

Abstract
Metformin is widely used in the treatment of type-2 diabetes. The pleotropic effects of metformin on glucose and lipid metabolism have been proposed to be mediated by the activation of AMP-activated protein kinase (AMPK) and the subsequent up-regulation of small heterodimer partner (SHP). SHP suppresses the functions of several nuclear receptors involved in the regulation of hepatic metabolism, including pregnane X receptor (PXR), which is referred to as a “master regulator” of drug/xenobiotic metabolism.
In this study, we hypothesize that metformin suppresses the expression of CYP3A4, a main detoxification enzyme and a target gene of PXR, due to SHP up-regulation.
We employed various gene reporter assays in cell lines and qRT-PCR in human hepatocytes and in Pxr−/− mice.
We show that metformin dramatically suppresses PXR-mediated expression of CYP3A4 in hepatocytes. Consistently, metformin significantly suppressed the up-regulation of Cyp3a11 mRNA in the liver and intestine of wild-type mice, but not in Pxr−/− mice. A mechanistic investigation of the phenomenon showed that metformin does not significantly up-regulate SHP in human hepatocytes. We further demonstrate that AMPK activation is not involved in this process. We show that metformin disrupts PXR’s interaction with steroid receptor coactivator-1 (SRC1) in a two-hybrid assay independently of the PXR ligand binding pocket. Metformin also inhibited vitamin D receptor-, glucocorticoid receptor- and constitutive androstane receptor (CAR)-mediated induction of CYP3A4 mRNA in human hepatocytes.
We show, therefore, a suppressive effect of metformin on PXR and other ligand-activated nuclear receptors in transactivation of the main detoxification enzyme CYP3A4 in human hepatocytes.
Keywords: metformin, cytochrome P450, induction, PXR, AMPK
1. INTRODUCTION
Metformin (N,N-dimethylimidodicarbonimidic diamide hydrochloride), a biguanide oral antihyperglycemic drug, is used as the first-line oral treatment for type 2 diabetes [1]. Despite the wide clinical use of metformin, its mechanisms of action have not been fully characterized. The main clinical effect of metformin, i.e., lowering serum glucose and lipids, is due to decreased hepatic gluconeogenesis, glycogenolysis and intestinal absorption of glucose. Metformin also increases the insulin sensitivity of peripheral tissues like muscle and adipocytes, thus resulting in increased peripheral glucose uptake [2–4]. Metformin is not metabolized by biotransformation enzymes in humans and no metabolites of metformin have been observed in the plasma, urine or feces. Metformin is not thought to cause drug-drug interactions (DDIs) with other medications [1]. Nevertheless, several reports indicate that metformin can affect the pharmacokinetics of co-administered drugs metabolized by liver cytochrome P450 enzymes such as topiramate (Micromedex 2.0 database), dofetilide [5], phenprocoumon [6] and itraconazole [7].
Recent studies suggest that activation of the AMP-activated protein kinase (AMPK) may play a central role in metformin’s actions [8]. AMPK is activated upon ATP depletion (e.g., due to ischemia, hypoxia, exercise and glucose deprivation), and in turn, AMPK switches off ATP utilization by inhibiting de novo glucose synthesis and cell growth while facilitating ATP production (e.g., via fatty acid oxidation, increased muscle glucose uptake and glycolysis) [9–11]. Metformin does not act as a direct allosteric activator of AMPK; however, it may act, in part, by inhibiting mitochondrial respiratory chain complex I, hence suppressing mitochondrial ATP synthesis and activating the AMPK pathway [12, 13]. Downstream targets of AMPK kinase include nuclear receptors and transcription factors (e.g., HNF4α), co-regulators (e.g., SRC1) and core proteins involved in gene transcription [14].
Several studies now implicate hepatic small heterodimer partner (SHP) in the metformin-mediated activation of AMPK. AMPK activation has been proposed to induce SHP expression, which inhibits gluconeogenesis by down-regulating phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase) gene expression [15, 16]. SHP is a member of the nuclear receptor family that lacks a DNA binding domain and, hence, acts through the direct binding and repression of nuclear receptors by co-regulator competition [17, 18]. Importantly, SHP has been found to repress the transcriptional activity of pregnane X nuclear receptor (PXR) in vitro and in vivo [19–21]. In a positive feedback loop, PXR inhibits SHP gene transcription [19]. Other nuclear receptors (NRs), such as constitutive androstane receptor (CAR) [18, 22, 23] and glucocorticoid receptor (GR) [24], also interact with SHP, which serves to function as a general corepressor (see review [17]). Significantly, these NRs are key regulators of xenobiotic-metabolizing enzymes of the cytochrome P450 superfamily (CYP) [25]. PXR is referred to as a “master regulator” or “xenosensor” for the metabolism and clearance of diverse endogenous and exogenous compounds [25–27]. When there is no ligand present, PXR remains repressed in terms of its gene transcription. However, when a ligand (any of a large number of structurally diverse xenobiotics, prescription drugs, herbal supplements, vitamins, and some endobiotics and bile acids) binds to PXR [25, 26, 28], target genes are up-regulated through transcriptional induction. CYP3A4 is the most important and well-studied target gene of PXR, which is the crucial drug/xenobiotic enzyme and metabolizes about 50% of all drugs and xenobiotics [29]. In addition, PXR regulates the expression of several other important drug/xenobiotic-metabolizing enzymes such as CYP2C9, CYP2B6, CYP2C19, CYP3A5, UGT1A1 and SULT2A1 as well as drug transporters such as MDR1(P-glycoprotein) and MRP2 [25].
Although SHP suppresses PXR, the effect of SHP up-regulation by metformin on PXR transcriptional regulation of its target genes has not been analyzed. Thus, the aim of this study was to examine the effect of metformin on PXR- and other nuclear receptor-mediated expression of the main drug-metabolizing enzyme, CYP3A4, in vitro in primary human hepatocytes or in vivo in Pxr wild-type and Pxr−/− knockout mice. In addition, mechanistic studies employing transient transfection gene reporter, two-hybrid and qRT-PCR assays were performed in several human liver- or intestine-derived cell lines.
We found that metformin suppressed PXR-mediated gene regulation of CYP3A4 in therapeutically relevant concentrations in primary human hepatocytes. Consistently, metformin diminished Pxr-mediated gene regulation of the mouse Cyp3a11 gene in wild-type but not in Pxr−/− knockout mice. We present mechanistic data suggesting that metformin abrogates the interaction of PXR with the SRC1 coactivator. Contrary to previous data, we cannot demonstrate consistent SHP induction by metformin in primary human hepatocytes at clinically relevant concentrations [16]. Therefore, in contrast to the currently accepted idea, we hypothesize that metformin might affect hepatic CYP enzyme activity in vivo.
