

Abstract
Reproduction is influenced by energy balance, but the physiological pathways mediating their relationship have not been fully elucidated. As the central regulators of fertility, gonadotropin-releasing hormone (GnRH) neurons integrate numerous physiological signals, including metabolic cues. Circulating glucose levels regulate GnRH release and may in part mediate the effects of negative energy balance on fertility. Existing evidence suggests that neural pathways originating in the hindbrain, as well as in the hypothalamic feeding nuclei, transmit information concerning glucose availability to GnRH neurons. Here we review recent evidence suggesting that GnRH neurons may directly sense changes in glucose availability by a mechanism involving adenosine monophosphate-activated protein kinase (AMPK). These findings expand our understanding of how metabolic signaling in the brain regulates reproduction.
Metabolic cues regulate reproductive function
Fertility is closely coupled to nutrition. The reproductive system must sense changes in bodily energy status to prevent reproduction during times of food scarcity, and to take advantage during times of plenty. Metabolic regulation of fertility is particularly important in females, in whom gestation and lactation have an exceptional energetic cost. More than three decades ago, based on observations in menarcheal adolescents and female athletes, a critical body composition hypothesis was proposed, positing that females must surpass a threshold level of adiposity to attain puberty and remain fertile1,2. The discovery of the adipocyte-derived hormone leptin seemed to substantiate this hypothesis, as leptin is permissive for fertility3. However, more recent observations indicate that in addition to being susceptible to metabolic signals reflecting long-term nutritional stores, the reproductive system monitors energy availability on a minute-to-minute basis by sensing levels of circulating nutrients, including glucose and fatty acids4,5.
GnRH neurons comprise the final common pathway by which the brain controls reproduction. These neurons secrete GnRH in discrete pulses that elicit corresponding pulses of luteinizing hormone (LH) and follicle-stimulating hormone (FSH) release from the pituitary. Signals that indicate fuel availability, and particularly deficiency, can be sensed centrally and transmitted to GnRH neurons to modulate their activity and thereby GnRH release. Metabolic control of GnRH pulsatility has been demonstrated primarily by measuring LH pulses as a surrogate for GnRH release6. These studies have found that food deprivation suppresses pulsatile LH secretion in rats7–9, sheep10,11, monkeys12, and humans13, consistent with inhibition of pulsatile GnRH release. In mice and hamsters, in which assessment of LH pulses is extremely difficult due to the small blood volume of these species, fasting suppresses estrous cyclicity14–16 and LH levels17. Similar findings were obtained from multiunit activity recordings of coordinated electrical discharges in the mediobasal hypothalamus; these discharges, which are associated with LH pulses and considered an electrophysiological correlate of GnRH pulses, are reduced in frequency by fasting in monkeys18 and goats19,20.
Glucose: a critical link between metabolism and reproduction
Numerous studies support the idea that glucose in particular mediates the effects of fasting to suppress GnRH-stimulated LH release. Reducing central glucose availability via intracerebroventricular (ICV) infusion of insulin or glucose antimetabolites (2-deoxyglucose, 2-DG, or 5-thioglucose, 5-TG) suppresses LH levels and pulse frequency in rats21,22, goats23, monkeys24, and sheep25. Additionally, 2-DG infusion increases the interval between bursts of multiunit activity in the mediobasal hypothalamus of goats, suggesting a slowing of GnRH pulse frequency23. Administration of glucose restored LH pulsatility in insulin-induced hypoglycemic rats26,27 and sheep28, suggesting that low glucose rather than high insulin mediates the suppression of LH. In addition to the negative effect of reduced glucose, increased glucose may positively influence GnRH/LH secretion. Goats provided with dietary supplementation exhibited parallel increases in serum glucose levels and LH pulse frequency, whereas pulses declined as food availability, and thus glucose, was reduced29. These studies provide a strong case that glucose can act as a signaling molecule in the brain to both positively and negatively modulate reproductive function.
Many of the aforementioned studies employed insulin to induce hypoglycemia, suggesting that insulin may participate in concert with glucose to regulate GnRH neuronal function. In reproductively normal women, insulin administration increases the LH pulse frequency, consistent with a stimulatory effect of insulin on GnRH pulsatility30; this finding argues against a potential effect of insulin to reduce GnRH pulse frequency during insulin-induced hypoglycemia. A recent study demonstrated that GnRH neuron-specific deletion of the insulin receptor had no effect on fertility31. In contrast, neuronal insulin receptor knockout mice exhibit subfertility attributable to reduced central stimulation of LH secretion32. Together, these studies suggest that insulin signaling in presynaptic neurons may be important for normal GnRH neuronal function. Here we will focus on the central reproductive effects of glucose, which have been characterized in greater detail.
Where and how glucose is detected in the brain, and how this information is conveyed to GnRH neurons, remain important questions with significant implications in the modern environment of overnutrition. Substantial evidence points to a system of hindbrain fuel detectors in the area postrema33, which, via intermediate signals that may include opioids24,34,35, catecholamines36, corticotropin-releasing hormone37,38, and gamma-aminobutryic acid (GABA)35,39, transmit information about metabolic status to GnRH neurons in the forebrain (reviewed in 4,5). Other potential sites for the relay of metabolic signals are the nutrient-sensing neurons of the arcuate, ventromedial, and lateral hypothalamus, some of which are known to project to and alter the function of GnRH neurons via neuropeptidergic signaling40. Lastly, several new studies suggest that GnRH neurons may directly sense information about glucose availability41,42. Here we review these studies, as well as evidence implicating AMPK in nutrient-sensing and metabolic control of GnRH neurons.
