

Abstract
Mycobacterium tuberculosis (Mtb) is an intracellular pathogen that infects 10 million worldwide and kills 2 million people every year. The uptake and utilization of nutrients by Mtb within the host cell is still poorly understood, although lipids play an important role in Mtb persistence. The recent identification of a large regulon of cholesterol catabolic genes suggests that Mtb can use host sterol for infection and persistence. In this review, we report on recent progress in elucidation of the Mtb cholesterol catabolic reactions and their potential utility as targets for tuberculosis therapeutic agents.
Tuberculosis: an old disease but still a continuing threat
Tuberculosis, which has been a scourge of mankind for thousands of years, continues to be a major cause of death in developing countries and in environments where drug resistance can develop rapidly. The threat of tuberculosis has become greater over the past decades with the expansion from single drug resistance to multiple drug resistant strains and, most recently, the emergence of extreme drug resistance that threatens to overwhelm all available drugs. The importance of tuberculosis in developing countries, the potential of its renewal as a major threat in the developed world, and the determination of the genome of Mycobacterium tuberculosis (Mtb) [1] have intensified the search for new avenues in the design of therapeutic agents against Mtb.
Many of the available drugs for tuberculosis target its lipid biosynthetic pathways. Mtb has highly unusual cell wall lipids (Figure 1) and a relatively large fraction of its genes are devoted to the synthesis of these lipids. Although Mtb, similar to most bacteria, does not make sterols, it has been shown that cholesterol is both required for infection of macrophages and that the mycobacteria can employ cholesterol as a sole source of carbon. In this review, we summarize the available information on the involvement of cholesterol in the virulence and growth of Mtb, as well as recent work suggesting that the utilization of cholesterol could provide new targets for the development of novel therapeutic agents.
Figure 1. Schematic representation of the cell envelope of Mtb with lipidomic analysis of the surface lipids and PDIM.

The cell envelope of Mtb cells, from the inside to the outside, consists of a plasma membrane; a cell wall composed of three covalently linked macromolecules (peptidoglycan, arabinogalactan and mycolic acids) and non-covalently linked lipids and proteins; and a capsule consisting of polysaccharides, proteins and lipids. For the mass spectrometric analysis, total lipids were extracted from cells grown in the presence of glycerol, cholesterol or heptadeuterated cholesterol. As shown, cholesterol metabolism in Mtb cells increases the average mass of the methyl-branched lipid virulence factor PDIM, presumably due to a higher metabolic flux of propionate derived from cholesterol side-chain catabolism [22]. Abbreviations: PIM, phosphatidylinositol mannosides; PDIM, phthiocerol dimycoserate; PAT, polyacyltrehaloses; DAT, diacyltrehaloses; SL-1, sulfolipid-1s; TDM, trehalose-6,6′-dimycolates; TMM, trehalose-6-monomycolates.
Mtb does not synthesize cholesterol
Cholesterol is a major structural component of animal cell membranes, where it is required to maintain proper membrane permeability and fluidity. In addition, cholesterol is an important anabolic precursor for the biosynthesis of bile acids, vitamin D and steroid hormones. Although cholesterol is mainly synthesized in animals, small quantities are also produced in plants and fungi. Sterol biosynthesis in bacteria has been controversial, with the only unambiguously equivalent enzymes found in the proteobacterium, Methylococcus capsulatus, and in the planctomycete, Gemmata obscuriglobus [2, 3]. In contrast, Mtb apparently lacks the squalene monooxygenase and oxidosqualene cyclase that are absolutely essential for sterol biosynthesis. Interestingly, the Mtb genome encodes the gene for CYP51B1, a cytochrome P450 enzyme that catalyzes the 14α-demethylation of lanosterol to give the 8,14-diene, a key step in cholesterol biosynthesis [4]. Although bacterial CYP51 enzymes are well conserved across actinomycetes species, it is relevant that CYP51 from Streptomyces coelicolor A3(2), unlike its eukaryotic counterparts, is not essential for cell viability under in vitro growth conditions [5]. The physiological function of CYP51B1 enzymes in Mtb and other bacteria remains obscure [5].
Cholesterol is important for Mtb infection and persistence
Mtb can infect, grow and survive in the harsh environment of the macrophage and other host cells using mechanisms that are not yet well understood [6, 7]. Host cholesterol levels are thought to be involved in the development of Mtb infection [8], with high levels of cholesterol in the diet significantly enhancing the bacterial burden in the lung [9] and impairing immunity to Mtb [10]. Specifically, cholesterol is required for the phagocytosis of mycobacteria into macrophages [11, 12]. In fact, mycobacteria enter phagocytes through cholesterol-rich membrane microdomains [13]. Additionally, cholesterol is required to hold the host protein coronin 1 (TACO) on Mtb-infected phagosomes, leading to the inhibition of phagosome-lysozyme fusion [14]. This experimental evidence suggests an important role for cholesterol during Mtb infection and persistence.
Mtb imports and utilizes cholesterol
The acquisition and utilization of nutrients during Mtb infection is poorly understood, although it has been proposed that host lipids play an important role in Mtb survival [15]. Several lines of evidence suggest that pathogenic mycobacteria primarily use fatty acids rather than carbohydrates as carbon substrates during infection [16]. Respiration of Mtb in mouse lungs is strongly stimulated by fatty acids but is unresponsive to carbohydrates [16, 17]. An ABC-like transport system, mce4, was recently identified in Mtb that is involved in cholesterol import into Mtb [18]. Deletion of the mce4 operon in Mtb resulted in a growth defect when cholesterol was used as the primary source of carbon [18]. Mce4 loci are also found in many other actinomycetes species, including Rhodococcus jostii [19, 20], Mycobacterium smegmatis [21] and Mycobacterium bovis BCG [19], and these can also utilize cholesterol for growth. Using 14C-labeled cholesterol derivatives, it has been elegantly demonstrated that cholesterol is degraded by Mtb, with the carbon atoms from the sterol framework and the aliphatic side-chain going to energy production and lipid synthesis, respectively [18]. Using a lipidomic approach, Yang et al. showed that cholesterol metabolism in Mtb cells increases the average mass of the lipid virulence factor phthiocerol dimycocerosate (PDIM), presumably due to a higher metabolic flux of propionate derived from cholesterol catabolism [22]. Subsequently, heptadeuterated [25, 26, 26, 26, 27, 27, 27-D7]-cholesterol was used to establish the role of the cytochrome P450 encoded by the Mtb cyp125 gene in the degradation of cholesterol and incorporation of the cholesterol side-chain into the virulence factor PDIM [23] (Figure 1). Additionally, the block of cholesterol import in Δmce4 cells markedly attenuated infection in both activated macrophages and the mouse model of infection, confirming that cholesterol metabolism is important for the chronic phase of infection [18]. Screening of a transposon mutant library allowed the identification of another locus, igr, which is essential for growth in unactivated macrophages [24]. The igr operon consists of six genes, including a cytochrome P450 (cyp125), two probable acyl-CoA dehydrogenases (fadE28 and fadE29), two conserved hypothetical proteins (Rv3541-2c) and a putative lipid carrier protein (ltp2) (Figure 2). In a follow-up study, inactivation of the whole igr operon was shown to result in a marked defect in growth on cholesterol alone or in combination with glycerol, suggesting some form of cell intoxication by cholesterol or cholesterol-derived metabolites [25]. Moreover, Δigr cells exhibited a growth defect in the early phase of the mouse model of infection, an effect that is suppressed by mutating the sterol uptake Mce4 system. Together, these results indicate that cholesterol is important throughout infection and is crucial for Mtb persistence, but additionally show that ineffective breakdown of this sterol in the early phase of disease gives rise to a different form of attenuation.
