

Abstract
K2P6.1, a member of the Two-Pore Domain K channel family, is highly expressed in the vascular system; however its function is unknown. We tested the following hypotheses. K2P6.1 regulates (1) systemic blood pressure, (2) the contractile state of arteries, (3) vascular smooth muscle cell migration, (4) proliferation, and/or (5) volume regulation. Mice lacking K2P6.1 (KO) were generated by deleting exon 1 of Kcnk6. Mean arterial blood pressure in both anesthetized and awake KO mice was increased by 17 ± 2 and 26 ± 3 mmHg respectively (p<0.05). The resting membrane potential in freshly dispersed vascular smooth muscle cells was depolarized by 17 ± 2 mV in the KO compared to WT littermates (p<0.05). The contractile responses to KCl (p<0.05) and BAY K 8644 (p<0.01), an activator of L-type calcium channels, were enhanced in isolated segments of aorta from KO mice. However there was no difference in current density of L-type calcium channels. Responses to U46619, an agent which activates rho kinase, showed an enhanced contraction in aorta from KO mice (p<0.001). The BAY K 8644-mediated increase in contraction was decreased to WT levels when treated with Y27632, a rho kinase inhibitor, (p<0.05). K2P6.1 does not appear to be involved with migration, proliferation, or volume regulation in cultured vascular smooth muscle cells. We conclude that K2P6.1 deficiency induces vascular dysfunction and hypertension through a mechanism that may involve smooth muscle cell depolarization and enhanced rho kinase activity.
Keywords: Two-pore domain potassium channels (K2P), Hypertension, Resting membrane potential, TWIK-2, Kcnk6, K2P6.1, Rho kinase
Introduction
In the mid to late 1990’s, researchers took advantage of the fact that known K channels had a highly conserved sequence of amino acids in the K+ selective pore1. A search for mammalian genes that carried homology to this signature sequence revealed a new, previously unrecognized family of K channels named “Two-Pore Domain” K channels (K2P, gene name Kcnk) on the basis that each protein subunit contained two pore forming regions of the K+ selectivity filter2,3. Unlike other K channels, which require four protein subunits, each contributing one pore domain to form a fully functioning channel, K2P channels only require two. Since the discovery of the first mammalian K2P in 1996, fifteen genes for this family have been identified3.
One of the K2P channels without a known function is K2P6.1 (Kcnk6 or TWIK-2). Unlike most other K2P, K2P6.1 is conspicuously absent from neuronal and glial cells but is expressed in a number of other tissues such as stomach, spleen, lung and pancreas4-6. Of interest to us is the fact that K2P6.1 is highly expressed in all blood vessels studied to date including resistance sized vessels2,7-10. Thus, K2P6.1 is a prime candidate for physiological regulation in the vascular system. In the present study, we tested the following hypotheses. (1) K2P6.1 regulates systemic blood pressure. (2) K2P6.1 in vascular smooth muscle cells (VSMC) regulates the contractile state of arteries. Since K channels in the vascular system are involved with other important physiological processes, we also tested the following hypotheses: (3) K2P6.1 in VSMC is involved with cell migration11 and/or proliferation12, (4) K2P6.1 in VSMC is involved in volume regulation during osmotic stress13 and/or apoptosis14. Since there are no selective activators or inhibitors for K2P6.1, we generated a K2P6.1 knockout mouse for these studies. We report that K2P6.1 deficient mice have depolarized plasma membranes of VSMC, increased vascular contractility and hypertension. A mechanism that accounts for these changes may involve K2P6.1 setting the resting membrane potential of VSMC.
Methods
For Methods see http://hyper.ahajournals.org. All studies were approved by the Institutional Animal Care and Use Committee at the Baylor College of Medicine.
Results
KO mice were viable and fertile, obeyed the Mendelian ratio, and showed no changes in gross body composition at 8-12 weeks of age (http://hyper.ahajournals.org, Figure S1 for knockout strategy and Table S1 for body composition analysis). Figure 1 shows the results of RNA detection by real-time PCR. The mRNA expression is shown as a percent of the reference mRNA, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Figures 1A and 1B). A panel of six K2P gene family members was probed in the thoracic aorta (A) and whole heart (B). A second panel of three non-K2P but vascular-related targets is also shown.
Figure 1.
