underlying defects in NAMPT-mediated NAD+ biosynthesis play an important role in the pathogenesis of diet- and age-induced T2D. Although the short-term NMN administration was unable to achieve a significant improvement of fasted glucose levels, NMN is still effective at normalizing multiple metabolic pathways, such as oxidative stress, inflammatory response and circadian rhythm, and GSIS, in T2D. Whereas how NMN is transported into cells currently remains unknown, metabolic tissues and organs seem to utilize NMN and convert it to NAD+ efficiently. Therefore, an adequate and consistent supply of this key NAD+ intermediate must be critical to maintain normal hepatic insulin sensitivity and GSIS in pancreatic β cells. To assess beneficial and possible adverse effects of NMN more comprehensively, we are currently conducting long-term NMN supplementation experiments in different dietary conditions.
Inflammation and/or oxidative stress caused by hepatosteatosis or aging appear to trigger the reduction in NAMPT-mediated NAD+ biosynthesis and contribute to the pathogenesis of T2D. Given that NMN is capable of reversing changes in gene expression related to oxidative stress, inflammatory response, and circadian rhythm, NAMPT-mediated NAD+ biosynthesis regulates homeostatic, protective mechanisms against nutritional perturbations, such as HFD. Our results also indicate that SIRT1 is at least one of the mediators for these beneficial effects of NMN on hepatic insulin sensitivity. In addition to SIRT1, there might be other NAD+-sensitive or NAD+-consuming factors that also contribute to the effects of NMN. For example, other sirtuin family members (SIRT2–7) also likely play some roles in the metabolic effects of NMN. Particularly, the functions of mitochondrial sirtuins (SIRT3–5) might be affected by deficits in NAMPT-mediated NAD+ biosynthesis, potentially resulting in the mitochondrial dysfunction observed in T2D (Lowell and Shulman, 2005). The sex difference that we observed in this study is also interesting. Given that estrogen signaling is critical for the regulation of hepatic insulin sensitivity and GSIS (Meyer et al., 2011), estrogen might play a role in the effects of NMN under HFD feeding. Further investigation will be required to identify detailed mechanisms for the efficacy of NMN.
Our results provide an interesting implication that NMN supplementation might also be effective in human T2D patients if they have defects in NAMPT-mediated NAD+ biosynthesis. Because NMN is an endogenous compound, this is rather a nutriceutical approach to T2D. Nonetheless, it will be of great interest to examine whether the effects of NMN are synergistic with those of small chemical SIRT1 activators (Haigis and Sinclair, 2010), particularly in aged, diabetic individuals. Taken together, we anticipate that long-term NMN administration might be a highly effective way to sustain enhanced SIRT1 activity in tissues and organs where NAMPT-mediated NAD+ biosynthesis is compromised and to combat against the disconcerting epidemic of T2D.
Experimental Procedures
Animal Experimentation
For the HFD-induced T2D model, mice at 3–6 months of age were fed a HFD containing 42% of the total calories from fat (TD88137; Harlan Taklad). We defined diabetes as mice having fasted blood glucose levels > 120 mg/dl or blood glucose levels at 2-hr time point in IPGTTs ≥ 200 mg/dl. To obtain age-induced diabetic mice, we screened male and female mice at 15–26 months of age following the criteria. For NMN treatment, we intraperitoneally administered NMN (Sigma) at the dose of 500 mg/kg body weight/day as indicated in the text and figure legends. This dose was determined in our previous studies (Ramsey et al., 2008). All animal studies were approved by the Washington University Animal Studies Committee. Details are available in the Supplemental Experimental Procedure.
NAD+ and NMN measurements
NAD+ and NMN levels were determined using a HPLC system (Shimadzu) with a Supelco LC-18-T column (15cm × 4.6cm; Sigma) and a Hypercarb column (15cm × 4.6cm; Thermo Scientific), respectively. Details are available in the Supplemental Experimental Procedure.
Primary hepatocyte isolation
Primary hepatocytes were isolated and cultured as described previously (Grimm et al., 2011). For treatments with NMN, enzyme inhibitors (FK866, gallotannin, EX527), TNF-α, and menadione, hepatocytes were cultured in DMEM containing 1.0% FBS and each reagent or their combinations as indicated in the text.