Figure 2. The cholesterol catabolic genes of Mtb.

The transcriptional repressor KstR controls the expression of the majority of the genes clustered in two chromosomal segments (Rv3494c -Rv3547 and Rv3566c - Rv3574). Many other KstR-regulated genes are scattered throughout the Mtb genome, including Rv1106c. KstR2 controls the expression of a second cholesterol regulon in M. smegmatis [28], with the corresponding orthologs in Mtb genome (Rv3548c - Rv3565). Genes in the map are color-coded according to their assigned or proposed catalytic activity: red, uptake; side-chain degradation, magenta; degradation of rings A and B, green; β-oxidation, blue; transcriptional regulator, orange; unassigned function, black; not regulated by KstR or KstR2, grey. The genes encoding for enzymes involved specifically in the degradation of rings C and D are not unambiguously assigned yet. The catalytic functions of HsaEFG (Rv3534c-Rv3536c) in Mtb have not been investigated yet. Genes predicted [30, 31] or proven [18, 24, 27, 46, 57] to be essential for survival in macrophage cells and in the mouse model of infection are indicated by * and #, respectively. Abbreviations: ABC-transporter, ATP-binding cassette transporter; PE, protein containing Pro-Glu motifs; PE-PGRS, protein consisting of the PE domain followed by a C-terminal extension with multiple tandem repetitions of Gly-Gly-Ala or Gly-Gly-Asn; CHP, conserved hypothetical protein; HP, hypothetical protein; PPE, protein containing Pro-Pro-Glu motifs.
Mtb cholesterol degradation pathway
A growing body of evidence suggests that Mtb uses host cholesterol during infection as a source of carbon and energy. In their breakthrough study, Van der Geize et al. identified a complete suite of genes required for steroid degradation in R. jostii [19]. Interestingly, their bioinformatic analysis found that the 51 genes specifically expressed during growth on cholesterol, including those predicted to break down the sterol A and B rings, are likewise found within an 82-gene cluster in the Mtb and M. bovis BCG genomes. One of the Mtb genes thus identified, kstR (Rv3574), encodes a TetR-like transcriptional repressor. Inactivation of its homolog in the related saprophyte M. smegmatis derepressed the expression of 83 genes, as assayed by quantitative reverse transcriptase (RT)-PCR and microarrays, with 74 orthologs predicted to be similarly regulated in Mtb (Figure 2) [26]. The role of KstR as a cholesterol-responsive regulator in Mtb was later demonstrated by performing transcriptional profiling of cholesterol gene inactivation and in a kstR mutant of Mtb, corroborating the results obtained from the other actinomycetes [27]. More recently, a second transcriptional repressor, kstR2 (Rv3557c), was identified and shown to control the expression of a subset of 15 genes (Rv3548 – Rv3565) within the kstR regulon (Figure 2) [28]. Moreover, the observation that a number of genes in the kstR/kstR2 and cholesterol regulons are induced in macrophages [29] or are essential for infection [30, 31] strongly suggests that cholesterol catabolism is central to Mtb survival in vivo. In the following sections we summarize the cholesterol catabolic pathway in Mtb, which can be divided into two major phases: (i) initial degradation of the aliphatic side-chain, and (ii) subsequent degradation of the A–D rings (Figure 3). It remains unclear whether there is an obligatory order of the degradation reactions in Mtb. The evidence obtained from the literature on rhodococcal sterol catabolism suggests that intermediates of ring and side-chain degradation can be exchanged between the two pathway branches [32]. However, the situation seems to differ in Mtb, as blockage of the side-chain degradation resulted in the accumulation of cholest-4-en-3-one as a major metabolite [23], suggesting that the ring-degrading enzymes (e.g. KsaAB and HsaA-C) optimally act after the side-chain is removed.
Figure 3. Proposed degradation pathways and flux of metabolites derived from catabolism of the cholesterol aliphatic side-chain and ring nucleus.

All the enzymes for which a catalytic activity has been identified are labeled in red, whereas the putative β-oxidation enzymes are labeled in magenta. The carbon atoms derived from the ring nucleus are converted to CO2 via the tricarboxylic acid cycle (TCA), whereas the propionyl-CoA produced from the degradation of the side-chain is assimilated into mycobacterial lipids, including the virulence factor PDIM. The enzymes and the cholesterol metabolites are described in the text. The methylmalonyl pathway enzymes include a propionyl-CoA carboxylase (PCC) and a vitamin B12-dependent methylmalonyl-CoA mutase (MCM). The fate of DOHNAA is still unknown.
Side-chain degradation
Less is known about the degradation of the aliphatic side-chain of cholesterol than about the degradation of the sterol framework. The side-chain, as found also for the biosynthesis of bile acids in mammals, is generally accepted to be shortened by β-oxidation reactions (Figure 3). This is consistent with the identification of genes encoding putative β-oxidation enzymes in the cholesterol regulons of Mtb and Rhodococcus rhodochrous DSM43269. These genes include Mtb fadA5. Indeed, the thiolase enzyme encoded by fadA5 catalyzes the thiolysis of acetoacetyl-CoA in vitro and is required for growth on cholesterol [27]. This thiolase activity, which is consistent with removal of the side-chain by β-oxidation to yield androsterone metabolites [e.g. 4-androstenedione (AD) and 1,4-androstenedione (ADD)] [27], is required for virulence, especially during the late stage of mouse infection. Very recently, the roles of several genes [fadD17, fadD19, fadE26, fadE27, and ro04690 (DSM43269)] putatively involved in cholesterol side-chain degradation were investigated in the actinomycete R. rhodococcus DSM43269 [33]. Among those genes, fadD19 encodes a steroid-coenzyme A (CoA) ligase with an essential in vivo role in degradation of the side chains of C-24 branched-chain sterols, but interestingly not cholesterol. The high similarity (67%) between the Rhodococcus FadD19 (DSM43269) and Mtb FadD19 enzymes suggests that FadD19 also has an in vivo role in sterol metabolism in Mtb [33].