2. MATERIALS AND METHODS
2.1. Cell lines and primary human hepatocytes
LS174T cells (purchased from ECCAC, Salisbury, UK) cell line was maintained in antibiotic-free DMEM medium supplemented with 10% FBS and 1% nonessential amino acids phenol red-free media, which were all purchased from Invitrogen (Carlsbad, CA). LS174T cells express high levels of endogenous and functional PXR [30, 31]. The human MZ-Hep-1 hepatocarcinoma cell line (kindly donated by Dr. Ramiro Jover, Hospital La Fe, Valencia, Spain) was maintained in antibiotic-free DMEM supplemented with 10% FBS and 1 mM sodium pyruvate. Fetal bovine serum (FBS) was purchased from PAA (Pasching, Austria). CITCO was purchased from BIOMOL (Plymouth Meeting, PA). Other chemicals and cell culture media were purchased from Sigma-Aldrich (St. Louis, MO). The final concentration of DMSO in the culture media was 0.1% (v/v) in all experiments.
Long-term human hepatocytes in monolayer were purchased from Biopredic International, Rennes, France, and were maintained according to the protocols provided by Biopredic. The medium was exchanged for serum-free medium the day after delivery, and the culture was allowed to stabilize for an additional 6–24 h prior to treatments.
LH28, LH29 and LH31 primary human hepatocyte preparations were isolated, cultivated and treated as described in our previous papers [32, 33]. Our tissue acquisition protocol was designed in accordance with the requirements issued by local ethical commissions in the Czech Republic. In addition, we used four commercial preparations of Long-term human hepatocytes in monolayer: Batch HEP220221 (86-year-old male), Batch HEP220466 (75-year-old female suffering from hepatocellular carcinoma), Batch HEP220465 (63-year-old male with liver metastases), and Batch HEP220492 (66-year-old female with hepatic metastases) (Biopredic International, Rennes, France).
2.2. DNA constructs
A chimeric p3A4-luc reporter construct [34] and expression plasmids for PXR, CAR, GRα, VDR and SHP receptors have been described in our previous reports [35]. Myc-tagged expression constructs for AMPKα2 and a catalytically inactive dominant negative AMPKα2K45R mutant were a generous gift from Dr. M. Birnbaum (University of Pennsylvania School of Medicine, Philadelphia, Pennsylvania).
2.3. Transient transfection and luciferase gene reporter assays
All transient transfection assays were carried out in MZ-Hep1 or LS174T cells, as described previously [35]. Because both human and mouse OCT1/Oct1 are responsible for the hepatic uptake of metformin and because the transporter is not expressed in hepatoma cell lines, we co-transfected MZ-Hep1 cells with the expression construct for the human OCT1(SLC22A1) transporter (50 or 60 ng per well) in our experiments [36, 37]. All firefly luciferase activities were normalized to the total cellular protein concentration and were expressed relative to basal control levels, which were assigned a value of 1. The bicinchoninic acid (BCA) assay was used to analyze the total cellular protein concentration of lysates according to the manufacturer’s protocol (Bio-Rad, Hercules, CA). We did not normalize firefly luciferase activities to Renilla luciferase activity because metformin falsely activates Renilla luciferase expression constructs with SV40 or CMV promoters. When required, an equivalent quantity of empty vectors (pcDNA3, pSG5 or pECE) was co-transfected to keep the amount of DNA constant. The small interfering RNA siSHP (sc-44101) was purchased from Santa Cruz Biotechnology, Inc.
2.4. Two-hybrid assays
Mammalian two-hybrid assays were carried out as described elsewhere with slight modifications [31]. We used the pM-GAL4-PXRwt fusion expression plasmid for wild-type PXR and the double mutant pM-GAL4-PXRmut (S247W/C284W) expression plasmid. The double PXR mutant (S247W/C284W) has replaced the serine at position 247 with the larger tryptophan, which effectively fills the ligand binding pocket of PXR [38]. This replacement blocks the ligand binding pocket-dependent activity of PXR, rendering the construct constitutively active independently of a ligand. The VP16-SRC1-RID fusion expression construct for SRC1 coactivator 1 has been described previously [39].
2.5. qRT-PCR
Total RNA isolation and semi-quantitative real-time RT-PCR (qRT-PCR) analyses of CYP3A4 (hCYP3A4_Q2), CYP2B6 (hCYP2B6_Q2), CYP2C9, MDR1 (hABCB1_Q1) and SHP (hNR0B2_Q2) mRNA expression in primary human hepatocytes and intestinal LS174T cells were performed by employing commercial assays from Generi-Biotech (Hradec Kralove, Czech Republic) as described elsewhere [31, 35, 40]. HPRT (hHPRT_Q3) and β2-microglobulin (B2M) housekeeping gene expression levels were used as normalization controls. The expression of mouse Cyp3a11 and Hprt1 was also analyzed using primers and TaqMan probes commercially designed by Generi-Biotech. Housekeeping gene mRNA expression was not affected by any experimental treatment in the study, as assessed by normalization to total RNA. All experiments were performed in triplicate.
2.6. Immunoblotting
The relative abundance of each specific protein in 25 to 50 μg of whole cell lysate was determined by Western blot analysis, as described previously [41]. Anti-SHP (H-160: sc-30169; dilution 1:250), anti-PXR (H-11: sc-48340; dilution 1:500), anti-human CYP3A4 (HL3: sc53850; 1:2000), mouse monoclonal IgG (1:2000) and goat polyclonal anti-actin antibodies (clone I-19: 1616; a loading control) were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, USA). An AMPK and ACC Antibody Sampler Kit No#9957 (Cell Signaling Technology, Danvers, MA, USA) was used to detect phospho-AMPKα (Thr172) and AMPKα. Densitometric analyses have been performed to semi-quantify the expression of the tested proteins.
2.7. Animal experiments and the loss of righting reflex (LORR) in C57BL/6 Pxr wild-type and Pxr−/− mice
To demonstrate the in vivo effects of metformin on the metabolism of tribromoethanol anesthesia (a substrate for enzymes induced by Pxr activation), six- to eight-week-old C57BL/6 Pxr wild-type and Pxr−/− mice received tail vein injections of vehicle (0.1% DMSO in 0.9% saline), metformin (150 mg/kg/d once per day for two days and then 100 mg/kg once per day for two days), PCN (a prototype mouse Pxr ligand, 100 mg/kg/d) or their combination in a final volume of 100 μl. Seventy-two hours after the first drug or vehicle injection, the mice were anesthetized with tribromoethanol (0.2 mg/g) administered by i.p. injection. The protocol for the LORR studies has been previously published [42]. Whole liver and intestine (duodenum and jejunum) samples were frozen on dry ice for mRNA isolation in Trizol® reagent. All animal experiments were approved by the Albert Einstein College of Medicine, Animal Institute Committee, and all experiments were conducted in accordance with institutional and national guidelines.
2.8. Statistical analyses
All data are expressed as the mean ± standard deviation (SD). Differences between the groups were compared with Student's unpaired two-tailed t-test. If more than two groups were examined, the data were analyzed by one-way analysis of variance (ANOVA) with Dunnett’s posthoc test. Vehicle(DMSO)- or rifampicin-treated cells were used as reference groups. Linear regression analyses were used to elucidate the relationship between mRNA expression and metformin concentration. All statistical analyses were performed and the coefficients of determination (r squared, r2) were determined using GraphPad Prism 4 Software. p values <0.05 were considered significant.