Neuronal glucosensing
The high metabolic activity of the brain requires a constant supply of glucose. Glucose enters the brain via facilitated transport through glucose transporters in the blood brain barrier, reaching an extracellular concentration of approximately 10–30 percent of the blood level. Specialized neurons in the brain translate changes in the ambient glucose concentration into changes in membrane potential and firing rate; this ability is distinct from the near ubiquitous neuronal inhibition that occurs in response to aglycemia, and is confined to specific neuronal subpopulations43. Such glucosensing neurons are subdivided into two classes: glucose-inhibited (GI) neurons, which hyperpolarize when the extracellular glucose concentration is increased, and glucose-excited (GE) neurons, which depolarize when glucose is increased. These neurons have been well characterized in the arcuate, ventromedial, and lateral hypothalamic nuclei, where they are involved in the control of glucoregulatory responses and potentially the regulation of feeding behavior44,45. Some of the most well-studied glucosensing neurons express neuropeptides in addition to classical neurotransmitters; for example, proopiomelanocortin (POMC) neurons are GE46, and orexin and neuropeptide Y (NPY)/agouti-related peptide (AgRP) neurons are GI47,48. Of interest, GnRH neurons contain receptors for and have been shown to be responsive to these neuropeptides40, suggesting that the glucosensing network responsible for maintaining peripheral metabolic homeostasis may also relay metabolic information to the reproductive system.
Mechanisms of neuronal glucosensing
It is widely held that the primary mechanism of glucosensing by GE neurons mirrors that of the pancreatic beta cell, in which ATP-sensitive potassium (KATP) channels and glucokinase play an integral role49. The KATP channel is comprised of four Kir6.2 pore-forming subunits and four sulfonylurea (SUR1) subunits; the latter confer sensitivity to sulfonylureas, such as tolbutamide, which close KATP channels. Extracellular glucose is transported into cells and serves as a substrate for ATP generation by glycolysis. The rate-limiting step in glycolysis is hexokinase-mediated conversion of glucose to glucose-6-phosphate; beta cells and some neurons express an isoform of this enzyme called glucokinase, which has a high Km and thus is not saturated at physiological glucose levels. ATP blocks membrane KATP channels, resulting in decreased potassium efflux and membrane depolarization. In beta cells, depolarization leads to insulin release; in neurons, it can increase the action potential firing rate. As greater glucose influx leads to increased ATP generation and KATP closure-mediated depolarization, neuronal firing rate is relatively proportional to the extracellular glucose concentration. Numerous studies support this model of neuronal glucosensing, although it is not without debate50. Reducing the extracellular glucose concentration around ventromedial hypothalamic neurons activates channels exhibiting properties consistent with KATP channels. Mice with deletion of the pore-forming subunit of the KATP channel lack functional glucose-excited neurons in the ventromedial hypothalamus and have impaired counterregulatory and feeding responses to glucoprivation51. Further, expression of a mutant, ATP-insensitive Kir6.2 subunit in POMC neurons abolished glucosensing in this cell type. With regard to a role for glucokinase, pharmacological inhibition of this kinase inhibits GE and stimulates GI neurons; conversely, its activation stimulates GE and inhibits GI neurons52,53. In addition to roles in glucosensing, KATP channels are involved in neuronal leptin and insulin signaling. Both leptin and insulin activate KATP channels in a phosphoinositide-3-kinase-dependent manner54. KATP channels are thus thought to perform diverse functions in transmitting metabolic information from the periphery.
Among other putative mechanisms of glucosensing is the adenosine monophosphate-activated protein kinase (AMPK), a cellular energy sensor that, like KATP channels, is postulated to play an integrative role in central metabolic signaling55. AMPK is activated allosterically by 5'AMP and via phosphorylation by AMPK kinase (AMPKK). AMP binding to AMPK makes it a better substrate for AMPKK, and a worse substrate for inactivating phosphatases; ATP antagonizes all effects of AMP. Thus, AMPK activation is determined by the intracellular ratio of AMP to ATP. AMPK consists of a catalytic α subunit, of which two isoforms have been identified, and regulatory beta and gamma subunits. In the central nervous system, AMPK α2 is expressed at higher levels than α1. AMPK subunit expression is mainly neuronal, although it has also been identified in astrocytes. In contrast to KATP channels, which are widely expressed, AMPK localization is restricted to distinct brain subregions55. In addition to regulation by intracellularly-generated adenine nucleotides, AMPK is amenable to regulation by peripheral hormones. Leptin inhibits, whereas ghrelin and adiponectin enhance, AMPK activation in the brain56. These findings have let to a surge of interest in AMPK as a regulator of energy homeostasis at both the cellular and whole-body levels.
GI neurons of the ventromedial hypothalamus sense glucose at least in part via activation of AMPK57. Low glucose activates AMPK, which phosphorylates and activates cystic fibrosis transmembrane regulator chloride channels, hyperpolarizing and inhibiting these cells. This process is amplified by AMPK-dependent generation of nitric oxide (NO). NO activates soluble guanylyl cyclase, increasing intracellular cyclic GMP and activating AMPK through some yet undetermined upstream kinase57. This mechanism differs from that of GI orexin neurons, which utilize a glucose-activated tandem-pore potassium current (distinct from the KATP current)47. A subset of orexin neurons exhibit time-dependent closure of these background channels, which may be an adaptive mechanism to maintain responsiveness to glucose changes over a wide range of baseline concentrations58. In addition to reported roles in GI neurons, one study has demonstrated a contribution of AMPK to glucosensing by GE neurons59. Genetic deletion of the AMPK α2 subunit from AgRP and POMC neurons completely abolished glucosensing in these cell types. Interestingly, AMPK α1 knockout mice are subfertile, but exhibit no apparent metabolic phenotype, indicating that subfertility in these mice is not secondary to metabolic dysregulation60. AMPK thus appears to play a role in both GE and GI neurons and has been independently linked with fertility.
Additional metabolism-independent mechanisms for glucosensing have been proposed, in light of the observation that, in some cells, responses to glucose can be dissociated from its metabolism to ATP50. One such mechanism is electrogenic glucose entry, whereby glucose transport into cells is coupled to ion movement. An example is the sodium-glucose cotransporter (SGLT). SGLTs concurrently transport sodium and glucose into cells, generating depolarizing inward currents. An exception is SGLT3, which generates inward current without concomitant glucose transport, acting as a “glucose receptor” on the cell surface61. However, this receptor has not yet been identified in glucosensing neurons. Glial cells have also been implicated in glucosensing. A rise in extracellular glucose increases glycolysis in astrocytes, which release lactate that can excite GE neurons via closure of KATP channels48. Further, a recent study has suggested a role for tanycytes, glial-like cells that line the cerebral ventricles and communicate with parenchymal cells, in glucosensing by hypothalamic neurons62. Glucose application to third ventricular tanycytes in a slice preparation evokes ATP release and the generation of ATP-dependent calcium waves that travel along the tanycyte processes. Due to their proximity and extension of processes to the arcuate and ventromedial nuclei, these tanycytes are poised to mediate the responses of glucosensing neurons. Interestingly, there is evidence for glial, and in particular tanycyte, modulation of GnRH neuronal function63.