Sterol ring degradation
The first step of sterol ring degradation is the conversion cholesterol to cholest-4-en-3-one catalyzed by either a 3β-hydroxysteroid dehydrogenase (3β-HSD) or a cholesterol oxidase (ChoD) (Figure 3). Mtb Rv1106c encodes for a 3β-HSD enzyme that is a member of the short-chain dehydrogenase superfamily, uses NAD+ as a cofactor, and oxidizes cholesterol, pregnenolone, and dehydroepiandrosterone to their respective 3-keto-4-ene products [34]. Mtb ChoD, encoded by Rv3409c, which shares only 24% amino acid identity with the well-characterized cholesterol oxidases from Streptomyces and Rhodococcus, is required for Mtb virulence [35]. A significant amount of cholest-4-en-3-one was shown to temporarily accumulate in cholesterol-fed M. smegmatis cells overexpressing Mtb ChoD [35], but the disruption of Rv3409c in Mtb cells did not result in a marked in vitro defect of growth on cholesterol [36], suggesting that ChoD does not play a major role in cholesterol oxidation. In contrast, Mtb ΔRv1106c cells cannot grow on cholesterol as a primary source of carbon, but they did not show an attenuated phenotype during infection of macrophages and guinea pigs [36]. The authors therefore concluded that cholesterol is not an essential nutrient during infection, but the existence of low levels of a redundant 3β-HSD or cholesterol oxidase activity expressed during infection and/or the presence of host cholest-4-en-3-one [37] cannot be ruled out definitively. Redundant 3β-HSD activities were also proposed for the initiation of cholesterol degradation in M. smegmatis, although the enzyme similar to Mtb Rv1106c and encoded by MSMEG_5228 also appears to constitute the major activity [38].
KstD is a flavoprotein that catalyzes the trans-axial elimination of the C1(α) and C2(β) hydrogen atoms of the 3-ketosteroid A-ring (Figure 3) [39, 40]. The Rv3537 gene encodes a putative and unique Δ1KstD enzyme that is part of the cholesterol regulon [19] and is essential for survival in macrophages [31]. Phylogenetic analysis indicates that the Rv3537 gene product is an ortholog of Δ1KstD3 of Rhodococcus erythropolis SQ1 [41]. Targeted disruption of the Rv3537 (Δ1KstD) gene inhibited growth on cholesterol and resulted in accumulation of 9-hydroxy-4-androstene-3,17-dione [42]. Interestingly, heterologously expressed Mtb Δ1KstD displayed a clear preference for 3-ketosteroids with a saturated A-ring, with little (e.g. progesterone) or no (e.g. 4-androstenedione and cholest-4-en-3-one) detectable activity for 3-keto-Δ4 steroids [41]. In contrast, biochemical characterization of the rhodococcal Δ1KstD enzymes revealed relatively high levels of activity with 3-keto-Δ4 steroids [41, 43, 44]. This data suggests that Mtb may possess an unidentified reductase activity that saturates 3-keto-Δ4 steroids.
9-Hydroxylation of the 3-ketosteroid together with the C1–C2 dehydrogenation catalyzed by Δ1KstD are key steps leading to opening of the B-ring and aromatization of the A-ring via the unstable intermediate, 9-hydroxy-1,4, androstene-3-17-dione (9OHADD) (Figure 3). In actinomycetes, the 9-hydroxylation of 3-ketosteroids is catalyzed by KshAB, a two component Rieske oxygenase [45–48]. KshA (Rv3526), the oxygenase component, is a homotrimer containing a Rieske [2Fe-2S] cluster and mononuclear ferrous iron, whereas the reductase component KshB (Rv3571) is a monomeric protein containing a plant-type [2Fe-2S] cluster and flavin adenine dinucleotide (FAD) [45]. Consistent with a role in cholesterol utilization, a transposon mutant of kshA exhibited a strongly attenuated phenotype in the infection of interferon-γ-activated macrophages [31]. Moreover, Hu et al. reported that targeted disruption of either kshA or kshB resulted in mycobacterial cell death in the mouse model of infection [46]. Interestingly, inactivation of kshB, but not kshA, alters the biosynthesis of penta-acylated trehalose (PAT), a multimethyl-branched lipid class, suggesting that the reductase of KshB is involved in several processes.
In R. jostii RHA1 and Mtb, the hsaACDB genes are part a single operon within the cholesterol regulon [19]. The hsaA and hsaB genes encode an oxygenase and a reductase, respectively [49]. The two proteins act together as a flavin-dependent monooxygenase that hydroxylates 3-hydroxy-9,10-seconandrost-1,3,5(10)-triene-9,17-dione (3-HSA) to the catechol 3,4-dihydroxy-9,10-seconandrost-1,3,5(10)-triene-9,17-dione (3,4-DHSA) (Figure 3). Analysis of the crystal structure of ligand-free HsaA at 2.5 Å resolution revealed that this enzyme displays the same fold, flavin-binding site, and catalytic residues as p-hydroxyphenyl acetate hydroxylase. Additionally, deletion of hsaA in R. jostii RHA1 caused a growth defect on cholesterol, while the screening of a transposon mutant library identified hsaA as one of the Mtb genes required for growth in activated macrophages [31].
HsaC, the next enzyme in the pathway, is an iron-dependent extradiol dioxygenase that efficiently oxygenates and cleaves the catecholic cholesterol metabolite 3,4-DHSA to 4,5–9,10-diseco-3-hydroxy-5,9,17-trioxoandrosta-1(10),2-diene-4-oic acid (4,9-DSHA) (Figure 3). The structures of HsaC:DHSA complexes at 2.1 Å revealed two catechol-binding modes with the position of the bicyclo-alkanone moiety of DHSA being very similar in the two binding models, suggesting that this interaction is a determinant of the initial substrate-binding step. The inactivation of Mtb hsaC resulted in cell death when the cells were grown in the presence of cholesterol, presumably due to cell poisoning by the accumulated catechol metabolites. The infection of immunocompromised mice and wild-type guinea pigs with ΔhsaC cells also revealed a significant attenuation phenotype. From these observations, the authors concluded that cholesterol metabolism is most important during the chronic stage of infection, but also contributes to pathogen dissemination throughout the infection.