3. RESULTS
3.1. Metformin suppresses CYP3A4 gene expression in primary human hepatocytes
In the first set of experiments, we examined whether metformin affects the expression of the main PXR target gene, CYP3A4, in primary human hepatocytes. Rifampicin was used as a prototypical ligand for human PXR. We found that metformin significantly suppressed both basal (constitutive) and rifampicin-induced expression of CYP3A4 mRNA after 24 h (Fig. 1A) and 60 h (p<0.05; Fig. 1B) of treatment in human hepatocytes.
Fig. 1. Metformin suppresses CYP3A4 mRNA expression in primary human hepatocytes.

Primary human hepatocyte cultures were treated with rifampicin (Rif, 10 μM), metformin (MET, 0.1, 0.5, 1.0 and 2.0 mM) or their combination for 24 h (A) or 60 h (B). Total RNA was isolated, and the mRNA levels were analyzed by qRT-PCR. The effects of compounds on the mRNA levels of the tested genes were normalized to housekeeping HPRT mRNA and are presented as the relative mRNA expression compared to control vehicle (DMSO)-treated cells (set to 1). Representative data from the HEP220465 preparation (A) and from the HEP220492 preparation (B) are shown.
(C) Immunodetection of CYP3A4, PXR, AMPKα, phospho-AMPKα (Thr172), SHP and actin proteins in the LH28 primary human hepatocyte preparation treated with rifampicin and metformin (2 mM) for 24 h. *p<0.05, **p<0.01 indicates a statistically significant effect of metformin on basal CYP3A4 mRNA expression compared to vehicle-treated controls; ƒp<0.05, ƒƒƒp<0.001 indicates a statistically significant effect of metformin on CYP3A4 mRNA expression compared to rifampicin-treated samples (ANOVA with Dunnett’s posthoc test).
To determine the effect of metformin on CYP3A4 and PXR protein expression in hepatocytes, we performed immunoblotting analyses. Rifampicin, as expected, qualitatively induced CYP3A4 protein expression (Fig. 1C). Metformin did not significantly decrease either CYP3A4 or PXR protein expression after 24 h (Fig. 1C); however, in the presence of rifampicin, metformin significantly (p<0.05) decreased CYP3A4 protein abundance to baseline levels (Fig. 1C). Metformin activated AMPKα phosphorylation, and the AMPKα kinase activation was further stimulated by rifampicin co-treatment (Fig. 1C). Neither metformin nor rifampicin significantly affected the level of AMPKα or SHP protein expression.
3.2. Metformin suppresses PXR-induced CYP3A4 expression and CYP3A4 gene reporter activity in the intestinal LS174T cell line
Next, we tested whether metformin would have similar effects in LS174T colon cancer cells, which are known to have high levels of endogenous and functional PXR expression. In those cells, metformin significantly suppressed both basal and inducible CYP3A4 mRNA expression in a concentration-dependent manner (Fig. 2A, regression analysis, r2=0.91, p<0.05 and r2=0.95, p<0.01 for basal and inducible CYP3A4, respectively).
Fig. 2. Metformin suppresses CYP3A4 mRNA expression and CYP3A4 luciferase reporter construct activation in intestinal LS174T cells.
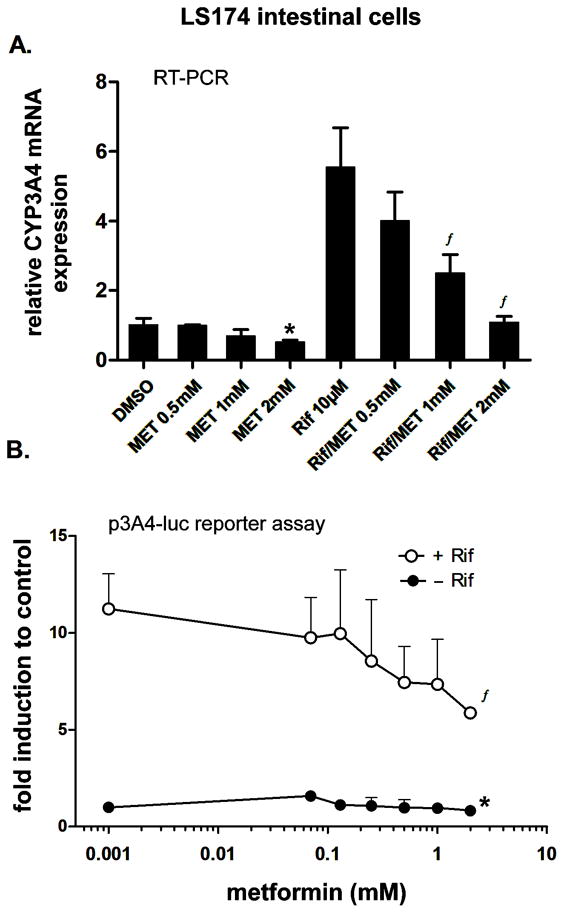
LS174T cells were pretreated with the indicated concentrations of metformin (MET) for 6 h, after which 10 μM rifampicin (Rif) and metformin in the noted concentrations were added to the culture media for an additional 48 h. Total RNA was isolated, and the mRNA levels were analyzed by qRT-PCR with specific primers for CYP3A4 (A). The effects of rifampicin and metformin or their combination on CYP3A4 mRNA levels are presented as the relative mRNA expression level relative to the control vehicle (DMSO)-treated cells (set to 1). Data represent the means of three independent experiments ± S.D.
(B) Transient transfection luciferase gene reporter assays with the p3A4-luc construct (200 ng/per well in 48-well plate) were performed in LS174T cells co-transfected with the OCT1 expression construct (50 ng/per well). After pretreatment with metformin and a 24-h incubation, cells were lysed and analyzed for firefly luciferase activity normalized to the total cellular protein concentration of the lysate as measured by BCA assay. Data represent the mean of three independent experiments and are reported as the fold activation of normalized luciferase activity relative to the solvent (0.1% DMSO) control.
*p<0.05 indicates a statistically significant effect of metformin on basal CYP3A4 mRNA expression or transactivation compared to the vehicle-treated control; ƒp<0.05 indicates a statistically significant effect of metformin on CYP3A4 mRNA expression or transactivation compared to rifampicin-treated samples (ANOVA with Dunnett’s posthoc test).
In transient transfection gene reporter experiments, we found that metformin significantly (p<0.01) suppressed rifampicin-induced CYP3A4 luciferase reporter activity in a concentration-dependent manner (Fig. 2B). In addition, 2mM metformin also significantly (p<0.05) suppressed luciferase reporter construct in the absence of rifampicin (Fig. 2B).
