

Abstract
RIP140 (Receptor-interacting protein 140) is highly expressed in mature adipocytes and functions as a co-repressor for gene expression involved in lipid and glucose metabolism. In adipocytes, activated PKCε (Protein kinase C epsilon) phosphorylates nuclear RIP140 which is then subsequently arginine methylated and exported to the cytoplasm. In the cytoplasm, RI140 can elicit additional activities. Here we report a new functional role for cytoplasmic RIP140 in adipocyte in regulating adiponectin secretion. Targeting cytoplasmic RIP140 by knocking down RIP140 itself or its nuclear export trigger, PKCε, promotes the secretion of adiponectin without affecting the production or oligomerization of adiponectin. Consequentially, conditioned media from either RIP140- or PKCε-silenced adipocytes, which contain higher levels of adiponectin, enhance glucose uptake in C2C12 cells and reduce gluconeogenesis in HepG2 cells. Further, these effects can be inhibited by an adiponectin-neutralizing antibody. The effect of cytoplasmic RIP140 in regulating adiponectin secretion is via interacting with AS160, a known RIP140-interacting protein. This study reveals a new functional role for cytoplasmic RIP140 in modulating adiponectin vesicle secretion, and suggests that targeting cytoplasmic RIP140 may be a potentially effective therapeutic strategy to improve adiponectin secretion and possibly to manage metabolic disorders.
Keywords: RIP140, cytoplasmic, Adiponectin, Insulin sensitivity, Glucose uptake, Gluconeogenesis
1. Introduction
In recent years obesity has become a worldwide epidemic. A major complication of obesity is type 2 diabetes mellitus (T2DM). The hallmark of this condition is the development of insulin resistance in adipose, muscle and liver [1]. In healthy individuals, adipocytes store lipid and secretes adipokines, and these adipokines are important in the regulation of metabolism. But in diabetic subjects, there is adipocyte dysfunction with changes in the profile of secreted adipokines, increase of lipolysis and reduction in glucose uptake. These changes may worsen the disease state leading to such complications as hypertension, atherosclerosis and cardiomyopathy [1, 2]. Although inflammation and endoplasmic reticulum (ER) stress could cause adipocyte dysfunction, the exact etiology of many of these pathophysiological processes is unclear [2–4].
RIP140 is a co-regulator for various nuclear receptors and transcription factors. It plays important roles in many biological activities and several disease conditions including the metabolic syndrome, especially T2DM [5–8]. In adipogenesis, both the expressions of RIP140 mRNA and protein are elevated. In this process, RIP140 acquires extensive post-translational modifications (PTMs) such as phosphorylation, acetylation, methylation and vitamin B6 conjugation. These PTMs can modulate RIP140’s gene-regulatory activity and alter its sub-cellular distribution [6, 9–11]. Recently, we found that activated nuclear PKCε phosphorylates RIP140 in adipocytes and the phosphorylated RIP140 is subsequently methylated at three arginine residues by protein arginine methyltransferase 1 (PRMT1). These modifications enhance the interaction of exportin 1 with RIP140 to promote RIP140’s nuclear export [9, 10]. We also demonstrated that the cytoplasmic RIP140 can blunt both basal and insulin-stimulated glucose uptake by interacting with AS160 to retard GLUT4 vesicle trafficking in adipocytes [7]. We found that interaction of RIP140 with AS160 maintains AS160’s activity by preventing Akt-mediated inactivation. In another study, we reported cytoplasmic RIP140 can reduce lipolysis through interacting with perilipin [11]. However, the full spectrum of functions of cytoplasmic RIP140 in adipocytes is not clear. The understanding of these functions and the regulatory mechanisms may reveal novel targets to treat adipocyte dysfunctions frequently observed in metabolic disorders.
Adiponectin is one of the most abundant adipokines and is known for its insulin-sensitizing effect. It also regulates systemic glucose and fatty acid metabolism and may also reduce inflammation [12–14]. Adiponectin’s targets include muscle, liver, macrophages, and the central nervous system [13]. There are three major oligomeric forms of adiponectin: trimer (low molecular weight: LMW), hexamer (medium molecular weight) and the high molecular weight form (HMW) which consists of 12- to 18-mer of adiponectin. These three oligomers of adiponectin have different affinities for adiponectin receptors in different cell types and they also show different biological activities [13]. Although disulfide bond formation in ER has been shown to control the oligomerization of adiponectin, the regulation of the secretion of adiponectin vesicle is not fully understood. In adipocytes of obese and diabetic patients and animals, secretion of adiponectin, as well as the translocation of GLUT4 vesicle, is reduced. It has been proposed that a reduction in circulating adiponectin levels in these subjects may lead to the development of cardiovascular diseases and other metabolic complications [15–17]. In adipocytes, insulin can stimulate both adiponectin and GLUT4 vesicles trafficking but they are two different secretory vesicles [18]. When adipocytes are dysfunctional, the trafficking of both GLUT4 and adiponectin vesicles is retarded, implying that these two vesicles may share some common mechanisms to regulate their transport.