The catabolic gene, hsaD, was identified as one of the Mtb genes required for survival in macrophages [31]. HsaD is a member of the α/β hydrolase family involved in the aerobic degradation of aromatic compounds in microbes. This enzyme, for which a crystal structure at 2.35 Å is available [50, 51], catalyzes hydrolytic carbon-carbon bond cleavage of 4,9-DSHA to yield 9,17-dioxo-1,2,3,4,10,19-hexanorandrostan-5-oic acid (DOHNAA) and 2-hydroxy-hexa-2,4-dienoic acid (HHD) (Figure 3) [51]. HHD is finally metabolized to tricarboxylic acid cycle intermediates and propionyl-CoA, whereas it is unknown if Mtb further degrades DOHNAA, which corresponds to the C and D-ring fragment. While much in known about the steps involved in sterol ring degradation, substantial gaps exist in our understanding of this pathway. Nevertheless, the structural data obtained for HsaA, HsaC and HsaD provides insights into the binding of steroid substrates that should facilitate inhibitor design.
Role of Mtb P450 in cholesterol catabolism
The saturated side-chain of cholesterol must be chemically functionalized at the ω-position before it can enter into the Mtb β-oxidation pathway (Figure 3). A minimum of four chemical steps are necessary to prepare the side-chain for β-oxidation. The first three steps, sequential oxidations of the cholesterol side-chain to the terminal alcohol, aldehyde, and acid, are catalyzed by cytochrome P450 enzymes. Three Mtb P450 enzymes, CYP125 (Rv3545c), CYP142 (Rv3518c) and CYP124 (Rv2266), are capable of oxidizing the side-chains of cholesterol and cholest-4-en-3-one. The final ATP-dependent step is catalyzed by a sterol-CoA ligase [52].
Cytochrome P450 enzymes catalyzing steroid side chain oxidations
The first oxidation introduces a hydroxy group onto the steroid side-chain, the second oxidizes the alcohol to the aldehyde, and the third converts the aldehyde to a carboxylic acid. In principle, the initial oxidation could occur at a primary, secondary or tertiary position, as all three are found in the cholesterol side-chain, although the ease of hydroxylation depends on the relative C-H bond dissociation energies and thus decreases in the order 3° > 2° ≫ 1°. Nevertheless, only oxidation of the primary C-H bond to give the ω-oxidation product is observed [53]. These P450 enzymes, which have evolved to selectively oxidize the relatively unreactive terminal methyl group, enforce a strict regiospecificity by preventing the more reactive carbon of the side-chain from being properly positioned relative to the heme iron atom [53]. However, as outlined below, there are distinct differences in the gene induction, protein expression, stereochemical preferences and catalytic efficiencies of the three enzymes able to oxidize cholesterol and cholest-4-en-3-one.
Which P450 enzymes are involved?
Comparison of the genome sequences of Mtb CDC1551 and H37Rv reveals key differences in the cytochrome P450 loci. In CDC1551, a 639-bp deletion encompassing the promoter region and the sequence encoding the first 18 amino acids of the protein is found upstream of the cyp142 (Rv3518c) gene, indicating that this strain does not produce CYP142. The cyp124 genes of H37Rv and CDC1551 only differ by two amino acids, G25D and Y75N, respectively. However, no notable differences in catalytic activity are observed between the CYP124 variants. In contrast, the cyp125 genes are identical in both strains. In vitro, CYP125, CYP142 and CYP124 catalyze the sequential oxidation of the hydrocarbon side-chain of cholesterol to the carboxylic acid. The 2nd order rate constants (kcat/KM) for terminal hydroxylation of the cholesterol and cholest-4-en-3-one side-chains establish that CYP125 is a slightly more efficient catalyst than CYP142 [54], and both are much more efficient than CYP124 [54]. The characterization of CYP125, reported independently by four groups, includes x-ray crystal structures [23, 55], biochemical studies [23, 32, 55, 56], and in vivo genetic analyses that probe the loss and gain of this P450 function in different virulent clinical strains and R. jostii RHA1 [23, 32, 56]. Two other Mtb P450 enzymes, CYP141 and CYP130, were found not to catalyze the oxidation of these sterols [54].
Oxidation can occur at either of the two terminal methyl groups, producing either the (25S)- and/or (25R)-26 hydroxy sterols. The stereochemical preferences of these enzymes differ: CYP125 preferentially forms the (25S)-26-hydroxy product, whereas CYP142 and CYP124 produce the (25R)-26-hydroxy configuration [54]. It is unclear whether there are any physiological consequences of these different stereochemical preferences.
Several lines of evidence indicate that CYP125 and CYP142 are induced when Mtb is grown on cholesterol as the sole carbon source [23, 27, 54]. Immunoblots confirmed that the CYP125 and CYP142 proteins are expressed and induced in cells grown on cholesterol, whereas CYP124 was not detectably expressed under these conditions [54]. Interestingly, cyp142 is able to fully complement the loss of cyp125 in CDC1551 Mtb cells when a chromosomal copy is introduced into the Δcyp125 knockout cells. CYP125 is the primary P450 responsible for the initiation of cholesterol side-chain degradation in Mtb. CYP142 can substitute for CYP125 in H37Rv but not CDC1551 cells, as the cyp142 gene in the latter is inactivated by a mutation. Similar to CYP125 and CYP142, CYP124 can catalyze sequential oxidation of the cholesterol and cholest-4-en-3-one side-chains, but CYP124 expression is not induced by cholesterol [54]. In agreement with the in vitro kinetic data, CYP124 can only partially reverse the abnormal growth phenotype of Δcyp125 cells grown on cholesterol. Concomitant with this partial growth restoration, cholest-4-en-3-one was found to accumulate in the Δcyp125 CYP124 overexpressing cells [54]. The presence of multiple cytochrome P450 isoforms capable of oxidizing the sterol side-chain and differences in their expression highlight some of the potential challenges of drug-targeting critical pathways of Mtb and the need for understanding the biochemical and genetic basis for the differences in clinically relevant strains.