3.3. Metformin suppresses Pxr-mediated regulation of the mouse Cyp3a11 gene and alters the loss of righting reflex (LORR) in vivo
Next, we examined if metformin suppresses basal and Pxr-stimulated expression of the mouse CYP3A4 ortholog, Cyp3a11, in wild-type and Pxr−/− mice. In Pxr wild-type mice, pregnenolone-16α-carbonitrile (PCN), a prototypical ligand of mouse Pxr, significantly (p<0.01) increased Cyp3a11 mRNA levels in the liver. Metformin alone had no effect on Cyp3a11 mRNA levels in the liver. However, in the presence of PCN, metformin suppressed Cyp3a11 mRNA expression (p<0.05, Student’s t-test) (Fig. 3A). In Pxr−/− mice, there was no significant effect of either PCN or metformin alone or of their combination on Cyp3a11 gene expression (Fig. 3B). Similar trends were observed in mouse intestinal tissue (Fig. 3C). Consistent data were observed in the LORR studies in mice monitoring Cyp3a enzymatic activity in an inverse manner. PCN significantly suppressed the duration of the LORR (p<0.01, Student's t-test), but metformin significantly attenuated this effect (p<0.05, Student's t-test) (Fig. 3D). No such effects were observed in Pxr−/− mice (Fig. 3E).
Fig. 3. Metformin suppresses Cyp3a11 mRNA expression and activity (LORR) in the liver and intestine of Pxr wild-type, but not Pxr−/− knockout mice.

Pxr wild-type (n=8) (A, C, D) and Pxr−/− knockout (B, E) mice (n=6) were injected with vehicle, PCN (150 and 100 mg/kg/day), metformin (MET; 150 and 100 mg/kg/day, see Methods) or their combination for four days. Mice were then assessed in the LORR assay, after which they were sacrificed and liver and intestinal samples were collected for mRNA isolation. The effects of the tested compounds on Cyp3a11 mRNA levels normalized to the mRNA levels of the Hprt1 housekeeping gene are presented as relative mRNA expression compared to control vehicle-treated cells (set to 1). The duration of righting after sleep onset (LORR) was measured in minutes and recorded by two investigators as described. *p<0.05, **p<0.01 indicate a statistically significant result compared to vehicle-treated animals; ƒp<0.05 indicates a statistically significant result compared to PCN-treated animals (Student's unpaired t-test).
3.4. SHP expression is not significantly influenced by metformin in primary human hepatocytes
To investigate whether the action of metformin on PXR-mediated gene expression is mediated through SHP [15, 16], we looked at the mRNA and protein expression of SHP in multiple cell models (primary human hepatocytes and LS174T and MZ-Hep1 cell lines) in the presence and absence of metformin. We observed no statistically significant effect of metformin on SHP mRNA after 24, 48 nor 60 h treatments in hepatocyte preparations from five different donors (Fig. 4A and 4B, data from representative preparations HEP220466 and HEP220492 are shown).
Fig. 4. The effect of metformin on SHP mRNA and protein expression in primary human hepatocytes and cell lines.

Primary human hepatocyte cultures, HEP220466 and HEP220492 (A, B, C), or LS174T and MZ-Hep1 cell lines (D) were treated with metformin (0.1, 0.5, 1.0 and 2.0 mM) for 24 h or 60 h. Total RNA was isolated, and SHP mRNA levels were analyzed by qRT-PCR (A,B,D). SHP protein levels were analyzed by Western blotting (C). The effect of metformin on SHP mRNA levels is presented as the relative SHP mRNA expression level normalized to HPRT housekeeping mRNA relative to control vehicle (DMSO)-treated cells (set to 1). Negative (−, untreated MZ-Hep1 cells) and positive (+, SHP -transfected MZ-Hep1 cells) controls were used for Western blotting analyses. *p<0.05; **p<0.01 indicates a statistically significant result compared to vehicle-treated controls (ANOVA with Dunnett’s posthoc test).
At the protein level, we did not observe a significant effect of metformin on SHP protein expression in two primary human preparations (Fig. 4B, Fig. 1C). In LS174T and MZ-Hep1 cell lines, we observed a statistically significant down-regulation of SHP mRNA upon treatment with 2 mM metformin (Fig. 4D). We consistently observed the down-regulation of mouse Shp mRNA in mouse primary hepatocytes after treatment with metformin (Aatsinki et al., manuscript in preparation).
Together these data demonstrate that SHP up-regulation is unlikely to be involved in the effect of metformin on CYP3A4 expression.
3.5. Metformin disrupts the interaction of PXR with its coactivator SRC1 in a two-hybrid assay
In the next experiment, we used pGAL4-SRC1-RID and VP16-PXR(LBD) constructs. Metformin significantly (p<0.05) and dose-dependently decreased the PXR-SRC1 interaction in the presence of rifampicin (10 μM), even though metformin alone had no significant effect (Fig. 5A). In addition, we observed the same suppressive effect of metformin on another prototypical PXR ligand, SR12813 (10 μM), in the assay (data not shown).
Fig. 5. Metformin disrupts the interaction of PXR with the SRC1 coactivator independently of the PXR ligand binding domain.

The mammalian two hybrid assay was performed in MZ-Hep1 cells transfected with the OCT1 expression construct (50 ng). (A) Cells were co-transfected with the pGl5-luc luciferase reporter construct (200 ng per 48-well plate), pGAL4-SRC1 (100 ng per well) and VP16-PXR LBD (100 ng per well) constructs. Cells were treated with rifampicin (Rif; 10 μM), metformin (MET; 1 or 2 mM) or their combination for 24 h, and cells were then lysed and analyzed for luciferase activity normalized to the total cellular protein concentration of the lysate, as measured by BCA assay. (B) Cells were co-transfected with the pGl5-luc luciferase reporter construct (200 ng per well in 48-well plate), the pGAL4 fusion construct with the wild-type PXR LBD domain or with the PXR LBD mutated at S247W/C284W (constitutively active form) (100 ng per well) and the VP16-SRC1 construct (100 ng per well). Cells were pretreated for 6 h with metformin, then the cells were treated with rifampicin (10 μM), metformin (1 or 2 mM) or their combinations for 24 h, and then cells were lysed and analyzed for luciferase activity normalized to total cellular protein concentration of lysate measured by BCA assay. Data represent the mean of three independent experiments and are shown as the fold activation of normalized luciferase activity relative to the solvent (0.1% DMSO) controls. ƒp<0.05 indicates a statistically significant result compared to rifampicin-treated cells co-transfected under the same experimental conditions with PXR wild-type construct; #p<0.05 indicates a statistically significant result compared to rifampicin-treated cells co-transfected under the same experimental conditions with the constitutively active pGAL4-PXRmut S247W/C284W construct; *p<0.05 indicates a statistically significant effect compared to the vehicle (DMSO)-treated cells (control) co-transfected with the wild-type PXR construct (ANOVA with Dunnett’s posthoc test).
We then studied whether metformin affects the recruitment of the SRC1 coactivator to either wild-type PXR or the constitutively active PXRmut(S247W/C284W), which encodes a ligand-binding pocket-defective mutant [38]. We found that metformin significantly inhibited the interaction of wild-type PXR ligand binding domain (LBD) with SRC1 after stimulation with rifampicin (Fig. 5B). Interestingly, we also found that metformin significantly (p<0.05) suppressed the interaction of the constitutively active ligand-binding pocket mutant PXR (S247W/C284W) with SRC1 regardless of the presence or absence of rifampicin (Fig. 5B). The data exclude the possibility that metformin is an antagonist of PXR in the ligand binding domain. Metformin (2 mM) alone significantly decreased luciferase reporter construct activity in the experiment if cells were pretreated with metformin for 6 h (Fig. 5B; Fig. 6C, D). These results correlate with the results obtained employing qRT-PCR (Fig. 1A, B; 2A; 6B and 7A) suggesting that metformin in high concentrations may also affect the basal expression of CYP3A4.