Glucosensing by GnRH neurons
Zhang et al. first demonstrated glucosensing in GnRH neurons41. Using extracellular recordings from GnRH neurons in brain slices exposed to high potassium to stimulate firing activity and high Ca2+/low Mg2+ solution to minimize presynaptic neurotransmitter release, over half of cells (5/9) demonstrated a reduction in firing frequency in response to a switch from 2.5 to 1mM glucose. This study also independently demonstrated Kir6.2 mRNA in 60% and glucokinase mRNA in 30% of GnRH neurons by single-cell quantitative PCR. In vivo treatment with estradiol potentiated the KATP current, demonstrated by application of the KATP antagonist tolbutamide and the agonist diazoxide. The demonstration of glucosensing, the cellular machinery for KATP-dependent glucosensing, and an intrinsic KATP current strongly suggest that glucosensing in GnRH neurons involves a similar mechanism as that reported for neurons of the arcuate and ventromedial hypothalamus, although this link was not directly established. This first evidence for direct sensing of glucose by GnRH neurons contributed a novel component to the model of fuel detection by the reproductive system.
A subsequent study by Huang et al. also addressed the role of KATP channels in the regulation of GnRH/LH secretion, particularly with regard to their role in mediating suppression of LH by fasting17. As predicted, LH was suppressed by a 48-hour fast. The KATP channel antagonist tolbutamide was administered intracerebroventricularly to either fed or fasted mice and evoked a 2-fold increase in LH in both groups. If KATP channel opening was the cause of suppressed LH, one would predict a greater LH increase in response to tolbutamide in fasted mice; thus, KATP channels did not appear to contribute to the response to fasting. The authors further corroborated this finding by demonstrating that there was no attenuation of fasting-induced LH suppression in sulfonylurea-1 null mice, which lack functional KATP channels. While supporting a role for KATP channels in GnRH neuronal regulation, these findings suggested KATP activation is not the mechanism by which fasting suppresses GnRH/LH secretion.
A recent report confirmed and extended the findings of Zhang and demonstrated that the majority of GnRH neurons (~70–80%) can be inhibited by reducing the extracellular glucose concentration42 (Figure 1A,C). This response persisted in the presence of blockers of receptors for fast GABAergic and glutamatergic neurotransmission, suggesting, as in the Zhang study, that glucosensing is intrinsic to the GnRH neuron. This study also attempted to directly link glucosensing and KATP channels by testing the response of GnRH neurons to low glucose in the presence of tolbutamide. Although acute application of tolbutamide evoked firing in a subset of GnRH neurons, confirming the presence of functional KATP channels, tolbutamide unexpectedly failed to attenuate the response to low glucose. This finding suggested that KATP channels are not the primary mediator of glucosensing in GnRH neurons. Moreover, the effects of different steroids (administered in vivo) on glucosensing were tested; estradiol did not appear to affect glucosensing properties, which would have been predicted by the previously reported estradiol enhancement of KATP current41. In contrast, the non-aromatizable androgen dihydrotestosterone (DHT) attenuated glucosensing; this finding may have implications for sex differences in responses to metabolic cues, or in the context of hyperandrogenemic infertility in females. While KATP channels may not be required for glucose responsiveness of GnRH neurons, they have been shown to have important functions beyond metabolic signaling, including a role in the mediation of steroid negative feedback64. What function glucokinase might serve in GnRH glucosensing remains to be fully explored; 30% of GnRH neurons express glucokinase mRNA, but this is less than the percentage that exhibited glucosensing42. Glucokinase-dependent glucosensing may act as a secondary mechanism in a subset of cells, as many of the signals controlling reproduction show physiological redundancy. Future studies could examine the role of glucokinase in GnRH neuronal function by employing glucokinase-modulating agents, or mice with targeted deletion of glucokinase from GnRH neurons.
Figure 1.