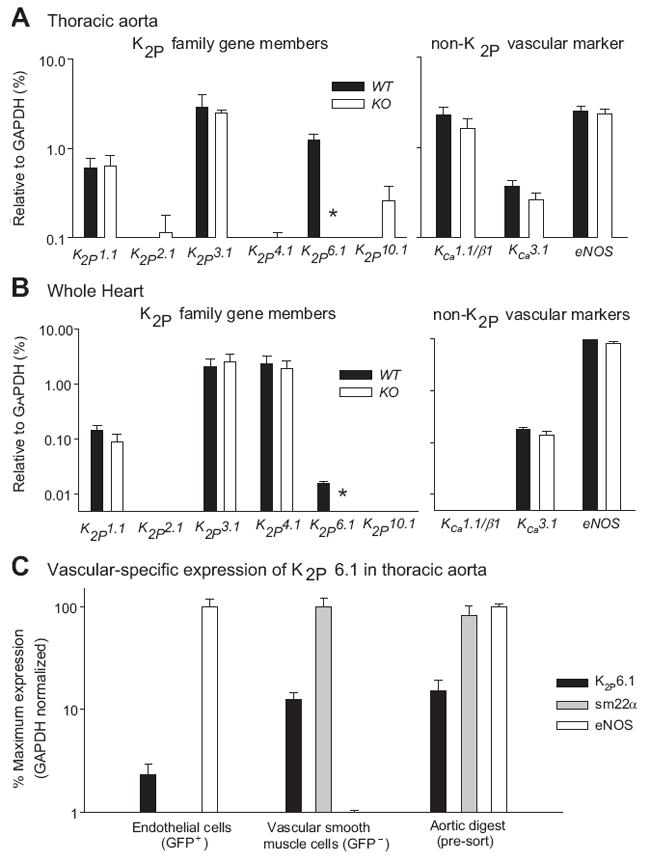
(A) Thoracic aorta and (B) whole heart RNA expressed as a percent of the reference mRNA, glyceraldehyde 3-phosphate dehydrogenase (GAPDH). The left panel shows expression of six K2P family gene members; the right panel shows expression levels of three non-K2P, but vascular related targets. (C) Expression of K2P6.1 in vascular smooth muscle and endothelial cells of Tie2-GFP aorta. GFP+ indicates Tie2-expressing cells as the endothelial fraction (RNA profile eNOS+/sm22α−). GFP− indicates non-Tie2 expressing cells as the vascular smooth muscle fraction (RNA profile eNOS−/sm22α+). Data in C are normalized to GADPH and expressed as the % of the gene with greatest expression for each individual fraction. *p<0.05, n=4-10 for each experimental group.
Gene expression of K2P1.1 (TWIK-1), the closest relative to K2P6.1, showed no change between genotypes. Other genes that are involved with vascular function including the K channels, KCa1.1/β1 and KCa3.1, and eNOS showed no change in expression. K2P6.1 mRNA is absent in the KO aorta and heart (p<0.05, n=4-10), and there is no evidence of gene profile remodeling at the level of mRNA in selected genes as a result of this gene deletion. However, this conclusion does not account for genes not studied or post-translational modifications as a result of K2P6.1 deletion.
For cell-specific expression of K2P6.1, aorta of Tie2-GFP (endothelial expressing) mice were digested and sorted into GFP+ (RNA profile eNOS+/sm22α−) as the endothelial fraction and GFP− (RNA profile eNOS−/sm22α+) as the VSMC fraction. K2P6.1 RNA expression levels were detectable to a greater extent in the VSMC of the thoracic aorta, however, RNA was also detected in the endothelial fraction (Figure 1C, n=4).
Figure 2A shows blood pressures in awake mice obtained using tail cuff plethysmography. Systolic and mean arterial pressures were increased in the KO mice by 34 ± 3 and 26 ± 3 mmHg respectively (n=6 pairs). Blood pressures in anesthetized mice confirm that KO mice were hypertensive (Figure 2B and http://hyper.ahajournals.org, Table S2). Systolic (+17 ± 3 mmHg), diastolic (+15 ± 2 mmHg), and mean arterial (+17 ± 2 mmHg) blood pressure measurements were significantly increased in the anesthetized KO mice (n=6 pairs) [systolic 115 ± 6 and 98 ± 3 mmHg (p<0.05); diastolic 80 ± 5 and 64 ± 5 mmHg (p=0.05); and mean arterial 98 ± 6 and 81 ± 4 mmHg (p<0.05) in KO and WT respectively]. Peripheral vascular resistance (PVR) was significantly increased in anesthetized KO mice (p=0.02, Figure 2B). For a complete table of cardiovascular indices in anesthetized mice see http://hyper.ahajournals.org, Table S2.
Figure 2.
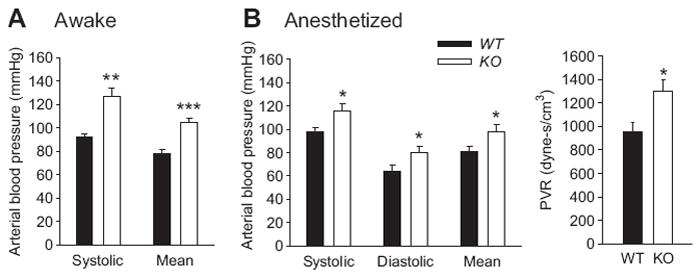
(A) Blood pressure in awake mice (tail plethysmography). (B) Blood pressure and peripheral vascular resistance (ratio of mean arterial pressure and mean aortic velocity) in Nembutal-anesthetized mice. All mice were between 8 and 12 weeks of age. n=6 animals per experimental group. *p≤ 0.05 **p<0.01 ***p<0.001.