Fig. 6. The AMPK pathway is not involved in the effect of metformin on CYP3A4 gene expression.

The AMPK activators metformin and AICAR have different effects on CYP3A4 gene reporter construct transactivation and on CYP3A4 mRNA expression in LS174T. (A) LS174T cells were transfected with the p3A4-luc reporter construct (200 ng per 48-well plate) and the OCT1 expression construct (50 ng per well). Cells were pretreated with metformin for 6 h, after which the cells were treated with the indicated compounds rifampicin (Rif, 10 μM), BML275 (BML, 25 μM), AICAR (500 μM) and metformin (MET, 2 mM) or their combination for an additional 24 h. Cells were lysed and analyzed for luciferase activity normalized to the total cellular protein concentration of the lysate, as measured by BCA assay. Data represent the mean of three independent experiments and are shown as the fold activation of normalized luciferase activity relative to the solvent (0.1% DMSO) control. **p<0.01 indicates a statistically significant effect compared to vehicle (DMSO)-treated cells co-transfected under the same experimental conditions; ƒp<0.05; ƒƒp<0.01 indicates a statistically significant effect compared to rifampicin-treated cells co-transfected under the same experimental conditions (ANOVA with Dunnett’s posthoc test).
(B) LS174T cells were pretreated with metformin for 6 h, after which the cells were treated with the indicated compounds or their combination for an additional 24 h. Cells were harvested into Trizol® reagent for mRNA isolation, and CYP3A4 mRNA was analyzed via qRT-PCR. Data represent the mean of three independent experiments and demonstrate the up-regulation of CYP3A4 mRNA expression normalized to housekeeping HPRT mRNA expression relative to the solvent (0.1% DMSO) control. *p<0.05 indicates a statistically significant effect compared to vehicle-treated cells; ƒp<0.05 indicates a statistically significant effect compared to rifampicin-treated cells (ANOVA with Dunnett’s posthoc test).
(C, D) The AMPK inhibition does not abolish the effect of metformin on PXR-SCR1 interaction in MZ-Hep1 cells. A two-hybrid assay in MZ-Hep1 cells was performed with pG5luc, pGAL4-SRC1 and VP16-PXR LBD constructs (all 100 ng per well in a 48-well plate) and the OCT1 expression construct (60 ng per well). Either the pcDNA3-AMPKα2K45R dominant negative mutant construct or the pcDNA3 empty construct (60 ng per well) were used in panel C. Cells were pretreated with metformin for 6 h, after which cells were treated with the indicated compounds or their combination for an additional 24 h. In panel D, cells were pretreated with metformin for 6 h, after which cells were treated with BML275 (compound C, 25 μM or 0.01% DMSO vehicle) and the indicated compounds or their combination for an additional 24 h. Cells were lysed and analyzed for luciferase activity normalized to the total cellular protein concentration of the lysate, as measured by BCA assay. Data represent the mean of three independent experiments and are shown as the fold activation of normalized luciferase activity relative to the vehicle (0.1% DMSO)-treated control or to control cells transfected with the empty pcDNA3 construct. *p<0.05, **p<0.01 indicate a statistically significant effect compared to vehicle-treated cells co-transfected under the same experimental conditions; ƒp<0.05 indicates a statistically significant effect compared to rifampicin-treated cells co-transfected under the same experimental conditions (ANOVA with Dunnett’s posthoc test).
Fig. 7. Metformin suppresses CAR-, VDR- and GR-mediated up-regulation of CYP3A4 mRNA in primary human hepatocytes and in a luciferase gene reporter assay.

(A) Primary human hepatocyte preparations LH28 and LH29 were treated with metformin (2 mM) and prototypical ligands of the tested nuclear receptors 1,25OHvitD3 (10 nM, a VDR receptor ligand), CITCO (1 μM, CAR ligand), dexamethasone (100 nM, GR ligand), or their combination with metformin for 24 h. After incubation, cells were lysed and analyzed for CYP3A4 mRNA expression levels normalized to the housekeeping HPRT mRNA level as measured by qRT-PCR. (B) The transient transfection assay was performed in LS174T cells transfected with the p3A4-luc reporter construct (150 ng per well in 48-well plate) and 100 ng of the expression construct (VDR, CAR or GR) together with the OCT1 expression plasmid (50 ng). LS174T cells were treated with metformin (2 mM) and the prototypical ligands 1,25OHvitD3 (10 nM), CITCO (1 μM), or dexamethasone (100 nM) or their combination with metformin for 24 h. Data represent the means of three independent experiments and are shown as the fold activation of normalized luciferase activity relative to the solvent (0.1% DMSO) controls. ƒp<0.05 indicates a statistically significant effect compared to the effects of the prototypical ligand (Student's unpaired t-test); *p<0.05 indicates a statistically significant effect compared to vehicle-treated cells (ANOVA with Dunnett’s posthoc test).
These data indicate that one possible mechanism of metformin action might involve the direct disruption of activated PXR interaction with SRC1 independently of the PXR ligand binding pocket although the detailed mechanism should be elucidated in our ongoing research.
3.6. Inhibition of the AMPK cascade does not abolish the effect of metformin on PXR-mediated CYP3A4 transactivation, PXR-SRC1 coactivation or CYP3A4 mRNA expression
To examine whether activation of the AMPK pathway by metformin results in the suppression of PXR-mediated regulation of the CYP3A4 gene and disruption of PXR-SRC1 coactivation, we performed a series of experiments with chemical modulators of the AMPK pathway, such as AICAR (AMPK activator) and BML275 (AMPK inhibitor), as well as with a dominant negative mutant of AMPKα2.
We found that AICAR (500 μM) and metformin (1–2 mM) have different effects on p3A4-luc reporter construct activation as well as on CYP3A4 mRNA expression in LS174T cells (Fig. 6A and B). AICAR did not significantly affect the rifampicin-stimulated transactivation and expression of CYP3A4 mRNA. BML275 (compound C) significantly suppressed rifampicin-induced transactivation and both basal and induced CYP3A4 mRNA expression (p<0.05; Fig. 6A and B). Moreover, BML275 did not abolish the effect of metformin on rifampicin-stimulated CYP3A4 expression (Fig. 6A and B).
In the next two-hybrid assays, we evaluated the effect of AMPK pathway inhibition on PXR-SRC-1 interaction. We used the expression construct for the dominant negative AMPKα2K45R mutant (Fig. 6C) and the AMPK inhibitor BML275 (Fig. 6D). We found that neither genetic nor chemical (pharmacological) inhibition of the AMPK pathway abolished the effect of metformin on CYP3A4-inducible expression. Importantly, AICAR, which has been reported to provoke changes in the AMP/ATP ratio leading to AMPK activation by LKB1[43], had the opposite effect in comparison with metformin (Fig. 6C and D).