Toxicity of cholesterol metabolites
Inhibition of the cholesterol catabolic pathways not only can cause carbon starvation, but blockage at certain steps has also been shown to result in cell death or bacteriostasis. For instance, the Mtb ΔhsaC mutant not only failed to grow on cholesterol, but the cells developed a pink color, indicating an accumulation of catechols and their conversion to colored o-benzoquinones and condensation products [19, 57]. Additionally, a loss of cell viability was observed when ΔhsaC cells were grown in the presence of cholesterol, suggesting that the catechol derivatives are toxic to Mtb cells [57]. A growth inhibitory effect of cholesterol metabolism that depends on cholesterol import was observed when Δigr cells were incubated with both cholesterol and glycerol [25]. The same inhibitory effect was observed when cyp125, which is part of the igr operon, was deleted in similarly cultured Mtb CDC1551 cells. Importantly, cholest-4-en-3-one, which was shown to accumulate in high amounts in cyp125 mutant cells [23, 56], was toxic to wild-type Mtb CDC1551, Erdman, and H37Rv strains grown in media containing a second carbon source [23].
A highly speculative mechanism for the toxicity associated with the accumulation of cholest-4-en-3-one is an alteration of the cell membrane integrity. In eukaryotic plasma membrane models, it was shown that cholesterol can participate in strong and close packing with saturated lipids, a critical feature required for the formation of lipid domains, with the latter implicated in numerous biological processes, including signal transduction events, and toxin entry into cells [58]. It was shown that cholest-4-en-3-one, for which the 3-OH group is replaced by a keto group, inhibits lipid domain formation, thus altering the plasma membranes properties [58, 59]. When cultivated in the presence of cholesterol and a second carbon source, Mtb is able to accumulate cholesterol in the free-lipid zone of its cell wall, resulting in a decreased permeability for the primary antitubercular drug, rifampin [42]. From these observations, it can be speculated that high levels of cholest-4-en-3-one might render the cell wall more permeable to drugs and xenobiotics. Whatever the mechanism, the identification of cholest-4-en-3-one as a toxic agent in Mtb opens up the intriguing possibility that elucidation of its site(s) of action could reveal new targets for antitubercular drug design.
Cholesterol catabolism inhibitors
In view of the importance of cholesterol catabolism in Mtb infection and persistence, targeting this pathway could represent a fruitful avenue for the development of new therapeutic agents. Some of the important questions that require attention are outlined in Box 1. The antifungal action of azole drugs has been shown to result from inhibition of sterol biosynthesis at the level of CYP51. Azole drugs, most notably econazole, exhibit antimycobacterial activities against both latent Mtb and multidrug-resistant strains [60, 61], a finding that implicates one or more cytochrome P450 enzymes as targets (Table 1). In this regard, all the Mtb cytochrome P450 enzymes characterized to date, including CYP125 and CYP142, bind azole drugs with low- to submicromolar affinities (Table 1) [62–67]. The crystal structure of CYP125 in complex with econazole, solved at a resolution of 2.2 Å, showed that although the ligand occupies most of the hydrophobic binding pocket, the azole group does not approach the heme iron due to the funnel shape of the active site [55]. Compound LP10 (α-[(4-methylcyclohexyl)carbonyl amino]-N-4-pyridinyl-1H-indole-3-propanamide), previously identified as a potent inhibitor of CYP51 from Trypanosoma cruzi co-crystallized with CYP125 and the crystal structure revealed that the pyridine group of the inhibitor is not coordinated to the heme iron, but instead stabilizes the coordination of a water molecule through H-bonding [68]. These last results provide insights into the structural requirements for developing selective CYP125 inhibitors.
Box 1. Outstanding questions.
Are the C- and D- rings and the side-chain of cholesterol completely degraded, and if so, how?
Besides leading to an increased mass of the methyl-branched lipid virulence factor, PDIM, does the utilization of host cholesterol result in the biosynthesis of unidentified but important lipids in vivo?
Does Mtb possess functionally redundant and as yet unidentified cholesterol oxidase or dehydrogenase activities? The apparent dispensability of the 3β-HSD encoded by Rv1106c for infection and the lack of detectable cholesterol oxidase activity catalyzed by ChoD (Rv3409c) in vitro suggest that cholesterol is not an essential nutrient during infection. Could cholest-4-en-3-one be also produced by the host at the site of infection?
Is cholesterol simply metabolized as a source of carbon and energy for growth or do cholesterol-derived metabolites secreted by Mtb cells have any physiological effects through host-pathogen interactions?
What is the target of cholest-4-en-3-one that results in a bacteriostatic effect? The elucidation of this inhibition effect could potentially lead to the identification of essential activities that could also be useful for the development of new therapeutics.
Is it possible to design agents that specifically target Mtb enzymes and not the host isoforms? Although antifungal azole drugs are known to target fungal cytochrome P450 enzymes most of them cannot be administered orally as it could result in serious side effects due to inhibition of the human cytochrome P450 complement.
Table 1.
Apparent dissociation constant (KD) for select azole drugs against Mtb sterol-oxidizing cytochrome P450 enzymes and minimum inhibitor concentration (MIC) values
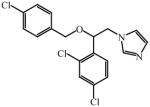
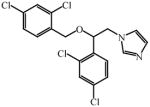
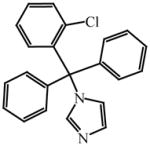
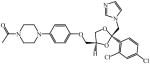
Very recently, a series of 6-azasteroid inhibitors were employed as a tool set to study the structure-activity relationships (SAR) for Mtb 3β-HSD encoded by Rv1106c [69]. Azasteroids are validated therapeutics that inhibit enzymes involved in steroid biosynthesis. For instance, finasteride inhibits the 5α-reductase-catalyzed production of dihydrotestosterone from testosterone, but also cross-reacts with human 3β-HSDs [70, 71]. Despite a low amino acid sequence identity (~29%) with the human isoforms, the SAR study indicated that the 6-aza version of cholesterol is the tightest binding competitive inhibitor (Ki = 100 nM) of the steroid substrate, an observation consistent with cholesterol being the preferred substrate for Mtb 3β-HSD [69].
Although the exact role of cholesterol metabolism during human infection remains to be determined, the development of new inhibitors targeting cholesterol degrading enzymes, especially of those whose inactivation results in the accumulation of toxic metabolites (e.g. igr, cyp125 and hsaC mutants), may provide a route to countering the emergence of multidrug resistant strains. For this, more structural and biochemical data are required to facilitate the design of new therapeutics.