Histological examination showed that the aortic wall was approximately 20% thicker in KO mice (http://hyper.ahajournals.org, Figure S2A and S2B); however, this increase did not reach statistical significance (p=0.26). Luminal diameters of the aortas were not significantly different between WT and KO mice. It is possible that major changes in vascular structure as a result of the hypertension requires longer than 8-12 week (the age studied) to develop.
Wire myography was performed to assess the contractile state of the aorta in the K2P6.1 KO and WT animals. Figure 3A shows the length-force relationship with the KO generating more force to the application of 60 mmol/L isotonic KCl (Na+ adjusted) with resting force between 15 and 40 mN (p<0.01, n=8). For all subsequent wire myography studies we chose a resting force of 15 mN for each aorta regardless of genotype. At this resting force, aortas from both KO and WT contracted to 80% of their respective maximums with addition of 60 mmol/L KCl. Figure 3B shows the concentration response curve for increases in KCl (isotonic by reducing Na+). Aortic rings from KO mice generated greater force at each K+ concentration compared to aortas from WT mice (p<0.01, n=6). At 60 mmol/L KCl aortas from the KO contracted with 54% greater force compared to the WT (p<0.01, n=6). The addition of 100 μmol/L LNAME to inhibit eNOS produced enhanced forces as a result of increasing extracellular K+ in rings from both genotypes. However, the forces generated by rings from KO mice were still greater after eNOS inhibition than WT rings with differences in force being the same as without eNOS inhibition (p<0.001, n=6, data not shown).
Figure 3.
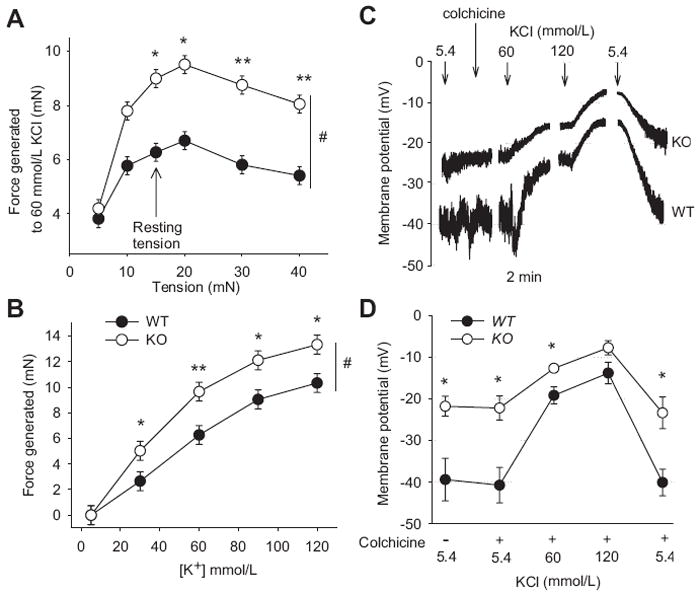
(A) Force generated by addition of 60 mmol/L KCl at different resting tensions in thoracic aortas. (B) Concentration response curves for KCl at a resting force of 15 mN. (C) Raw traces of membrane potential, as measured by current clamp, in response to KCl in aortic VSMC from KO (upper trace) versus WT (lower trace). (D) Summary data from membrane potential studies. n=5-8 for each experimental group. #p<0.05 using 2-way RM-ANOVA, *p<0.05 and **p<0.01 using the Holm-Sidak method for multiple comparison.
Figures 3C and 3D show electrical potentials across the membrane using the current clamp mode (i=0 pA) in freshly dispersed VSMC from the aorta. An individual cell of each genotype is shown in 3C with the summary data given in 3D. VSMC from KO mice were +17 ± 2 mV more depolarized compared to VSMC from WT mice (-39 mV ± 5 and -22 mV ± 2 respectively, p<0.05, n=7 for each genotype). Addition of 10 μmol/L colchicine, which prevented contraction of the cells and loss of the pipette seal, had no effect on membrane potential. Increasing KCl in the extracellular buffer from 5.4 mmol/L to 60 mmol/L (Na+ adjusted) depolarized the plasma membrane of both genotypes; however, the KO maintained a significantly more depolarized state compared to the WT (p<0.05, n=7, Figures 3C and 3D). With the addition of 120 mmol/L KCl, the difference in membrane potential between genotypes was no longer statistically different (p=0.23, n=7, Figure 3D). After washing with physiological KCl buffer, the initial resting membrane potentials were re-established. Cell size as determined by capacitance did not differ between genotypes (n=6) [WT 12.1 pF ± 1.6 and KO 12.5 pF ± 0.8 (p=0.85)].