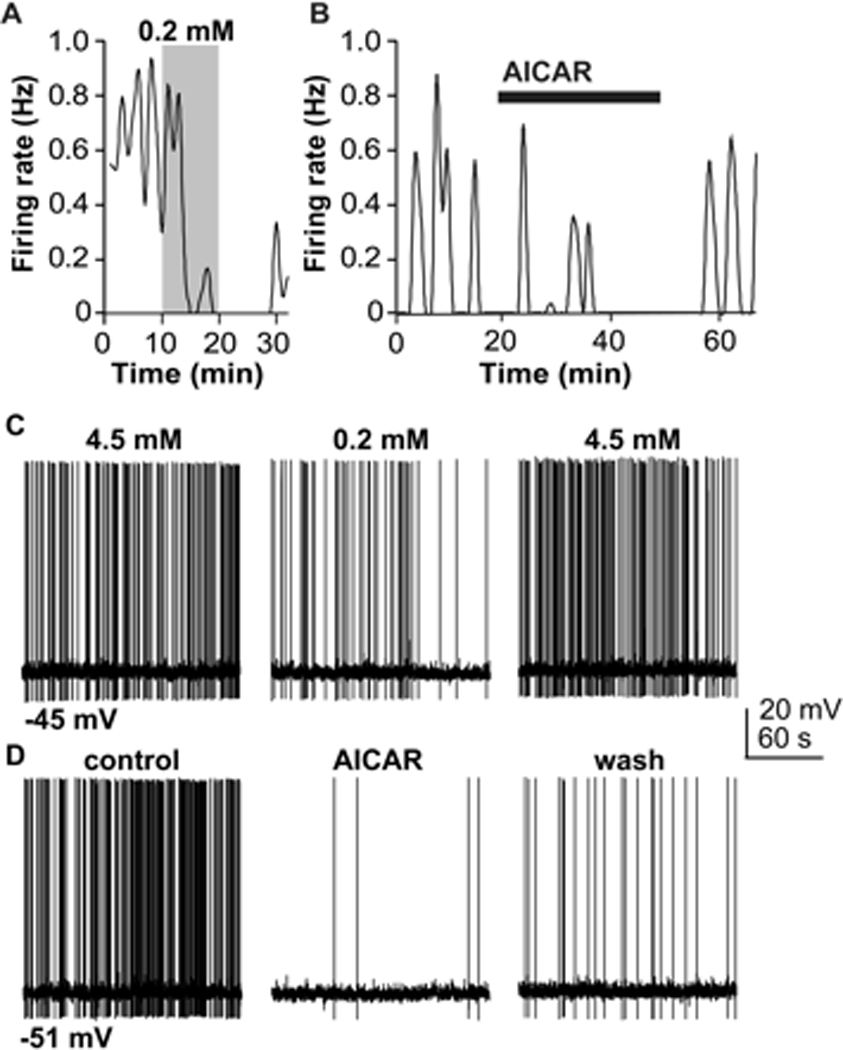
GnRH neurons are inhibited by both low glucose and AICAR. (a). Plot of firing rate (binned in 60-second intervals) of a GnRH neuron from an ovariectomized mouse in response to a reduction from 4.5 mM to 0.2 mM glucose. Shaded region indicates time of low glucose exposure. Low glucose slowly inhibits the firing rate; this effect is not immediately reversed upon restoration of 4.5 mM glucose. The decrease at the return to high glucose in this example was typical of many GnRH neurons; this observation was found to be a coincidental aspect of the temporal nature of the response to low glucose, rather than a further suppression by restoration to normal glucose. (b). AICAR, which activates AMPK, inhibits GnRH neuron firing in 4.5 mM glucose. (c, d). Whole-cell, current-clamp recordings demonstrating low glucose (c) and AICAR (d) suppression of GnRH neuron action potential firing rate. mV, millivolts. Adapted from reference 43 with permission.
AMPK: a novel regulator of GnRH neuronal function
Whereas blockade of KATP channels failed to alter glucosensing, pharmacological antagonism of AMPK using compound C attenuated glucosensing in GnRH neurons42. Acute application of the AMPK agonist AICAR inhibited GnRH neurons (Figure 1B, D), supporting the idea that inhibition by low glucose was due to AMPK-mediated inhibition. Metformin, an antihyperglycemic agent that activates AMPK through a mechanism distinct from that of AICAR, similarly inhibited GnRH neuron firing activity. GnRH neurons from DHT-treated mice were less sensitive to inhibition by AICAR, consistent with their attenuated response to low glucose. Mechanistically, AMPK and low glucose activated a similar current with a reversal potential of −50 mV; this current was unaffected by blockade of action-potential dependent synaptic transmission with tetrodotoxin, providing further evidence that glucosensing is intrinsic to the GnRH neuron. The possibility that this current was carried by chloride was ruled out, suggesting, rather, that it was a mixed cation current.
Several other recent studies have identified a role for AMPK in GnRH neuronal regulation. Three studies demonstrated the expression of AMPK in GT1–7 immortalized GnRH neurons 65–67. AICAR, metformin, and globular adiponectin were shown individually to inhibit GnRH release from these cells by an AMPK-dependent mechanism65–67. In GT1–7 cells, AMPK activation inhibited the hyperpolarization-activated cation current. The discrepancy between this finding, which involves inhibition of a cation current, and that mentioned above, which suggested activation of a cation current, may be related to the cell type; cultured immortalized GT1–7 cells may function differently than GnRH neurons recorded in acutely prepared brain slices. Two recent studies examined the effects of in vivo treatment with AMPK activators. The first examined estrous cyclicity and food intake following ICV infusion of AICAR65. AICAR caused AMPK phosphorylation in the hypothalamus as expected, and transiently increased food intake, consistent with reports that hypothalamic AMPK stimulates feeding68. Another study demonstrated effects of in vivo treatment with metformin on the firing activity of GnRH neurons from control and prenatally androgenized female mice, the latter of which have increased baseline GnRH neuron activity69. While metformin had no effect on the firing activity of GnRH neurons from control mice, it suppressed firing activity in the prenatally androgenized mice, restoring it to control levels. Further, GnRH neurons from metformin-treated (androgenized or control), but not untreated, mice were excited by the AMPK antagonist compound C in vitro, suggesting AMPK was activated in cells from these mice. Low glucose failed to inhibit firing in the majority of cells from metformin-treated mice. Together, these findings suggested that metformin administered in vivo activated AMPK in GnRH neurons, leading to a suppression of activity in mice in which GnRH neurons were excessively active. The above studies provide further support that AMPK activation has an inhibitory effect on GnRH neurons.
Concluding remarks
Several questions remain unanswered with regard to AMPK regulation of GnRH neuronal activity. Importantly, although AMPK expression was demonstrated in the GT1–7 cell line, it has not been assessed in GnRH neurons from adult mice. A limitation of the studies done in mice is that manipulations were solely pharmacological, and nonspecific actions of AICAR, metformin, and compound C are possible, although the observation that all three had experimentally consistent effects would argue against such non-specificity. The above studies imply that presynaptic neuronal activity is not required for the response of GnRH neurons to low glucose, and thus AMPK is likely expressed in GnRH neurons. However, it is also possible that astrocytes are involved in the GnRH neuronal response to low glucose. AMPK expression has been demonstrated in astrocytes70, and astrocytes may transmit metabolic information to alter neuronal function. Thus, identifying the site of AMPK localization is essential. Another remaining question is the identity of the specific subtype of mixed cation current activated by AICAR and low glucose, as well as the signaling pathways that lead to its activation.