Concluding remarks
Despite the fact that Mtb does not make any sterols, it retains one cytochrome P450 enzyme (CYP51) with high sequence identity and the ability to catalyze the same specific reaction as lanosterol 14α-demethylase, a key enzyme in sterol biosynthesis. Furthermore, roles for host cholesterol and its metabolism throughout the infection, particularly during the chronic phase of infection, are now well established. Cholesterol is required for the Mtb infection of macrophages and serves as a carbon source for growth of the mycobacteria. Degradation of the cholesterol molecule is initiated by hydroxylation of the sterol side-chain either by CYP125 or CYP142. In the absence of these enzymes, the cholesterol metabolite cholest-4-en-3-one accumulates and has a bacteriostatic effect. Catechol intermediates that are also toxic accumulate if sterol degradation is interrupted at the level of HsaC, the enzyme that fragments ring A of the sterol. The cholesterol degradation pathway thus provides targets for the endogenous generation of toxic molecules. Furthermore, azole drugs that normally target cytochrome P450 enzymes have been shown to inhibit the growth of Mtb in both the proliferating and resting states. These collective results suggest that the cholesterol degradation pathway could prove to be a fertile ground for the development of antituberculosis agents.
Acknowledgments
The preparation of this review and the work from the author’s laboratory were supported by National Institutes of Health Grant AI07824. J.B.J. was supported by a Heiser Postdoctoral Fellowship for Research in Leprosy and Tuberculosis sponsored by the New York Community Trust.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cole ST, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 2.Lamb DC, et al. Lanosterol biosynthesis in the prokaryote Methylococcus capsulatus: insight into the evolution of sterol biosynthesis. Mol Biol Evol. 2007;24:1714–1721. doi: 10.1093/molbev/msm090. [DOI] [PubMed] [Google Scholar]
- 3.Pearson A, et al. Phylogenetic and biochemical evidence for sterol synthesis in the bacterium Gemmata obscuriglobus. Proc Natl Acad Sci U S A. 2003;100:15352–15357. doi: 10.1073/pnas.2536559100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bellamine A, et al. Characterization and catalytic properties of the sterol 14alpha-demethylase from Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 1999;96:8937–8942. doi: 10.1073/pnas.96.16.8937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lamb DC, et al. Sterol 14alpha-demethylase activity in Streptomyces coelicolor A3(2) is associated with an unusual member of the CYP51 gene family. Biochem J. 2002;364:555–562. doi: 10.1042/BJ20011380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clark-Curtiss JE, Haydel SE. Molecular genetics of Mycobacterium tuberculosis pathogenesis. Annu Rev Microbiol. 2003;57:517–549. doi: 10.1146/annurev.micro.57.030502.090903. [DOI] [PubMed] [Google Scholar]
- 7.Koul A, et al. Interplay between mycobacteria and host signalling pathways. Nat Rev Microbiol. 2004;2:189–202. doi: 10.1038/nrmicro840. [DOI] [PubMed] [Google Scholar]
- 8.Kim MJ, et al. Caseation of human tuberculosis granulomas correlates with elevated host lipid metabolism. EMBO Mol Med. 2010;2:258–274. doi: 10.1002/emmm.201000079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schafer G, et al. The role of scavenger receptor B1 in infection with Mycobacterium tuberculosis in a murine model. PLoS One. 2009;4:e8448. doi: 10.1371/journal.pone.0008448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Martens GW, et al. Hypercholesterolemia impairs immunity to tuberculosis. Infect Immun. 2008;76:3464–3472. doi: 10.1128/IAI.00037-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gatfield J, Pieters J. Essential role for cholesterol in entry of mycobacteria into macrophages. Science. 2000;288:1647–1650. doi: 10.1126/science.288.5471.1647. [DOI] [PubMed] [Google Scholar]
- 12.Peyron P, et al. Nonopsonic phagocytosis of Mycobacterium kansasii by human neutrophils depends on cholesterol and is mediated by CR3 associated with glycosylphosphatidylinositol-anchored proteins. J Immunol. 2000;165:5186–5191. doi: 10.4049/jimmunol.165.9.5186. [DOI] [PubMed] [Google Scholar]
- 13.Munoz S, et al. Mycobacterium tuberculosis entry into mast cells through cholesterol-rich membrane microdomains. Scand J Immunol. 2009;70:256–263. doi: 10.1111/j.1365-3083.2009.02295.x. [DOI] [PubMed] [Google Scholar]
- 14.Nguyen L, Pieters J. The trojan horse: survival tactics of pathogenic mycobacteria in macrophages. Trends Cell Biol. 2005;15:269–276. doi: 10.1016/j.tcb.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 15.Savvi S, et al. Functional characterization of a vitamin B12-dependent methylmalonyl pathway in Mycobacterium tuberculosis: implications for propionate metabolism during growth on fatty acids. J B acteriol. 2008;190:3886–3895. doi: 10.1128/JB.01767-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Munoz-Elias EJ, McKinney JD. Mycobacterium tuberculosis isocitrate lyases 1 and 2 are jointly required for in vivo growth and virulence. Nat Med. 2005;11:638–644. doi: 10.1038/nm1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bloch H, Segal W. Biochemical differentiation of Mycobacterium tuberculosis grown in vivo and in vitro. J Bacteriol. 1956;72:132–141. doi: 10.1128/jb.72.2.132-141.1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci U S A. 2008;105:4376–4380. doi: 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van der Geize R, et al. A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc Natl Acad Sci U S A. 2007;104:1947–1952. doi: 10.1073/pnas.0605728104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mohn WW, et al. The actinobacterial mce4 locus encodes a steroid transporter. J Biol Chem. 2008;283:35368–35374. doi: 10.1074/jbc.M805496200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Av-Gay Y, Sobouti R. Cholesterol is accumulated by mycobacteria but its degradation is limited to non-pathogenic fast-growing mycobacteria. Can J Microbiol. 2000;46:826–831. [PubMed] [Google Scholar]