Figure 4 shows contractile responses of aortic rings to phenylephrine, U46619, and BAY K 8644. Contractions to phenylephrine, an α-adrenergic agonist, were similar in aortic rings from WT and KO mice (figure 4A, p=0.97). However, aortic rings from KO mice generated more force with the addition of U46619, a thromboxane mimetic (figure 4B, p=0.01 for genotype and p<0.001 for interaction between genotype and U46619 concentration) and BAY K 8644, an L-type calcium channel activator (figure 4C, p=0.04 for genotype and p<0.001 for interaction between genotype and BAY K 8644 concentration). Contractions with the addition 100 μmol/L LNAME were enhanced with all agonists in both genotypes compared to having no LNAME present. The contractions to phenylephrine were similar in aortic rings from WT and KO mice (n=8, p=0.75). However, contractions to U46619 and BAY K in the presence of LNAME were enhanced in rings from KO compared to WT mice (n=6 and 5 respectively; p<0.01 and p=0.01 respectively for interaction between genotype and concentration).
Figure 4.
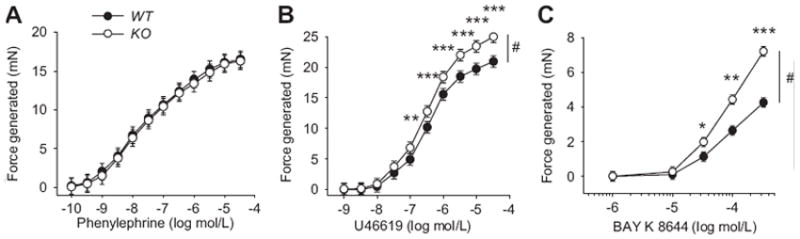
Concentration response curves for (A) phenylephrine, an α-adrenergic agonist, (B) U46619, a thromboxane mimetic, and (C) BAY K 8644, an L-type calcium channel activator in aortic rings from WT and KO mice. n=6-9 per experimental group. #p<0.01 using 2-way RM-ANOVA, *p<0.05, **p<0.01, and ***p<0.001 using the Holm-Sidak method for multiple comparison.
Having established differences in contractile properties of the aortic rings to KCl, U46619 and BAY K 8644, we sought to determine the role of calcium channels and rho kinase in these contractions (http://hyper.ahajournals.org, figure S3). After aortic rings were precontracted with 60 mmol/L KCl, 10-6 mol/L U46619, or 10-4 mol/L BAY K 8644, changes in force were recorded with the addition of the L-type calcium channel blocker, nifedipine, the T-type calcium channel blocker, mibefradil, or the rho kinase inhibitor, Y27632. Y27632 is greater than 100 times more potent for rho kinase than to other kinases including PKC, PKA and MLCK15. From the studies described in Figure S3, we determined that (1) the L-type calcium channel is the predominant channel in the mouse aorta, (2) U46619, a thromboxane mimetic, only constricts through activation of rho kinase; calcium channels are not involved, and (3) 10-6 mol/L nifedipine relaxed KO aortas 62% less than WT when precontracted with BAY K 8644, an L-type calcium channel activator.
In the next series of experiments, we determined if the densities of calcium channels were different in aortic VSMC from WT and KO mice. Studies were conducted by measuring whole cell currents using ruptured patches in the voltage clamp configuration. Ba2+, which is conducted by calcium channels, was used as a surrogate for Ca2+ since calcium channels do not inactivate as rapidly and conduct up to 4 times greater with Ba2+ compare to Ca2+ 16. Figure S4 shows raw traces and summary data for currents at different voltage steps from VSMC of WT and KO mice. There were no statistical differences in current densities between genotype at baseline (p=0.46), after administration of BAY K 8644 (p=0.80) or after the addition of nifedipine (p=1.0). Cell size as determined by capacitance did not differ among genotypes (n=6) [WT 12.2 pF ± 1.7 and KO 12.7 pF ± 1.0 (p=0.8)].
We conducted a study where the rho kinase inhibitor was added prior to the addition of BAY K 8644 (Figure 5). Pre-incubation with 10-6 mol/L Y27632 for 60 minutes had very little effect on the contractions in aortic rings from WT mice. However, Y27632 significantly reduced the contractions to BAY K 8644 in aortas from KO mice as shown in Figure 5A (p<0.05, RM ANOVA, n=6). In the presence of the rho kinase inhibitor, contractions produced by BAY K 8644 in aortas from WT and KO mice were similar. Figure 5B shows the maximum contractions elicited by 10-4 mol/L BAY K 8644 in the presence and absence of 10-6 mol/L Y27632. The data in Figure 5B is a portion of the data shown in Figure 5A but in bar chart format to emphasize effects of inhibition of rho kinase. Pre-incubation with Y27632 significantly decreased the contraction (-42% ± 3) in KO (n=6, p<0.001) but not in aortas from WT mice (-25% ± 8, n=6, p=0.302). These results suggest that the KO aortas have increased rho kinase activity in the presence of BAY K 8644 compared to aortas from WT mice.