A role for AMPK in glucosensing by GnRH neurons could be confirmed using mice deficient in various AMPK subunits. Further, in order to fully integrate direct glucosensing by GnRH neurons into a model of fuel-sensing, we must establish the physiological relevance of this phenomenon. Presumably, the sensitivity of GnRH neurons to low glucose contributes to the suppression of GnRH pulsatility by central or peripheral glucoprivation, but these hypotheses must be tested empirically through pharmacological or genetic manipulation of AMPK together with fasting, insulin, or glucose anti-metabolites administered to the whole animal. Drawing once again from the energy homeostasis field, 2-deoxyglucose administration and insulin-induced hypoglycemia have been shown to activate central AMPK, and compound C inhibits glucoprivic feeding68. These findings suggest that the same stimuli that have previously been shown to suppress LH (and therefore GnRH) pulses also activate central AMPK, consistent with the proposed working model. Lastly, as AMPK is known to be the target of ghrelin, adiponectin, and other peripheral metabolic hormones56, the effects of these cues on GnRH neurons should be examined, as well as the AMPK-dependence of their actions.
In summary, the worked described here suggests that direct glucosensing may contribute to the response of the reproductive system to fluctuations in nutrient availability. Combined with previous work implicating distal brain regions in the metabolic control of GnRH neurons, these findings suggest a model (Figure 2) in which direct glucosensing functions together with afferent signals to ensure that reproduction occurs only in the presence of an adequate nutrient supply.
Figure 2.
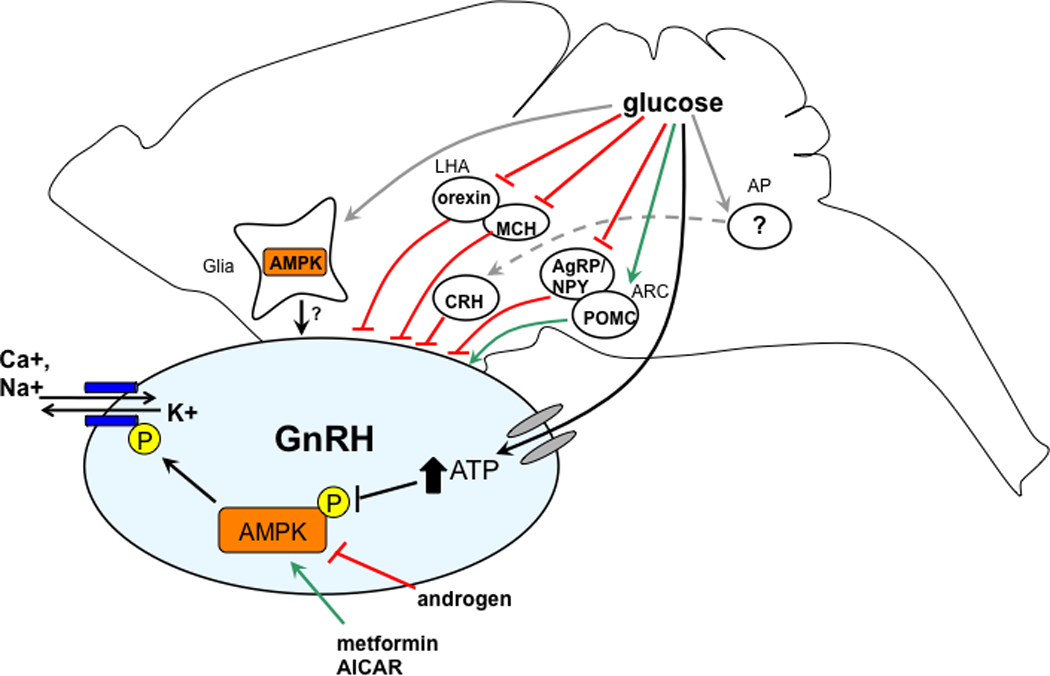
Putative sites of glucose action in the brain to regulate GnRH neuronal function. Glucose may act on distal brain regions in the hindbrain area postrema (AP), and in the arcuate (ARC) and lateral hypothalamus (LHA), which project (both directly and indirectly) to GnRH neurons. In addition, glucose may alter the function of GnRH neurons through direct action mediated by AMPK, or through effects on proximal glial cells.
Acknowledgements
We thank Beth Wagenmaker and Laura Berger for their helpful editorial comments.
Supported by National Institute of Health/Eunice Kennedy Shriver National Institute of Child Health and Human Development U54 HD28934 and F31 NS62646.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Frisch RE. The right weight: body fat, menarche and ovulation. Baillieres Clin Obstet Gynaecol. 1990;4:419–439. doi: 10.1016/s0950-3552(05)80302-5. [DOI] [PubMed] [Google Scholar]
- 2.Frisch RE, McArthur JW. Menstrual cycles: fatness as a determinant of minimum weight for height necessary for their maintenance or onset. Science. 1974;185:949–951. doi: 10.1126/science.185.4155.949. [DOI] [PubMed] [Google Scholar]
- 3.Barash IA, et al. Leptin is a metabolic signal to the reproductive system. Endocrinology. 1996;137:3144–3147. doi: 10.1210/endo.137.7.8770941. [DOI] [PubMed] [Google Scholar]
- 4.Wade GN, Schneider JE, Li HY. Control of fertility by metabolic cues. American Journal of Physiology. 1996;270:E1–E19. doi: 10.1152/ajpendo.1996.270.1.E1. [DOI] [PubMed] [Google Scholar]
- 5.Wade GN, Jones JE. Neuroendocrinology of nutritional infertility. American journal of physiology. Regulatory, integrative and comparative physiology. 2004;287:R1277–R1296. doi: 10.1152/ajpregu.00475.2004. [DOI] [PubMed] [Google Scholar]
- 6.Wildt L, et al. Frequency and amplitude of gonadotropin-releasing hormone stimulation and gonadotropin secretion in the rhesus monkey. Endocrinology. 1981;109:376–385. doi: 10.1210/endo-109-2-376. [DOI] [PubMed] [Google Scholar]
- 7.Badger TM, Lynch EA, Fox PH. Effects of fasting on luteinizing hormone dynamics in the male rat. J Nutr. 1985;115:788–797. doi: 10.1093/jn/115.6.788. [DOI] [PubMed] [Google Scholar]
- 8.Cagampang FR, Maeda K, Yokoyama A, Ota K. Effect of food deprivation on the pulsatile LH release in the cycling and ovariectomized female rat. Horm Metab Res. 1990;22:269–272. doi: 10.1055/s-2007-1004900. [DOI] [PubMed] [Google Scholar]
- 9.Nagatani S, et al. Reduction of glucose availability suppresses pulsatile luteinizing hormone release in female and male rats. Endocrinology. 1996;137:1166–1170. doi: 10.1210/endo.137.4.8625885. [DOI] [PubMed] [Google Scholar]
- 10.Foster DL, Olster DH. Effect of restricted nutrition on puberty in the lamb: patterns of tonic luteinizing hormone (LH) secretion and competency of the LH surge system. Endocrinology. 1985;116:375–381. doi: 10.1210/endo-116-1-375. [DOI] [PubMed] [Google Scholar]
- 11.Thomas GB, et al. Effect of restricted feeding on the concentrations of growth hormone (GH), gonadotropins, and prolactin (PRL) in plasma, and on the amounts of messenger ribonucleic acid for GH, gonadotropin subunits, and PRL in the pituitary glands of adult ovariectomized ewes. Endocrinology. 1990;126:1361–1367. doi: 10.1210/endo-126-3-1361. [DOI] [PubMed] [Google Scholar]