- 22.Yang X, et al. Cholesterol metabolism increases the metabolic pool of propionate in Mycobacterium tuberculosis. Biochemistry. 2009;48:3819–3821. doi: 10.1021/bi9005418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ouellet H, et al. Mycobacterium tuberculosis CYP125A1, a steroid C27 monooxygenase that detoxifies intracellularly generated cholest-4-en-3-one. Mol Microbiol. 2010;77:730–742. doi: 10.1111/j.1365-2958.2010.07243.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang JC, et al. Identification of mycobacterial genes that alter growth and pathology in macrophages and in mice. J Infect Dis. 2007;196:788–795. doi: 10.1086/520089. [DOI] [PubMed] [Google Scholar]
- 25.Chang JC, et al. igr genes and Mycobacterium tuberculosis cholesterol metabolism. J Bacteriol. 2009;191:5232–5239. doi: 10.1128/JB.00452-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kendall SL, et al. A highly conserved transcriptional repressor controls a large regulon involved in lipid degradation in Mycobacterium smegmatis and Mycobacterium tuberculosis. Mol Microbiol. 2007;65:684–699. doi: 10.1111/j.1365-2958.2007.05827.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nesbitt NM, et al. A thiolase of Mycobacterium tuberculosis is required for virulence and production of androstenedione and androstadienedione from cholesterol. Infect Immun. 2010;78:275–282. doi: 10.1128/IAI.00893-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kendall SL, et al. Cholesterol utilization in mycobacteria is controlled by two TetR-type transcriptional regulators: kstR and kstR2. Microbiology. 2010;156:1362–1371. doi: 10.1099/mic.0.034538-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schnappinger D, et al. Transcriptional adaptation of Mycobacterium tuberculosis within macrophages: Insights into the phagosomal environment. J Exp Med. 2003;198:693–704. doi: 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sassetti CM, Rubin EJ. Genetic requirements for mycobacterial survival during infection. Proc Natl Acad Sci U S A. 2003;100:12989–12994. doi: 10.1073/pnas.2134250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rengarajan J, et al. Genome-wide requirements for Mycobacterium tuberculosis adaptation and survival in macrophages. Proc Natl Acad Sci U S A. 2005;102:8327–8332. doi: 10.1073/pnas.0503272102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosloniec KZ, et al. Cytochrome P450 125 (CYP125) catalyses C26-hydroxylation to initiate sterol side-chain degradation in Rhodococcus jostii RHA1. Mol Microbiol. 2009;74:1031–1043. doi: 10.1111/j.1365-2958.2009.06915.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wilbrink MH, et al. FadD19 of Rhodococcus rhodochrous DSM43269, a steroid-coenzyme A ligase essential for egradation of C-24 branched sterol side chains. Appl Environ Microbiol. 77:4455–4464. doi: 10.1128/AEM.00380-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang X, et al. Rv1106c from Mycobacterium tuberculosis is a 3beta-hydroxysteroid dehydrogenase. Biochemistry. 2007;46:9058–9067. doi: 10.1021/bi700688x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brzostek A, et al. Cholesterol oxidase is required for virulence of Mycobacterium tuberculosis. FEMS Microbiol Lett. 2007;275:106–112. doi: 10.1111/j.1574-6968.2007.00865.x. [DOI] [PubMed] [Google Scholar]
- 36.Yang X, et al. Cholesterol is not an essential source of nutrition for Mycobacterium tuberculosis during infection. J Bacteriol. 2011;193:1473–1476. doi: 10.1128/JB.01210-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tabas I, et al. Acyl coenzyme A:cholesterol acyl transferase in macrophages utilizes a cellular pool of cholesterol oxidase-accessible cholesterol as substrate. J Biol Chem. 1988;263:1266–1272. [PubMed] [Google Scholar]
- 38.Uhia I, et al. Initial step in the catabolism of cholesterol by Mycobacterium smegmatis mc(2)155. Environ Microbiol. 2011;4:943–959. doi: 10.1111/j.1462-2920.2010.02398.x. [DOI] [PubMed] [Google Scholar]
- 39.Itagaki E, et al. Spectral properties of 3-ketosteroid-delta 1-dehydrogenase from Nocardia corallina. Biochim Biophys Acta. 1990;1040:281–286. doi: 10.1016/0167-4838(90)90088-w. [DOI] [PubMed] [Google Scholar]
- 40.Itagaki E, et al. Purification and characterization of 3-ketosteroid-delta 1-dehydrogenase from Nocardia corallina. Biochim Biophys Acta. 1990;1038:60–67. doi: 10.1016/0167-4838(90)90010-d. [DOI] [PubMed] [Google Scholar]
- 41.Knol J, et al. 3-Keto-5alpha-steroid Delta(1)-dehydrogenase from Rhodococcus erythropolis SQ1 and its orthologue in Mycobacterium tuberculosis H37Rv are highly specific enzymes that function in cholesterol catabolism. Biochem J. 2008;410:339–346. doi: 10.1042/BJ20071130. [DOI] [PubMed] [Google Scholar]
- 42.Brzostek A, et al. Mycobacterium tuberculosis is able to accumulate and utilize cholesterol. J Bacteriol. 2009;191:6584–6591. doi: 10.1128/JB.00488-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van der Geize R, et al. Molecular and functional characterization of the kstD2 gene of Rhodococcus erythropolis SQ1 encoding a second 3-ketosteroid Delta(1)-dehydrogenase isoenzyme. Microbiology. 2002;148:3285–3292. doi: 10.1099/00221287-148-10-3285. [DOI] [PubMed] [Google Scholar]
- 44.van der Geize R, et al. Unmarked gene deletion mutagenesis of kstD, encoding 3-ketosteroid Delta1-dehydrogenase, in Rhodococcus erythropolis SQ1 using sacB as counter-selectable marker. FEMS Microbiol Lett. 2001;205:197–202. doi: 10.1016/s0378-1097(01)00464-5. [DOI] [PubMed] [Google Scholar]
- 45.Capyk JK, et al. Characterization of 3-ketosteroid 9-alpha-hydroxylase, a Rieske oxygenase in the cholesterol degradation pathway of Mycobacterium tuberculosis. J Biol Chem. 2009;284:9937–9946. doi: 10.1074/jbc.M900719200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hu Y, et al. 3-Ketosteroid 9alpha-hydroxylase is an essential factor in the pathogenesis of Mycobacterium tuberculosis. Mol Microbiol. 2010;75:107–121. doi: 10.1111/j.1365-2958.2009.06957.x. [DOI] [PubMed] [Google Scholar]