Figure 5.
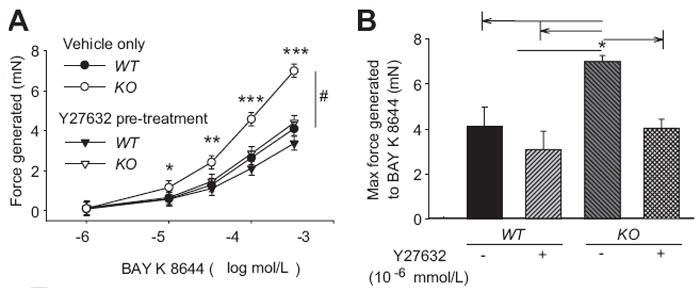
(A) Contraction to BAY K 8644, an activator of L-type calcium channels, in control and after pre-incubation of aortic rings with Y27632, a rho kinase inhibitor. (B) Summary data from maximum contractions obtained at 10-4 mol/L BAY K 8644. n=6 per experimental group. #p<0.05 using 2-way RM-ANOVA, *p<0.05, **p<0.01, and ***p<0.001 using the Holm-Sidak method for multiple comparison.
There were no significant differences between genotype in aortic VSM in studies involving migration (Figure S5A), proliferation (Figure S5B), osmotic volume regulation (Figure S5C), or apoptotic volume regulation (Figure S5D).
Discussion
Although human, rat, and mouse K2P6.1 have been cloned and the electrophysiological properties studied in heterologous expression systems4,5,17, the physiological function of K2P6.1 is presently not known. The fact that K2P6.1 is highly expressed in all blood vessels studied to date2,7-10 led us to ask questions regarding the functional role of K2P6.1 in the vascular system. Our studies reveal that (1) K2P6.1 influences systemic blood pressure, (2) K2P6.1 regulates the contractile state of vascular muscle, likely by setting its membrane potential and rho kinase activity, and (3) K2P6.1 does not appear to be involved with VSMC migration, proliferation, or volume regulation in the models tested.
We acknowledge the inherent limitations in the use of a mouse knockout model as pre- or post-translational changes in other genes can occur with gene deletion especially when the gene deletion leads to pathophysiological conditions such as hypertension. Further studies into the mechanisms of action of K2P6.1 in the vascular system will be needed to develop a clearer picture of its role during normal states and in disease states like hypertension.
At an age of 8-12 weeks, cardiac indices were not different between WT and KO mice (http://hyper.ahajournals.org, Table S2). However, blood pressure in KO mice was significantly increased in awake and anesthetized mice compared to WT (Figures 2A and 2B). The peripheral vascular resistance increased 42% in KO mice (Figure 2B and http://hyper.ahajournals.org, Table S2). The increase in peripheral vascular resistance, which is associated with essential hypertension18, is likely due to peripheral vasoconstriction. While the present study looked at the aorta, it is noteworthy that smaller vessels also express K2P6.12,7-10 and are likely responsible for the increased peripheral vascular resistance. Since neurons express little to no K2P6.14-6, a central mechanism for the hypertension in KO mice is not likely. Similarly, K2P6.1 appears to have little to no expression in mouse kidney6,19; reducing the possibility of a renal component to the observed hypertension in the KO mice.
Our data strongly suggest that rho kinase is responsible for the enhanced contractions to KCl, BAY K 8644, and U46619 (Figures 3A, 3B, 4B, and 4C). First, aortic segments from KO mice contracted more to U46619 than did corresponding segments from WT mice (Figure 4B). U46619 is an agonist that works exclusively through activation of rho kinase without activation of L-type calcium channels (Figure S3C-D). Therefore, rho kinase in aortic segments from KO mice must be either more active at rest, is activated more by U46619, or both. Second, pretreatment of aortic segments from KO mice with the rho kinase inhibitor, Y27632, normalized the contraction to BAY K 8644 (Figures 5A, and 5B). Third, the addition of nifedipine to arterial segments from KO mice that were precontracted with BAY K 8644 did not relax as much on a percentage basis at concentrations of 10-7 and 10-6 mol/L as did the segments from WT mice (Figure S3E). Since BAY K 8644 contracted aortic segments from KO mice more on an absolute scale (mN of force, see Figure 4C), then the enhanced force generated in the segments from KO mice is likely due to greater activation of rho kinase. When taken together, there are multiple lines of evidence demonstrating enhanced rho kinase activation in aortic rings from KO mice.