Micoroarrays
Total RNA was isolated from frozen liver samples of RC, HFD, and NMN-treated HFD mice and used for Illumina Mouse Ref 8 whole genome microarrays (version 2). Details are available in the Supplemental Experimental Procedure.
Quantitative Real-Time PCR
Real-time PCR was performed using the 7900HT Fast Real-Time PCR System (Applied Biosystems). Relative expression levels were determined based on the CT values and normalized to CT values for the Gapdh gene. P values for the differences of CT values were calculated using one-way ANOVA with the Fisher’s PLSD test.
Detection of acetylated NF-κB p65
Nuclear extracts were prepared from frozen liver samples as previously described (Rodgers and Puigserver, 2007). 100 μg of liver nuclear extract were analyzed by Western blotting with an anti-acetyl NF-κB p65 (Lys310) antibody (Cell Signaling Technology). The same blot was re-probed with anti-NF-κB p65 antibody (Santa Cruz). Signals were visualized using the ECL Plus detection system (Amersham).
Statistical Analyses
Differences between two groups were assessed using Student’s paired or unpaired t test. Comparisons among several groups were performed using one-way ANOVA with the Fisher’s PLSD post-hoc test. P values of less than 0.05 were considered statistically significant.
Supplementary Material
Highlights.
NAMPT-mediated NAD+ biosynthesis is compromised in metabolic organs by HFD.
NMN ameliorates defects in NAD+ biosynthesis and glucose metabolism in T2D mice.
NMN enhances hepatic insulin sensitivity by reversing gene expression caused by HFD.
NMN also ameliorates defects in glucose and lipid metabolism in age-induced T2D mice.
Acknowledgments
We thank Koji Kadota for the PAGE analysis, Trey Coleman for lipid and calorimetric measurements, Xuntian Jiang and Daniel Ory for mass spec analysis in the Metabolomics Facility at Washington University. We also thank Joe Bass, Yo-ichi Nabeshima, and members of the Imai lab for critical reading and suggestions on this manuscript. This work was supported in part by the National Institute on Aging (AG02150), the Ellison Medical Foundation, and the Longer Life Foundation to S.I. and by institutional support from the Washington University Nutrition Obesity Research Center (P30DK056341) and the Washington University Diabetes Research and Training Center (P60DK020579). J.Y. is supported by the Japan Research Foundation for Clinical Pharmacology, the Manpei Suzuki Diabetes Foundation, and the Kanae Foundation For the Promotion of Medical Science. S.I. serves as a scientific advisory board member for Sirtris, a GSK company.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Berger F, Lau C, Dahlmann M, Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem. 2005;280:36334–36341. doi: 10.1074/jbc.M508660200. [DOI] [PubMed] [Google Scholar]
- Canto C, Jiang LQ, Deshmukh AS, Mataki C, Coste A, Lagouge M, Zierath JR, Auwerx J. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 2010;11:213–219. doi: 10.1016/j.cmet.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croce MA, Eagon JC, LaRiviere LL, Korenblat KM, Klein S, Finck BN. Hepatic lipin 1beta expression is diminished in insulin-resistant obese subjects and is reactivated by marked weight loss. Diabetes. 2007;56:2395–2399. doi: 10.2337/db07-0480. [DOI] [PubMed] [Google Scholar]
- Evans JL, Maddux BA, Goldfine ID. The molecular basis for oxidative stress-induced insulin resistance. Antioxid Redox Signal. 2005;7:1040–1052. doi: 10.1089/ars.2005.7.1040. [DOI] [PubMed] [Google Scholar]
- Fulco M, Cen Y, Zhao P, Hoffman EP, McBurney MW, Sauve AA, Sartorelli V. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev Cell. 2008;14:661–673. doi: 10.1016/j.devcel.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm AA, Brace CS, Wang T, Stormo GD, Imai SI. A nutrient-sensitive interaction between Sirt1 and HNF-1alpha regulates Crp expression. Aging Cell. 2011;10:305–317. doi: 10.1111/j.1474-9726.2010.00667.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 2010;5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasmann M, Schemainda I. FK866, a highly specific noncompetitive inhibitor of nicotinamide phosphoribosyltransferase, represents a novel mechanism for induction of tumor cell apoptosis. Cancer Res. 2003;63:7436–7442. [PubMed] [Google Scholar]