These data clearly indicate that AMPK activation by metformin is unlikely to be involved in the effect of metformin on CYP3A4 expression.
3.7. Metformin suppresses CYP3A4 mRNA induction by CAR, VDR and GR receptors in primary human hepatocytes and transactivation of p3A4-luc reporter construct in LS174T cells
In subsequent experiments, we found that metformin (2 mM) significantly (p<0.05) suppressed the ligand-induced expression of CYP3A4 mRNA after treatment with prototype ligands of all of the tested nuclear receptors in primary human hepatocytes (Fig. 7A). The experiments were repeated in two primary human hepatocyte preparations with the same results (data not shown). Consistently, prototype ligand-induced transactivations of the p3A4-luc reporter construct were significantly suppressed (p<0.05) by metformin in LS174T cells cotransfected with nuclear receptors CAR, VDR and GR (Fig. 7B). Metformin also significantly suppressed basal transactivation of the construct and decreased basal expression of CYP3A4 mRNA in human hepatocytes (Fig. 7; and Fig. 1A,B; 2B, 6A, B).
These data suggest a similar effect of metformin on ligand-activated PXR-, CAR-, VDR-, and GR-mediated transactivation of the CYP3A4 gene.
4. DISCUSSION
In this study, we present novel data demonstrating a dramatic effect of metformin on hepatic and intestinal expression of CYP3A4/Cyp3a11 mRNA. CYP3A4 is a key drug-metabolizing enzyme, as it metabolizes about 50% of drugs and xenobiotics. We show that metformin represses its induction via the major xenobiotic- and hormone-dependent nuclear receptors PXR, CAR, VDR and GR in human hepatocytes. We present data indicating that in the case of PXR the phenomenon is not due to metformin acting as an antagonist in the PXR ligand binding pocket, a putative effect of metformin on SHP expression or metformin-stimulated activation of AMPK pathway. We suppose instead that metformin disrupts the coactivation of PXR with SRC1 independently of the PXR ligand binding domain, thus resulting in the inhibition of PXR transcriptional activity and the suppression of its target gene expression.
Recently, we have shown that ketoconazole disrupts the PXR-SRC1 interaction and consequently suppresses the PXR-mediated induction of its target genes, including the CYP3A4/Cyp3a11 genes [31, 42]. Ketoconazole was believed to interact with the coactivator binding domain because it inhibits the constitutively active PXR mutant with the inactive LBD [42]. In this paper, we observed a similar phenomenon with metformin. We show that metformin inhibits the constitutively active PXR mutant with the mutated LBD (Fig. 5B). These data strongly indicate that metformin disrupts PXR-SRC1 coactivation, which may be a reasonable cause of the metformin-mediated suppressive effect on the PXR–controlled induction of CYP3A4 as well as on the VDR, CAR and GR nuclear receptors, which are also coactivated with SRC1 in transactivation of CYP3A4 [44]. Further experiments in our laboratory will address in detail the mechanistic explanation for the effects of metformin on CYP3A4 gene induction and on PXR-SRC1 coactivation. Our current projects address the effect of metformin on additional coactivators (such as TIF2, PGC1α, HNF4α), the corepressor SMRT, and their posttranslational modification, and on other target genes (CYP2C9, CYP2B6) of the nuclear receptors in terms of both basal and induced expression levels (Aatsinki et al., manuscript in preparation).
The mechanism of metformin action is still unclear. It has been suggested that this drug activates the AMPK pathway [8], a major regulator of cell energy homeostasis. Activation of the AMPK cascade by metformin is likely mediated by the suppression of mitochondrial ATP synthesis via the inhibition of mitochondrial respiratory chain complex I [12, 13]. Recently, another molecular mechanism of metformin action related to AMPK activation has been proposed in the liver. Metformin has been reported to up-regulate SHP in HepG2 hepatoma cells and in human and rat hepatocytes [15, 16]. SHP suppresses the transcriptional activity of PXR both in vitro and in vivo [19–21]. Thus, we hypothesized that metformin suppresses the PXR-mediated induction of its major target gene, CYP3A4, via SHP up-regulation. However, in contrast to published data [15, 16], we observed no consistent up-regulation of SHP mRNA by metformin in clinically relevant concentrations in five primary human hepatocyte preparations (Fig.1 and 4, data from LH29 and LH31 hepatocyte preparations are not shown). Similarly, we observed no consistent induction of SHP mRNA in either MZ-Hep1 or LS174T cells (Fig. 4D). In addition, the silencing of SHP had no effect on the metformin-mediated suppression of induced CYP3A4 expression (unpublished results), again suggesting that SHP up-regulation is not involved in the phenomenon. Finally, we observed the down-regulation of mouse Shp mRNA in mouse primary hepatocytes after treatment with metformin (Aatsinki et al., manuscript in preparation). This discrepancy prompts further study on SHP regulation by metformin.
An important link between energy metabolism and xenobiotic biotransformation has been proposed via AMPK. There is evidence that the regulation of hepatic CYPs is affected in diabetes and that the regulation of some CYP genes depends on the nutritional status of cells [45]. Cyp2b10 and Car genes are induced in the livers of fasted mice [46]. It has also been shown that AMPK activation and the inhibition of mitochondrial function are connected with the effect of the prototype inducer, phenobarbital, on hepatic CYP genes [45, 47]. A pharmacological inhibitor of AMPK or a dominant negative form of AMPK abolished the response to phenobarbital [47, 48]. Consistently, phenobarbital failed to induce the expression of CYPs in AMPKα1α2LS−/− mice [48].
In this paper, we show that AMPK activation is unlikely to be involved in the suppression of CYP3A4 induction by metformin. This conclusion is supported by the fact that AICAR does not display the same effect as metformin on either CYP3A4 transactivation in the gene reporter assay or on CYP3A4 mRNA expression (Fig. 6A and B). Moreover, the dominant negative mutant AMPKα2K45R did not abolish the inhibitory effect of metformin on CYP3A4 transactivation (Fig. 6C). Interestingly, we found that inhibition of the AMPK cascade by BML275 resulted in a significant reduction of both basal and induced CYP3A4 transactivation (Fig. 6A, B). These data suggest that the activation of AMPK does not suppress CYP3A4 expression and that activated AMPK is necessary for the transactivation of the CYP3A4 gene.
Another matter for consideration is whether the suppressive effect of metformin on detoxification genes is clinically relevant in humans. No clinical study has evaluated in detail the effect of metformin on CYP3A4 catalytic activity and expression in humans after treatment with a potent ligand of PXR.
We can conclude that clinical investigation is needed to confirm the dramatic effect of metformin on the activity of the main detoxification enzyme, CYP3A4, as well as on other CYP enzymes, such as CYP2B6 and CYP2C9, which are regulated primarily by PXR in the human liver under standard pharmacotherapeutic conditions and dosage. In addition, further experiments should comprehensively elucidate the proposed novel molecular mechanism of drug-mediated CYP enzyme repression.
Acknowledgments
This work was supported by the Czech Scientific Agency (Grant Nos. 303/07/0128 and No. 304/10/0149 to P. Pavek), the Damon Runyon Clinical Investigator Award CI 15-02, NIH R01CA 127231 to SM and by SVV-2011-263-003 project to L. Stejskalova.