If, as our data suggests, K2P6.1 channels regulate membrane potential in VSM, then the depolarized state of VSM may account for the increased rho kinase activity, the increased peripheral vascular resistance, and hypertension observed in the KO mice (Figure 6). Given that K2P6.1 is expressed throughout the vascular system including resistance vessels2,7-10, it is highly likely that in addition to aorta, vascular muscle in smaller resistance arteries are also depolarized. While our data show that there are no differences in densities of L-type calcium channels, the depolarized state of the VSMC from KO mice confers an increased open state probability. At the resting membrane potential of -39 mV in VSMC of WT mice, there is very little activation of the L-type calcium channels and influx of extracellular Ca2+ through these channels is minimal. However, at a membrane potential of -22 mV in VSMC of KO mice, L-type calcium channels are more activated and do not fully inactivate (window current)20. Thus VSMC from KO mice should have an increased intracellular Ca2+ and as a consequence an enhanced contractile state. The depolarized state of VSMC from KO mice and/or the increased Ca2+ from L-type calcium channels serves to activate rho kinase and render the cell more sensitive to intracellular free Ca2+ 21-23 (Figure 6). The net results should be an enhanced contractile state of VSM, increased peripheral vascular resistance, and hypertension18.
Figure 6.
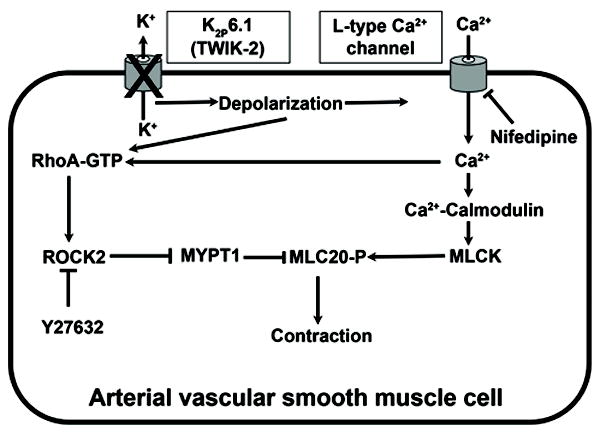
Proposed model for activation of rho kinase in VSM with K2P6.1 dysfunction. ROCK2 (rho-associated, coiled-coil containing protein kinase 2), MYPT1 (myosin phosphatase target subunit 1), MLC20-P [phosphorylated (active) state of myosin regulatory light chain 20 kD subunit], MLCK (myosin light chain kinase).
Perspectives
If our observations are translatable to the human, then mutations in the K2P6.1 gene that produce a loss of channel function and/or expression could be a genetic contributor to essential hypertension. The NIH single nucleotide polymorphism (SNP) database (dbSNP) currently lists 107 human SNP’s associated with K2P6.1. Additionally, K2P6.1 has a promoter polymorphism that affects both transcription and functional expression24. Given that K2P6.1 is highly expressed in the vascular system2,9,10,25-27, it is reasonable to speculate that a link between K2P6.1 variants and cardiovascular disease including hypertension may exist. Polymorphisms in K2P6.1 are strongly associated with severity of sickle cell anemia28. Of note, sickle cell patients presenting with severe symptoms also experience pulmonary hypertension compared to those patients characterized as having mild symptoms29. K2P6.1 is among a set of genes whose altered expression patterns appear to predict acute rejection of transplanted hearts30. Whether this rejection involves a vascular component of K2P6.1 variants is not known. Finally, K2P6.1 is included in a gene network which is under overall control of estrogen31. Although this association with estrogen may or may not have a link to cardiovascular disease, the fact that estrogen has important regulatory roles in the cardiovascular system is provocative. Given the results of our study in mice and the paucity of human studies dealing with K2P6.1 variants, it will be important to take a close look at K2P6.1variants in cardiovascular pathologies.
In summary, functional loss of K2P6.1 results in depolarization of vascular smooth muscle that may trigger a cascade of events including enhanced activation of rho kinase. K2P6.1 may exert a dilator influence on the vasculature during normal conditions. The loss of function as a result of alterations in K2P6.1 expression and/or function may contribute to the pathogeneses of cardiovascular disease.
Supplementary Material
Acknowledgments
None
Sources of Funding This research was supported by National Institutes of Health Grants RO1 NS46666 (RMB), P01 NS038660 (RMB), R21 HL098921 (RMB), RO1 HL088435 (SPM).