- Imai S. “Clocks” in the NAD World: NAD as a metabolic oscillator for the regulation of metabolism and aging. Biochim Biophys Acta. 2010;1804:1584–1590. doi: 10.1016/j.bbapap.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Guarente L. Ten years of NAD-dependent SIR2 family deacetylases: implications for metabolic diseases. Trends Pharmacol Sci. 2010;31:212–220. doi: 10.1016/j.tips.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeoung NH, Harris RA. Pyruvate dehydrogenase kinase-4 deficiency lowers blood glucose and improves glucose tolerance in diet-induced obese mice. Am J Physiol Endocrinol Metab. 2008;295:E46–54. doi: 10.1152/ajpendo.00536.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Volsky DJ. PAGE: parametric analysis of gene set enrichment. BMC Bioinformatics. 2005;6:144. doi: 10.1186/1471-2105-6-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazar MA. How obesity causes diabetes: not a tall tale. Science. 2005;307:373–375. doi: 10.1126/science.1104342. [DOI] [PubMed] [Google Scholar]
- Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307:384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp Gerontol. 2009;44:625–633. doi: 10.1016/j.exger.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer MR, Clegg DJ, Prossnitz ER, Barton M. Obesity, insulin resistance and diabetes: sex differences and role of oestrogen receptors. Acta Physiol (Oxf) 2011 doi: 10.1111/j.1748-1716.2010.02237.x. Epub on Feb 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moller N, Gormsen L, Fuglsang J, Gjedsted J. Effects of ageing on insulin secretion and action. Horm Res. 2003;60:102–104. doi: 10.1159/000071233. [DOI] [PubMed] [Google Scholar]
- Moynihan KA, Grimm AA, Plueger MM, Bernal-Mizrachi E, Ford E, Cras-Meneur C, Permutt MA, Imai S. Increased dosage of mammalian Sir2 in pancreatic β cells enhances glucose-stimulated insulin secretion in mice. Cell Metab. 2005;2:105–117. doi: 10.1016/j.cmet.2005.07.001. [DOI] [PubMed] [Google Scholar]
- Nemeth J, Stein I, Haag D, Riehl A, Longerich T, Horwitz E, Breuhahn K, Gebhardt C, Schirmacher P, Hahn M, Ben-Neriah Y, Pikarsky E, Angel P, Hess J. S100A8 and S100A9 are novel nuclear factor kappa B target genes during malignant progression of murine and human liver carcinogenesis. Hepatology. 2009;50:1251–1262. doi: 10.1002/hep.23099. [DOI] [PubMed] [Google Scholar]
- Nov O, Kohl A, Lewis EC, Bashan N, Dvir I, Ben-Shlomo S, Fishman S, Wueest S, Konrad D, Rudich A. Interleukin-1beta may mediate insulin resistance in liver-derived cells in response to adipocyte inflammation. Endocrinology. 2010;151:4247–4256. doi: 10.1210/en.2010-0340. [DOI] [PubMed] [Google Scholar]
- Peraldi P, Spiegelman B. TNF-alpha and insulin resistance: summary and future prospects. Mol Cell Biochem. 1998;182:169–175. [PubMed] [Google Scholar]
- Ramsey KM, Mills KF, Satoh A, Imai S. Age-associated loss of Sirt1-mediated enhancement of glucose-stimulated insulin secretion in β cell-specific Sirt1-overexpressing (BESTO) mice. Aging Cell. 2008;7:78–88. doi: 10.1111/j.1474-9726.2007.00355.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revollo JR, Körner A, Mills KF, Satoh A, Wang T, Garten A, Dasgupta B, Sasaki Y, Wolberger C, Townsend RR, Milbrandt J, Kiess W, Imai S. Nampt/PBEF/visfatin regulates insulin secretion in β cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007;6:363–375. doi: 10.1016/j.cmet.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers JT, Puigserver P. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc Natl Acad Sci USA. 2007;104:12861–12866. doi: 10.1073/pnas.0702509104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yach D, Stuckler D, Brownell KD. Epidemiologic and economic consequences of the global epidemics of obesity and diabetes. Nat Med. 2006;12:62–66. doi: 10.1038/nm0106-62. [DOI] [PubMed] [Google Scholar]
- Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.