- 12.Cameron JL, Nosbisch C. Suppression of pulsatile luteinizing hormone and testosterone secretion during short term food restriction in the adult male rhesus monkey (Macaca mulatta) Endocrinology. 1991;128:1532–1540. doi: 10.1210/endo-128-3-1532. [DOI] [PubMed] [Google Scholar]
- 13.Cameron JL, Weltzin TE, McConaha C, Helmreich DL, Kaye WH. Slowing of pulsatile luteinizing hormone secretion in men after forty-eight hours of fasting. Journal of Clinical Endocrinology & Metabolism. 1991;73:35–41. doi: 10.1210/jcem-73-1-35. [DOI] [PubMed] [Google Scholar]
- 14.Sullivan SD, Howard LC, Clayton AH, Moenter SM. Serotonergic activation rescues reproductive function in fasted mice: does serotonin mediate the metabolic effects of leptin on reproduction? Biology of Reproduction. 2002;66:1702–1706. doi: 10.1095/biolreprod66.6.1702. [DOI] [PubMed] [Google Scholar]
- 15.Ahima RS, et al. Role of leptin in the neuroendocrine response to fasting. Nature. 1996;382:250–252. doi: 10.1038/382250a0. [DOI] [PubMed] [Google Scholar]
- 16.Schneider JE, Wade GN. Decreased availability of metabolic fuels induces anestrus in golden hamsters. American Journal of Physiology. 1990;258:R750–R755. doi: 10.1152/ajpregu.1990.258.3.R750. [DOI] [PubMed] [Google Scholar]
- 17.Huang W, Acosta-Martinez M, Horton TH, Levine JE. Fasting-induced suppression of LH secretion does not require activation of ATP-sensitive potassium channels. Am J Physiol Endocrinol Metab. 2008;295:E1439–E1446. doi: 10.1152/ajpendo.90615.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen MD, O'Byrne KT, Chiappini SE, Hotchkiss J, Knobil E. Hypoglycemic 'stress' and gonadotropin-releasing hormone pulse generator activity in the rhesus monkey: role of the ovary. Neuroendocrinology. 1992;56:666–673. doi: 10.1159/000126291. [DOI] [PubMed] [Google Scholar]
- 19.Ichimaru T, Mori Y, Okamura H. A possible role of neuropeptide Y as a mediator of undernutrition to the hypothalamic gonadotropin-releasing hormone pulse generator in goats. Endocrinology. 2001;142:2489–2498. doi: 10.1210/endo.142.6.8002. [DOI] [PubMed] [Google Scholar]
- 20.Matsuyama S, et al. Simultaneous observation of the GnRH pulse generator activity and plasma concentrations of metabolites and insulin during fasting and subsequent refeeding periods in Shiba goats. J Reprod Dev. 2004;50:697–704. doi: 10.1262/jrd.50.697. [DOI] [PubMed] [Google Scholar]
- 21.Howland BE. Effect of glucoprivation induced by 2-deoxy-D-glucose on serum gonadotropin levels, pituitary response to GnRH and progesterone-induced release of luteinizing hormone in rats. Horm Metab Res. 1980;12:520–523. doi: 10.1055/s-2007-999190. [DOI] [PubMed] [Google Scholar]
- 22.Murahashi K, et al. Suppression of luteinizing hormone pulses by restriction of glucose availability is mediated by sensors in the brain stem. Endocrinology. 1996;137:1171–1176. doi: 10.1210/endo.137.4.8625886. [DOI] [PubMed] [Google Scholar]
- 23.Ohkura S, Ichimaru T, Itoh F, Matsuyama S, Okamura H. Further evidence for the role of glucose as a metabolic regulator of hypothalamic gonadotropin-releasing hormone pulse generator activity in goats. Endocrinology. 2004;145:3239–3246. doi: 10.1210/en.2003-1516. [DOI] [PubMed] [Google Scholar]
- 24.Lado-Abeal J, et al. Hypoglycemia-induced suppression of luteinizing hormone (LH) secretion in intact female rhesus macaques: role of vasopressin and endogenous opioids. Stress. 2002;5:113–119. doi: 10.1080/10253890290027886. [DOI] [PubMed] [Google Scholar]
- 25.Bucholtz DC, Vidwans NM, Herbosa CG, Schillo KK, Foster DL. Metabolic interfaces between growth and reproduction. V. Pulsatile luteinizing hormone secretion is dependent on glucose availability. Endocrinology. 1996;137:601–607. doi: 10.1210/endo.137.2.8593808. [DOI] [PubMed] [Google Scholar]
- 26.Rodriguez M, Arias P, Refojo D, Feleder C, Moguilevsky J. Arrest of pulsatile luteinizing hormone (LH) secretion during insulin-induced hypoglycemia (IIH): improvement by intrahypothalamic perfusion with glucose. Exp Clin Endocrinol Diabetes. 1999;107:257–261. doi: 10.1055/s-0029-1212109. [DOI] [PubMed] [Google Scholar]
- 27.He D, et al. Effects of glucose and related substrates on the recovery of the electrical activity of gonadotropin-releasing hormone pulse generator which is decreased by insulin-induced hypoglycemia in the estrogen-primed ovariectomized rat. Brain research. 1999;820:71–76. doi: 10.1016/s0006-8993(98)01358-4. [DOI] [PubMed] [Google Scholar]
- 28.Clarke IJ, Horton RJ, Doughton BW. Investigation of the mechanism by which insulin-induced hypoglycemia decreases luteinizing hormone secretion in ovariectomized ewes. Endocrinology. 1990;127:1470–1476. doi: 10.1210/endo-127-3-1470. [DOI] [PubMed] [Google Scholar]
- 29.Zabuli J, Tanaka T, Lu W, Kuroiwa T, Kamomae H. Responses of gonadotropin secretion to short-term dietary supplementation in ovariectomized goats with different body weights. Anim Reprod Sci. 2009;116:274–281. doi: 10.1016/j.anireprosci.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 30.Moret M, et al. Insulin modulation of luteinizing hormone secretion in normal female volunteers and lean polycystic ovary syndrome patients. Neuroendocrinology. 2009;89:131–139. doi: 10.1159/000160911. [DOI] [PubMed] [Google Scholar]