Footnotes
Conflict(s) of interest/Disclosure(s) Statement None
References
- 1.Hille B. Ionic Channels of Excitable Membranes. 3. Sunderland, Mass: 2001. [Google Scholar]
- 2.Bryan RM, Jr, Joseph BK, Lloyd E, Rusch NJ. Starring TREK-1: the next generation of vascular K+ channels. Circ Res. 2007;101:119–121. doi: 10.1161/CIRCRESAHA.107.157412. [DOI] [PubMed] [Google Scholar]
- 3.Goldstein SA, Bayliss DA, Kim D, Lesage F, Plant LD, Rajan S. International Union of Pharmacology. LV. Nomenclature and molecular relationships of two-P potassium channels. Pharmacol Rev. 2005;57:527–540. doi: 10.1124/pr.57.4.12. [DOI] [PubMed] [Google Scholar]
- 4.Chavez RA, Gray AT, Zhao BB, Kindler CH, Mazurek MJ, Mehta Y, Forsayeth JR, Yost CS. TWIK-2, a new weak inward rectifying member of the tandem pore domain potassium channel family. J Biol Chem. 1999;274:7887–7892. doi: 10.1074/jbc.274.12.7887. [DOI] [PubMed] [Google Scholar]
- 5.Patel AJ, Maingret F, Magnone V, Fosset M, Lazdunski M, Honore E. TWIK-2, an inactivating 2P domain K+ channel. J Biol Chem. 2000;275:28722–28730. doi: 10.1074/jbc.M003755200. [DOI] [PubMed] [Google Scholar]
- 6.Salinas M, Reyes R, Lesage F, Fosset M, Heurteaux C, Romey G, Lazdunski M. Cloning of a new mouse two-P domain channel subunit and a human homologue with a unique pore structure. J Biol Chem. 1999;274:11751–11760. doi: 10.1074/jbc.274.17.11751. [DOI] [PubMed] [Google Scholar]
- 7.Blondeau N, Petrault O, Manta S, Giordanengo V, Gounon P, Bordet R, Lazdunski M, Heurteaux C. Polyunsaturated fatty acids are cerebral vasodilators via the TREK-1 potassium channel. Circ Res. 2007;101:176–184. doi: 10.1161/CIRCRESAHA.107.154443. [DOI] [PubMed] [Google Scholar]
- 8.Bryan RM, Jr, You J, Phillips SC, Andresen JJ, Lloyd EE, Rogers PA, Dryer SE, Marrelli SP. Evidence for two-pore domain potassium channels in rat cerebral arteries. Am J Physiol Heart Circ Physiol. 2006;291:H770–H780. doi: 10.1152/ajpheart.01377.2005. [DOI] [PubMed] [Google Scholar]
- 9.Gurney AM, Osipenko ON, MacMillan D, McFarlane KM, Tate RJ, Kempsill FE. Two-pore domain K channel, TASK-1, in pulmonary artery smooth muscle cells. Circ Res. 2003;93:957–964. doi: 10.1161/01.RES.0000099883.68414.61. [DOI] [PubMed] [Google Scholar]
- 10.Olschewski A, Li Y, Tang B, Hanze J, Eul B, Bohle RM, Wilhelm J, Morty RE, Brau ME, Weir EK, Kwapiszewska G, Klepetko W, Seeger W, Olschewski H. Impact of TASK-1 in human pulmonary artery smooth muscle cells. Circ Res. 2006;98:1072–1080. doi: 10.1161/01.RES.0000219677.12988.e9. [DOI] [PubMed] [Google Scholar]
- 11.Potier M, Joulin V, Roger S, Besson P, Jourdan ML, Leguennec JY, Bougnoux P, Vandier C. Identification of SK3 channel as a new mediator of breast cancer cell migration. Mol Cancer Ther. 2006;5:2946–2953. doi: 10.1158/1535-7163.MCT-06-0194. [DOI] [PubMed] [Google Scholar]
- 12.Neylon CB. Potassium channels and vascular proliferation. Vascul Pharmacol. 2002;38:35–41. doi: 10.1016/s1537-1891(02)00124-6. [DOI] [PubMed] [Google Scholar]
- 13.Hoffmann EK, Lambert IH, Pedersen SF. Physiology of cell volume regulation in vertebrates. Physiol Rev. 2009;89:193–277. doi: 10.1152/physrev.00037.2007. [DOI] [PubMed] [Google Scholar]
- 14.Remillard CV, Yuan JX. Activation of K+ channels: an essential pathway in programmed cell death. Am J Physiol Lung Cell Mol Physiol. 2004;286:L49–L67. doi: 10.1152/ajplung.00041.2003. [DOI] [PubMed] [Google Scholar]
- 15.Ishizaki T, Uehata M, Tamechika I, Keel J, Nonomura K, Maekawa M, Narumiya S. Pharmacological properties of Y-27632, a specific inhibitor of rho-associated kinases. Mol Pharmacol. 2000;57:976–983. [PubMed] [Google Scholar]
- 16.Liu X, Rusch NJ, Striessnig J, Sarna SK. Down-regulation of L-type calcium channels in inflamed circular smooth muscle cells of the canine colon. Gastroenterology. 2001;120:480–489. doi: 10.1053/gast.2001.21167. [DOI] [PubMed] [Google Scholar]
- 17.Lloyd EE, Marrelli SP, Namiranian K, Bryan RM., Jr Characterization of TWIK-2, a two-pore domain K+ channel, cloned from the rat middle cerebral artery. Exp Biol Med (Maywood) 2009;234:1493–1502. doi: 10.3181/0903-RM-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carretero OA, Oparil S. Essential hypertension. Part I: definition and etiology. Circulation. 2000;101:329–335. doi: 10.1161/01.cir.101.3.329. [DOI] [PubMed] [Google Scholar]