- 47.van der Geize R, et al. Characterization of a second Rhodococcus erythropolis SQ1 3-ketosteroid 9alpha-hydroxylase activity comprising a terminal oxygenase homologue, KshA2, active with oxygenase-reductase component KshB. Appl Environ Microbiol. 2008;74:7197–7203. doi: 10.1128/AEM.00888-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Andor A, et al. Generation of useful insertionally blocked sterol degradation pathway mutants of fast-growing mycobacteria and cloning, characterization, and expression of the terminal oxygenase of the 3-ketosteroid 9alpha-hydroxylase in Mycobacterium smegmatis mc(2)155. Appl Environ Microbiol. 2006;72:6554–6559. doi: 10.1128/AEM.00941-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dresen C, et al. A flavin-dependent monooxygenase from Mycobacterium tuberculosis involved in cholesterol catabolism. J Biol Chem. 2010;285:22264–22275. doi: 10.1074/jbc.M109.099028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lack N, et al. Structure of HsaD, a steroid-degrading hydrolase, from Mycobacterium tuberculosis. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2008;64:2–7. doi: 10.1107/S1744309107065931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lack NA, et al. Characterization of a carbon-carbon hydrolase from Mycobacterium tuberculosis involved in cholesterol metabolism. J Biol Chem. 2010;285:434–443. doi: 10.1074/jbc.M109.058081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kunau WH, et al. Beta-oxidation of fatty acids in mitochondria, peroxisomes, and bacteria: a century of continued progress. Prog Lipid Res. 1995;34:267–342. doi: 10.1016/0163-7827(95)00011-9. [DOI] [PubMed] [Google Scholar]
- 53.Johnston JB, et al. Structural control of cytochrome P450-catalyzed omega-hydroxylation. Arch Biochem Biophys. 2011;507:86–94. doi: 10.1016/j.abb.2010.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Johnston JB, et al. Functional redundancy of steroid C26-monooxygenase activity in Mycobacterium tuberculosis revealed by biochemical and genetic analyses. J Biol Chem. 2010;285:36352–36360. doi: 10.1074/jbc.M110.161117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McLean KJ, et al. The structure of Mycobacterium tuberculosis CYP125: molecular basis for cholesterol binding in a P450 needed for host infection. J Biol Chem. 2009;284:35524–35533. doi: 10.1074/jbc.M109.032706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Capyk JK, et al. Mycobacterial cytochrome p450 125 (cyp125) catalyzes the terminal hydroxylation of c27 steroids. J Biol Chem. 2009;284:35534–35542. doi: 10.1074/jbc.M109.072132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yam KC, et al. Studies of a ring-cleaving dioxygenase illuminate the role of cholesterol metabolism in the pathogenesis of Mycobacterium tuberculosis. PLoS Pathog. 2009;5:e1000344. doi: 10.1371/journal.ppat.1000344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu X, London E. The effect of sterol structure on membrane lipid domains reveals how cholesterol can induce lipid domain formation. Biochemistry. 2000;39:843–849. doi: 10.1021/bi992543v. [DOI] [PubMed] [Google Scholar]
- 59.Beattie ME, et al. Sterol structure determines miscibility versus melting transitions in lipid vesicles. Biophys J. 2005;89:1760–1768. doi: 10.1529/biophysj.104.049635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ahmad Z, et al. The potential of azole antifungals against latent/persistent tuberculosis. FEMS Microbiol Lett. 2006;258:200–203. doi: 10.1111/j.1574-6968.2006.00224.x. [DOI] [PubMed] [Google Scholar]
- 61.Ahmad Z, et al. Antimycobacterial activity of econazole against multidrug-resistant strains of Mycobacterium tuberculosis. Int J Antimicrob Agents. 2006;28:543–544. doi: 10.1016/j.ijantimicag.2006.07.028. [DOI] [PubMed] [Google Scholar]
- 62.Driscoll MD, et al. Expression and characterization of Mycobacterium tuberculosis CYP144: common themes and lessons learned in the Mycobacterium tuberculosis P450 enzyme family. Biochim Biophys Acta. 2011;1814:76–87. doi: 10.1016/j.bbapap.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 63.Driscoll MD, et al. Structural and biochemical characterization of Mycobacterium tuberculosis CYP142: evidence for multiple cholesterol 27-hydroxylase activities in a human pathogen. J Biol Chem. 2010;285:38270–38282. doi: 10.1074/jbc.M110.164293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Johnston JB, et al. Biochemical and structural characterization of CYP124: a methyl-branched lipid omega-hydroxylase from Mycobacterium tuberculosis. Proc Natl Acad Sci U S A. 2009;106:20687–20692. doi: 10.1073/pnas.0907398106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McLean KJ, et al. Expression, purification and spectroscopic characterization of the cytochrome P450 CYP121 from Mycobacterium tuberculosis. J Inorg Biochem. 2002;91:527–541. doi: 10.1016/s0162-0134(02)00479-8. [DOI] [PubMed] [Google Scholar]
- 66.McLean KJ, et al. Azole antifungals are potent inhibitors of cytochrome P450 mono-oxygenases and bacterial growth in mycobacteria and streptomycetes. Microbiology. 2002;148:2937–2949. doi: 10.1099/00221287-148-10-2937. [DOI] [PubMed] [Google Scholar]
- 67.Ouellet H, et al. Mycobacterium tuberculosis CYP130: crystal structure, biophysical characterization, and interactions with antifungal azole drugs. J Biol Chem. 2008;283:5069–5080. doi: 10.1074/jbc.M708734200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ouellet H, et al. Reverse type I inhibitor of Mycobacterium tuberculosis CYP125A1. Bioorg Med Chem Lett. 2011;21:332–337. doi: 10.1016/j.bmcl.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thomas ST, et al. Inhibition of the Mycobacterium tuberculosis 3beta-hydroxysteroid dehydrogenase by azasteroids. Bioorg Med Chem Lett. 2011;21:2216–2219. doi: 10.1016/j.bmcl.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Frye SV, et al. 6-Azasteroids: potent dual inhibitors of human type 1 and 2 steroid 5 alpha-reductase. J Med Chem. 1993;36:4313–4315. doi: 10.1021/jm00078a022. [DOI] [PubMed] [Google Scholar]
- 71.Frye SV, et al. 6-Azasteroids: structure-activity relationships for inhibition of type 1 and 2 human 5 alpha-reductase and human adrenal 3 beta-hydroxy-delta 5-steroid dehydrogenase/3-keto-delta 5-steroid isomerase. J Med Chem. 1994;37:2352–2360. doi: 10.1021/jm00041a014. [DOI] [PubMed] [Google Scholar]
- 72.McLean KJ, et al. Characterization of active site structure in CYP121. A cytochrome P450 essential for viability of Mycobacterium tuberculosis H37Rv. J Biol Chem. 2008;283:33406–33416. doi: 10.1074/jbc.M802115200. [DOI] [PMC free article] [PubMed] [Google Scholar]