- 31.Divall SA, et al. Divergent roles of growth factors in the GnRH regulation of puberty in mice. The Journal of clinical investigation. 2010;120:2900–2909. doi: 10.1172/JCI41069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bruning JC, et al. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000;289:2122–2125. doi: 10.1126/science.289.5487.2122. [DOI] [PubMed] [Google Scholar]
- 33.Cates PS, O'Byrne KT. The area postrema mediates insulin hypoglycaemia-induced suppression of pulsatile LH secretion in the female rat. Brain research. 2000;853:151–155. doi: 10.1016/s0006-8993(99)02301-x. [DOI] [PubMed] [Google Scholar]
- 34.Cagampang FR, et al. Hypoglycaemia-induced inhibition of pulsatile luteinizing hormone secretion in female rats: role of oestradiol, endogenous opioids and the adrenal medulla. Journal of neuroendocrinology. 1997;9:867–872. doi: 10.1046/j.1365-2826.1997.00653.x. [DOI] [PubMed] [Google Scholar]
- 35.Singh SR, Sylvester PW, Briski KP. Caudal hindbrain glucoprivation enhances gamma-aminobutyric acid release in discrete septopreoptic structures in the steroid-primed ovariectomized rat brain: role of mu opioid receptors. Neuroendocrinology. 2004;80:201–209. doi: 10.1159/000082544. [DOI] [PubMed] [Google Scholar]
- 36.I'Anson H, Sundling LA, Roland SM, Ritter S. Immunotoxic destruction of distinct catecholamine neuron populations disrupts the reproductive response to glucoprivation in female rats. Endocrinology. 2003;144:4325–4331. doi: 10.1210/en.2003-0258. [DOI] [PubMed] [Google Scholar]
- 37.Chen MD, et al. The insulin hypoglycemia-induced inhibition of gonadotropin-releasing hormone pulse generator activity in the rhesus monkey: roles of vasopressin and corticotropin-releasing factor. Endocrinology. 1996;137:2012–2021. doi: 10.1210/endo.137.5.8612542. [DOI] [PubMed] [Google Scholar]
- 38.Tsukahara S, Tsukamura H, Foster DL, Maeda KI. Effect of corticotropin-releasing hormone antagonist on oestrogen-dependent glucoprivic suppression of luteinizing hormone secretion in female rats. Journal of neuroendocrinology. 1999;11:101–105. doi: 10.1046/j.1365-2826.1999.00312.x. [DOI] [PubMed] [Google Scholar]
- 39.Sullivan SD, DeFazio RA, Moenter SM. Metabolic regulation of fertility through presynaptic and postsynaptic signaling to gonadotropin-releasing hormone neurons. Journal of Neuroscience. 2003;23:8578–8585. doi: 10.1523/JNEUROSCI.23-24-08578.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crown A, Clifton DK, Steiner RA. Neuropeptide signaling in the integration of metabolism and reproduction. Neuroendocrinology. 2007;86:175–182. doi: 10.1159/000109095. [DOI] [PubMed] [Google Scholar]
- 41.Zhang C, Bosch M, Leinve J, Ronnekleiv O, Kelly M. GnRH neurons express K-ATP channels that are regulated by estrogen and responsive to glucose and metabolic inhibition. Journal of Neuroscience. 2007 doi: 10.1523/JNEUROSCI.1657-07.2007. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Roland AV, Moenter SM. Glucosensing by GnRH Neurons: Inhibition by Androgens and Involvement of AMP-Activated Protein Kinase. Molecular endocrinology. 2011;25:847–858. doi: 10.1210/me.2010-0508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mobbs CV, Kow LM, Yang XJ. Brain glucose-sensing mechanisms: ubiquitous silencing by aglycemia vs. hypothalamic neuroendocrine responses. Am J Physiol Endocrinol Metab. 2001;281:E649–E654. doi: 10.1152/ajpendo.2001.281.4.E649. [DOI] [PubMed] [Google Scholar]
- 44.Levin BE. Metabolic sensing neurons and the control of energy homeostasis. Physiol Behav. 2006;89:486–489. doi: 10.1016/j.physbeh.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 45.Watts AG, Donovan CM. Sweet talk in the brain: glucosensing, neural networks, and hypoglycemic counterregulation. Front Neuroendocrinol. 2010;31:32–43. doi: 10.1016/j.yfrne.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ibrahim N, et al. Hypothalamic proopiomelanocortin neurons are glucose responsive and express K(ATP) channels. Endocrinology. 2003;144:1331–1340. doi: 10.1210/en.2002-221033. [DOI] [PubMed] [Google Scholar]
- 47.Burdakov D, et al. Tandem-pore K+ channels mediate inhibition of orexin neurons by glucose. Neuron. 2006;50:711–722. doi: 10.1016/j.neuron.2006.04.032. [DOI] [PubMed] [Google Scholar]
- 48.Belgardt BF, Okamura T, Bruning JC. Hormone and glucose signalling in POMC and AgRP neurons. The Journal of physiology. 2009;587:5305–5314. doi: 10.1113/jphysiol.2009.179192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Levin BE, Dunn-Meynell AA, Routh VH. Brain glucosensing and the K(ATP) channel. Nat Neurosci. 2001;4:459–460. doi: 10.1038/87405. [DOI] [PubMed] [Google Scholar]