- 19.Pountney DJ, Gulkarov I, Vega-Saenz de Miera E, Holmes D, Saganich M, Rudy B, Artman M, Coetzee WA. Identification and cloning of TWIK-originated similarity sequence (TOSS): a novel human 2-pore K+ channel principal subunit. FEBS Lett. 1999;450:191–196. doi: 10.1016/s0014-5793(99)00495-0. [DOI] [PubMed] [Google Scholar]
- 20.Cheng X, Pachuau J, Blaskova E, Asuncion-Chin M, Liu J, Dopico AM, Jaggar JH. Alternative splicing of Cav1.2 channel exons in smooth muscle cells of resistance-size arteries generates currents with unique electrophysiological properties. Am J Physiol Heart Circ Physiol. 2009;297:H680–H688. doi: 10.1152/ajpheart.00109.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Urban NH, Berg KM, Ratz PH. K+ depolarization induces RhoA kinase translocation to caveolae and Ca2+ sensitization of arterial muscle. Am J Physiol Cell Physiol. 2003;285:C1377–C1385. doi: 10.1152/ajpcell.00501.2002. [DOI] [PubMed] [Google Scholar]
- 22.Sakurada S, Takuwa N, Sugimoto N, Wang Y, Seto M, Sasaki Y, Takuwa Y. Ca2+-dependent activation of Rho and Rho kinase in membrane depolarization-induced and receptor stimulation-induced vascular smooth muscle contraction. Circ Res. 2003;93:548–556. doi: 10.1161/01.RES.0000090998.08629.60. [DOI] [PubMed] [Google Scholar]
- 23.Fernandez-Tenorio M, Porras-Gonzalez C, Castellano A, Del Valle-Rodriguez A, Lopez-Barneo J, Urena J. Metabotropic Regulation of RhoA/Rho-Associated Kinase by L-type Ca2+ Channels: New Mechanism for Depolarization-Evoked Mammalian Arterial Contraction. Circ Res. 108:1348–1357. doi: 10.1161/CIRCRESAHA.111.240127. [DOI] [PubMed] [Google Scholar]
- 24.Buckland PR, Hoogendoorn B, Coleman SL, Guy CA, Smith SK, O’Donovan MC. Strong bias in the location of functional promoter polymorphisms. Hum Mutat. 2005;26:214–223. doi: 10.1002/humu.20207. [DOI] [PubMed] [Google Scholar]
- 25.Garry A, Fromy B, Blondeau N, Henrion D, Brau F, Gounon P, Guy N, Heurteaux C, Lazdunski M, Saumet JL. Altered acetylcholine, bradykinin and cutaneous pressure-induced vasodilation in mice lacking the TREK1 potassium channel: the endothelial link. EMBO Rep. 2007;8:354–359. doi: 10.1038/sj.embor.7400916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gurney A, Manoury B. Two-pore potassium channels in the cardiovascular system. Eur Biophys J. 2009;38:305–318. doi: 10.1007/s00249-008-0326-8. [DOI] [PubMed] [Google Scholar]
- 27.Gonczi M, Szentandrassy N, Johnson IT, Heagerty AM, Weston AH. Investigation of the role of TASK-2 channels in rat pulmonary arteries; pharmacological and functional studies following RNA interference procedures. Br J Pharmacol. 2006;147:496–505. doi: 10.1038/sj.bjp.0706649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sebastiani P, Solovieff N, Hartley SW, Milton JN, Riva A, Dworkis DA, Melista E, Klings ES, Garrett ME, Telen MJ, Ashley-Koch A, Baldwin CT, Steinberg MH. Genetic modifiers of the severity of sickle cell anemia identified through a genome-wide association study. Am J Hematol. 85:29–35. doi: 10.1002/ajh.21572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vichinsky EP. Pulmonary hypertension in sickle cell disease. N Engl J Med. 2004;350:857–859. doi: 10.1056/NEJMp038250. [DOI] [PubMed] [Google Scholar]
- 30.Morgun A, Shulzhenko N, Perez-Diez A, Diniz RV, Sanson GF, Almeida DR, Matzinger P, Gerbase-DeLima M. Molecular profiling improves diagnoses of rejection and infection in transplanted organs. Circ Res. 2006;98:e74–e83. doi: 10.1161/01.res.0000228714.15691.8a. [DOI] [PubMed] [Google Scholar]
- 31.Cicatiello L, Mutarelli M, Grober OM, Paris O, Ferraro L, Ravo M, Tarallo R, Luo S, Schroth GP, Seifert M, Zinser C, Chiusano ML, Traini A, De Bortoli M, Weisz A. Estrogen receptor alpha controls a gene network in luminal-like breast cancer cells comprising multiple transcription factors and microRNAs. Am J Pathol. 176:2113–2130. doi: 10.2353/ajpath.2010.090837. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.