The authors would like to thank to all of our kind donors for providing us with the necessary cDNA constructs to complete this work. The mouse strains used were the kind gift of Dr. Wen Xie, University of Pittsburgh, PA.
Footnotes
The data have been presented as a poster at the 60th Czech and Slovak Pharmacological Conference (October 2010).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bosi E. Metformin--the gold standard in type 2 diabetes: what does the evidence tell us? Diabetes Obes Metab. 2009;11 (Suppl 2):3–8. doi: 10.1111/j.1463-1326.2008.01031.x. [DOI] [PubMed] [Google Scholar]
- 2.Witters LA. The blooming of the French lilac. J Clin Invest. 2001;108:1105–7. doi: 10.1172/JCI14178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diamanti-Kandarakis E, Christakou CD, Kandaraki E, Economou FN. Metformin: an old medication of new fashion: evolving new molecular mechanisms and clinical implications in polycystic ovary syndrome. Eur J Endocrinol. 2010;162:193–212. doi: 10.1530/EJE-09-0733. [DOI] [PubMed] [Google Scholar]
- 4.Correia S, Carvalho C, Santos MS, Seica R, Oliveira CR, Moreira PI. Mechanisms of action of metformin in type 2 diabetes and associated complications: an overview. Mini Rev Med Chem. 2008;8:1343–54. doi: 10.2174/138955708786369546. [DOI] [PubMed] [Google Scholar]
- 5.Pfizer TUSPPI. TIKOSYN US Patient Product Information Pfizer. New York, NY: Pfizer Inc; 2011. [Google Scholar]
- 6.Ohnhaus EE, Berger W, Duckert F, Oesch F. The influence of dimethylbiguanide on phenprocoumon elimination and its mode of action. A drug interaction study. Klin Wochenschr. 1983;61:851–8. doi: 10.1007/BF01537460. [DOI] [PubMed] [Google Scholar]
- 7.Choi YH, Lee U, Lee BK, Lee MG. Pharmacokinetic interaction between itraconazole and metformin in rats: competitive inhibition of metabolism of each drug by each other via hepatic and intestinal CYP3A1/2. Br J Pharmacol. 2010;161:815–29. doi: 10.1111/j.1476-5381.2010.00913.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–74. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fogarty S, Hardie DG. Development of protein kinase activators: AMPK as a target in metabolic disorders and cancer. Biochim Biophys Acta. 2010;1804:581–91. doi: 10.1016/j.bbapap.2009.09.012. [DOI] [PubMed] [Google Scholar]
- 10.Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 11.Hardie DG. AMPK: a key regulator of energy balance in the single cell and the whole organism. Int J Obes (Lond) 2008;32 (Suppl 4):S7–12. doi: 10.1038/ijo.2008.116. [DOI] [PubMed] [Google Scholar]
- 12.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348(Pt 3):607–14. [PMC free article] [PubMed] [Google Scholar]
- 13.El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275:223–8. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- 14.Leff T. AMP-activated protein kinase regulates gene expression by direct phosphorylation of nuclear proteins. Biochem Soc Trans. 2003;31:224–7. doi: 10.1042/bst0310224. [DOI] [PubMed] [Google Scholar]
- 15.Kim YD, Park KG, Lee YS, Park YY, Kim DK, Nedumaran B, et al. Metformin inhibits hepatic gluconeogenesis through AMP-activated protein kinase-dependent regulation of the orphan nuclear receptor SHP. Diabetes. 2008;57:306–14. doi: 10.2337/db07-0381. [DOI] [PubMed] [Google Scholar]
- 16.Lee JM, Seo WY, Song KH, Chanda D, Kim YD, Kim DK, et al. AMPK-dependent repression of hepatic gluconeogenesis via disruption of CREB/CRTC2 complex by orphan nuclear receptor SHP. J Biol Chem. 2010 doi: 10.1074/jbc.M110.134890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bavner A, Sanyal S, Gustafsson JA, Treuter E. Transcriptional corepression by SHP: molecular mechanisms and physiological consequences. Trends Endocrinol Metab. 2005;16:478–88. doi: 10.1016/j.tem.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 18.Seol W, Choi HS, Moore DD. An orphan nuclear hormone receptor that lacks a DNA binding domain and heterodimerizes with other receptors. Science. 1996;272:1336–9. doi: 10.1126/science.272.5266.1336. [DOI] [PubMed] [Google Scholar]
- 19.Li T, Chiang JY. Mechanism of rifampicin and pregnane X receptor inhibition of human cholesterol 7 alpha-hydroxylase gene transcription. Am J Physiol Gastrointest Liver Physiol. 2005;288:G74–84. doi: 10.1152/ajpgi.00258.2004. [DOI] [PubMed] [Google Scholar]
- 20.Ourlin JC, Lasserre F, Pineau T, Fabre JM, Sa-Cunha A, Maurel P, et al. The small heterodimer partner interacts with the pregnane X receptor and represses its transcriptional activity. Mol Endocrinol. 2003;17:1693–703. doi: 10.1210/me.2002-0383. [DOI] [PubMed] [Google Scholar]
- 21.Wang L, Lee YK, Bundman D, Han Y, Thevananther S, Kim CS, et al. Redundant pathways for negative feedback regulation of bile acid production. Dev Cell. 2002;2:721–31. doi: 10.1016/s1534-5807(02)00187-9. [DOI] [PubMed] [Google Scholar]
- 22.Bae Y, Kemper JK, Kemper B. Repression of CAR-mediated transactivation of CYP2B genes by the orphan nuclear receptor, short heterodimer partner (SHP) DNA Cell Biol. 2004;23:81–91. doi: 10.1089/104454904322759894. [DOI] [PubMed] [Google Scholar]
- 23.Baes M, Gulick T, Choi HS, Martinoli MG, Simha D, Moore DD. A new orphan member of the nuclear hormone receptor superfamily that interacts with a subset of retinoic acid response elements. Mol Cell Biol. 1994;14:1544–52. doi: 10.1128/mcb.14.3.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Borgius LJ, Steffensen KR, Gustafsson JA, Treuter E. Glucocorticoid signaling is perturbed by the atypical orphan receptor and corepressor SHP. J Biol Chem. 2002;277:49761–6. doi: 10.1074/jbc.M205641200. [DOI] [PubMed] [Google Scholar]
- 25.Pavek P, Dvorak Z. Xenobiotic-induced transcriptional regulation of xenobiotic metabolizing enzymes of the cytochrome P450 superfamily in human extrahepatic tissues. Curr Drug Metab. 2008;9:129–43. doi: 10.2174/138920008783571774. [DOI] [PubMed] [Google Scholar]
- 26.Tirona RG, Kim RB. Nuclear receptors and drug disposition gene regulation. J Pharm Sci. 2005;94:1169–86. doi: 10.1002/jps.20324. [DOI] [PubMed] [Google Scholar]