- 50.Gonzalez JA, Reimann F, Burdakov D. Dissociation between sensing and metabolism of glucose in sugar sensing neurones. The Journal of physiology. 2009;587:41–48. doi: 10.1113/jphysiol.2008.163410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miki T, et al. ATP-sensitive K+ channels in the hypothalamus are essential for the maintenance of glucose homeostasis. Nature neuroscience. 2001;4:507–512. doi: 10.1038/87455. [DOI] [PubMed] [Google Scholar]
- 52.Dunn-Meynell AA, Routh VH, Kang L, Gaspers L, Levin BE. Glucokinase is the likely mediator of glucosensing in both glucose-excited and glucose-inhibited central neurons. Diabetes. 2002;51:2056–2065. doi: 10.2337/diabetes.51.7.2056. [DOI] [PubMed] [Google Scholar]
- 53.Kang L, Routh VH, Kuzhikandathil EV, Gaspers LD, Levin BE. Physiological and molecular characteristics of rat hypothalamic ventromedial nucleus glucosensing neurons. Diabetes. 2004;53:549–559. doi: 10.2337/diabetes.53.3.549. [DOI] [PubMed] [Google Scholar]
- 54.Mirshamsi S, et al. Leptin and insulin stimulation of signalling pathways in arcuate nucleus neurones: PI3K dependent actin reorganization and KATP channel activation. BMC Neurosci. 2004;5:54. doi: 10.1186/1471-2202-5-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ramamurthy S, Ronnett GV. Developing a head for energy sensing: AMP-activated protein kinase as a multifunctional metabolic sensor in the brain. J Physiol. 2006;574:85–93. doi: 10.1113/jphysiol.2006.110122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kola B, Boscaro M, Rutter GA, Grossman AB, Korbonits M. Expanding role of AMPK in endocrinology. Trends in endocrinology and metabolism: TEM. 2006;17:205–215. doi: 10.1016/j.tem.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 57.Murphy BA, Fakira KA, Song Z, Beuve A, Routh VH. AMP-activated protein kinase and nitric oxide regulate the glucose sensitivity of ventromedial hypothalamic glucose-inhibited neurons. Am J Physiol Cell Physiol. 2009;297:C750–C758. doi: 10.1152/ajpcell.00127.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Williams RH, Alexopoulos H, Jensen LT, Fugger L, Burdakov D. Adaptive sugar sensors in hypothalamic feeding circuits. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:11975–11980. doi: 10.1073/pnas.0802687105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Claret M, et al. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J Clin Invest. 2007;117:2325–2336. doi: 10.1172/JCI31516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jorgensen SB, et al. Knockout of the alpha2 but not alpha1 5'-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. The Journal of biological chemistry. 2004;279:1070–1079. doi: 10.1074/jbc.M306205200. [DOI] [PubMed] [Google Scholar]
- 61.Diez-Sampedro A, et al. A glucose sensor hiding in a family of transporters. Proc Natl Acad Sci U S A. 2003;100:11753–11758. doi: 10.1073/pnas.1733027100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Frayling C, Britton R, Dale N. ATP-mediated glucosensing by hypothalamic tanycytes. The Journal of physiology. 2011;589:2275–2286. doi: 10.1113/jphysiol.2010.202051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rodriguez E, et al. Hypothalamic tanycytes: a key component of brain-endocrine interaction. Int Rev Cytol. 2005;247:89–164. doi: 10.1016/S0074-7696(05)47003-5. [DOI] [PubMed] [Google Scholar]
- 64.Zhang C, Kelly MJ, Ronnekleiv OK. 17Beta-estradiol rapidly increases K(ATP) activity in GnRH via a protein kinase signaling pathway. Endocrinology. 2010;151:4477–4484. doi: 10.1210/en.2010-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Coyral-Castel S, et al. The effect of AMP-activated kinase activation on gonadotrophin-releasing hormone secretion in GT1–7 cells and its potential role in hypothalamic regulation of the oestrous cyclicity in rats. J Neuroendocrinol. 2008;20:335–346. doi: 10.1111/j.1365-2826.2007.01643.x. [DOI] [PubMed] [Google Scholar]
- 66.Wen JP, et al. Globular adiponectin inhibits GnRH secretion from GT1–7 hypothalamic GnRH neurons by induction of hyperpolarization of membrane potential. Biochem Biophys Res Commun. 2008;371:756–761. doi: 10.1016/j.bbrc.2008.04.146. [DOI] [PubMed] [Google Scholar]
- 67.Cheng XB, et al. GnRH secretion is inhibited by adiponectin through activation of AMP-activated protein kinase and extracellular signal-regulated kinase. Endocrine. 2011;39:6–12. doi: 10.1007/s12020-010-9375-8. [DOI] [PubMed] [Google Scholar]
- 68.Kim MS, Lee KU. Role of hypothalamic 5'-AMP-activated protein kinase in the regulation of food intake and energy homeostasis. J Mol Med. 2005;83:514–520. doi: 10.1007/s00109-005-0659-z. [DOI] [PubMed] [Google Scholar]
- 69.Roland AV, Moenter SM. Prenatal androgenization of female mice programs an increase in firing activity of gonadotropin-releasing hormone (GnRH) neurons that is reversed by metformin treatment in adulthood. Endocrinology. 2011;152:618–628. doi: 10.1210/en.2010-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Blazquez C, Woods A, de Ceballos ML, Carling D, Guzman M. The AMP-activated protein kinase is involved in the regulation of ketone body production by astrocytes. J Neurochem. 1999;73:1674–1682. doi: 10.1046/j.1471-4159.1999.731674.x. [DOI] [PubMed] [Google Scholar]