- 27.Biswas A, Mani S, Redinbo MR, Krasowski MD, Li H, Ekins S. Elucidating the 'Jekyll and Hyde' nature of PXR: the case for discovering antagonists or allosteric antagonists. Pharm Res. 2009;26:1807–15. doi: 10.1007/s11095-009-9901-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orans J, Teotico DG, Redinbo MR. The nuclear xenobiotic receptor pregnane X receptor: recent insights and new challenges. Mol Endocrinol. 2005;19:2891–900. doi: 10.1210/me.2005-0156. [DOI] [PubMed] [Google Scholar]
- 29.Martinez-Jimenez CP, Jover R, Donato MT, Castell JV, Gomez-Lechon MJ. Transcriptional regulation and expression of CYP3A4 in hepatocytes. Curr Drug Metab. 2007;8:185–94. doi: 10.2174/138920007779815986. [DOI] [PubMed] [Google Scholar]
- 30.Burk O, Arnold KA, Nussler AK, Schaeffeler E, Efimova E, Avery BA, et al. Antimalarial artemisinin drugs induce cytochrome P450 and MDR1 expression by activation of xenosensors pregnane X receptor and constitutive androstane receptor. Mol Pharmacol. 2005;67:1954–65. doi: 10.1124/mol.104.009019. [DOI] [PubMed] [Google Scholar]
- 31.Svecova L, Vrzal R, Burysek L, Anzenbacherova E, Cerveny L, Grim J, et al. Azole antimycotics differentially affect rifampicin-induced pregnane X receptor-mediated CYP3A4 gene expression. Drug Metab Dispos. 2008;36:339–48. doi: 10.1124/dmd.107.018341. [DOI] [PubMed] [Google Scholar]
- 32.Bachleda P, Vrzal R, Pivnicka J, Cvek B, Dvorak Z. Examination of Zolpidem effects on AhR- and PXR-dependent expression of drug-metabolizing cytochromes P450 in primary cultures of human hepatocytes. Toxicol Lett. 2009;191:74–8. doi: 10.1016/j.toxlet.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 33.Dvorak Z, Vrzal R, Starha P, Klanicova A, Travnicek Z. Effects of dinuclear copper(II) complexes with 6-(benzylamino)purine derivatives on AhR and PXR dependent expression of cytochromes P450 CYP1A2 and CYP3A4 genes in primary cultures of human hepatocytes. Toxicol In Vitro. 2010;24:425–9. doi: 10.1016/j.tiv.2009.10.012. [DOI] [PubMed] [Google Scholar]
- 34.Cerveny L, Svecova L, Anzenbacherova E, Vrzal R, Staud F, Dvorak Z, et al. Valproic acid induces CYP3A4 and MDR1 gene expression by activation of constitutive androstane receptor and pregnane X receptor pathways. Drug Metab Dispos. 2007;35:1032–41. doi: 10.1124/dmd.106.014456. [DOI] [PubMed] [Google Scholar]
- 35.Pavek P, Pospechova K, Svecova L, Syrova Z, Stejskalova L, Blazkova J, et al. Intestinal cell-specific vitamin D receptor (VDR)-mediated transcriptional regulation of CYP3A4 gene. Biochem Pharmacol. 2010;79:277–87. doi: 10.1016/j.bcp.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 36.Wang DS, Jonker JW, Kato Y, Kusuhara H, Schinkel AH, Sugiyama Y. Involvement of organic cation transporter 1 in hepatic and intestinal distribution of metformin. J Pharmacol Exp Ther. 2002;302:510–5. doi: 10.1124/jpet.102.034140. [DOI] [PubMed] [Google Scholar]
- 37.Shu Y, Sheardown SA, Brown C, Owen RP, Zhang S, Castro RA, et al. Effect of genetic variation in the organic cation transporter 1 (OCT1) on metformin action. J Clin Invest. 2007;117:1422–31. doi: 10.1172/JCI30558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang H, Li H, Moore LB, Johnson MD, Maglich JM, Goodwin B, et al. The phytoestrogen coumestrol is a naturally occurring antagonist of the human pregnane X receptor. Mol Endocrinol. 2008;22:838–57. doi: 10.1210/me.2007-0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takeshita A, Taguchi M, Koibuchi N, Ozawa Y. Putative role of the orphan nuclear receptor SXR (steroid and xenobiotic receptor) in the mechanism of CYP3A4 inhibition by xenobiotics. J Biol Chem. 2002;277:32453–8. doi: 10.1074/jbc.M111245200. [DOI] [PubMed] [Google Scholar]
- 40.Pavek P, Cerveny L, Svecova L, Brysch M, Libra A, Vrzal R, et al. Examination of Glucocorticoid receptor alpha-mediated transcriptional regulation of P-glycoprotein, CYP3A4, and CYP2C9 genes in placental trophoblast cell lines. Placenta. 2007;28:1004–11. doi: 10.1016/j.placenta.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 41.Pospechova K, Rozehnal V, Stejskalova L, Vrzal R, Pospisilova N, Jamborova G, et al. Expression and activity of vitamin D receptor in the human placenta and in choriocarcinoma BeWo and JEG-3 cell lines. Mol Cell Endocrinol. 2009;299:178–87. doi: 10.1016/j.mce.2008.12.003. [DOI] [PubMed] [Google Scholar]
- 42.Huang H, Wang H, Sinz M, Zoeckler M, Staudinger J, Redinbo MR, et al. Inhibition of drug metabolism by blocking the activation of nuclear receptors by ketoconazole. Oncogene. 2007;26:258–68. doi: 10.1038/sj.onc.1209788. [DOI] [PubMed] [Google Scholar]
- 43.Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. Embo J. 2004;23:833–43. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu J, Wu RC, O'Malley BW. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat Rev Cancer. 2009;9:615–30. doi: 10.1038/nrc2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Blattler SM, Rencurel F, Kaufmann MR, Meyer UA. In the regulation of cytochrome P450 genes, phenobarbital targets LKB1 for necessary activation of AMP-activated protein kinase. Proc Natl Acad Sci U S A. 2007;104:1045–50. doi: 10.1073/pnas.0610216104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maglich JM, Watson J, McMillen PJ, Goodwin B, Willson TM, Moore JT. The nuclear receptor CAR is a regulator of thyroid hormone metabolism during caloric restriction. J Biol Chem. 2004;279:19832–8. doi: 10.1074/jbc.M313601200. [DOI] [PubMed] [Google Scholar]
- 47.Rencurel F, Stenhouse A, Hawley SA, Friedberg T, Hardie DG, Sutherland C, et al. AMP-activated protein kinase mediates phenobarbital induction of CYP2B gene expression in hepatocytes and a newly derived human hepatoma cell line. J Biol Chem. 2005;280:4367–73. doi: 10.1074/jbc.M412711200. [DOI] [PubMed] [Google Scholar]
- 48.Rencurel F, Foretz M, Kaufmann MR, Stroka D, Looser R, Leclerc I, et al. Stimulation of AMP-activated protein kinase is essential for the induction of drug metabolizing enzymes by phenobarbital in human and mouse liver. Mol Pharmacol. 2006;70:1925–34. doi: 10.1124/mol.106.029421. [DOI] [PubMed] [Google Scholar]