Other studies have also suggested that receptor interacting protein 140 (RIP140) may be involved in the regulation of adipocyte function [7, 19]. In our preliminary screening for RIP140-related adipocyte dysfunctions, we found that cytoplasmic RIP140 can regulate adiponectin secretion. This current study presents evidence for this new functional role of cytoplasmic RIP140. We also determine the underlying mechanism, and provide a proof-of-concept that targeting RIP140 itself or PKCε to prevent RIP140’s nuclear export can promote the secretion of functional adiponectin. This can be beneficial for several adiponectin’s target cells, such as improving glucose uptake in C2C12 muscle cells and reducing glucose production in HepG2 hepatocytes.
2. Materials and methods
2.1. Cell culture, reagents and transfection
3T3-L1 cells and RIP140-null mouse embryonic fibroblasts were maintained and differentiated as described [20]. C2C12 cells were maintained in DMEM with 10% FBS, and differentiated in DMEM with 2% horse serum for six days. HepG2 was maintained in DMEM with 10%FBS. 293TN (System Biosciences) were maintained in low glucose DMEM with 10%FBS. siRNAs in this study were from Qiagen. Adiponectin ELISA kit was from Invitrogen. DeliverX Plus siRNA transfection kit (Panomics) was used for siRNA transfection. Plasmid transfection in 3T3-L1 adipocytes was done by electroporation with GenePluser (Bio-Rad). Lentivirus transduction was conducted for RIP140-null adipocytes.
2.2. Plasmid and reagents
The wild type and phosphor-deficient form (CN) of Flag-RIP140 fragments were amplified by PCR from Flag-RIP140 vectors [7] and cloned into pCDH-CMV-EF1 (System Biosciences). The wild type and non-phosphorylatable form (4P) of AS160 were amplified from AS160 vectors (gifts of Gustav Lienhard) and cloned into pCMV-PL1 containing HA-tag. Adiponectin ELISA kit was from Invitrogen. Endothelin-1 ELISA kit was from Assay Designs. Mouse/Rat insulin ELISA kit was from Millipore.
2.3. Lentivirus production and transduction
Lentivirus was produced in 293TN cells using lentivirus packing system (System Biosciences) and concentrated with lenti-X concentrator (Clontech). For transduction, RIP140-null adipocytes were differentiated with concentrated lentivirus for 4 days, and changed to a polybrene-containing medium (8 μg/ml) on day 5. After 24 h, medium was refreshed using a normal medium with the differentiation cocktail.
2.4. Treatment and protein precipitation
Differentiated 3T3-L1 adipocyte cultures were washed with PBS twice and incubated with DMEM without serum for another 6 hr. Secreted proteins in the medium were precipitated by TCA. Medium and cell lysate were both collected for Western blotting and ELISA. To quantify adiponectin complex, media were collected and subjected into SDS-PAGE in a non-reducing loading buffer. Immunoblots were quantified by Image J.
2.5. Antibodies, Western blotting and cellular fractionation
Anti-actin, anti-HA and anti-lamin antibodies were from Santa Cruz. Anti-RIP140 antibody and anti-alpha-tubulin antibody were purchased from Abcam and Sigma-Aldrich, respectively. Isolation of nuclear and cytoplasmic fractions was conducted as described [7]. Briefly, cells were gently lysed by hypotonic buffer and then, after centrifugation for 10 min at 1,000g, supernatants (cytosolic fraction) were collected. Pellets were further lysed in RIPA buffer by sonication. Lysates were centrifuged and supernatants were nuclear fraction.
2.6. Glucose uptake and glucose production assays
Conditioned media were collected from mature adipocytes cultured in DMEM with 10% FBS for 48 hr. Glucose uptake assay was conducted as described [21] with modification. Differentiated C2C12 was serum-starved for 3 hrs and then treated, in the presence or absence of 100 nM insulin, with conditioned medium. After 30 min, cultures were refreshed with KRPH buffer containing 25 μM 2-deoxy-glucose for 5 min. To determine glucose production, HepG2 was serum-starved for 3 hr and then treated with conditioned media for another 1 hr. Cells were washed with PBS and then incubated in a glucose production medium in the absence or presence of 100 nM insulin for 4 hr. Glucose levels were determined using a glucose assay kit (Sigma Aldrich) and normalized to the protein concentrations of cell lysates. To neutralize the activity of adiponectin, 200 μg anti-mouse adiponectin antibody (AF1119, R&D Systems) was added to a 5 ml conditioned medium for 2 hr at 4°C [22]. The neutralized conditioned media were used in glucose uptake and glucose production assays.
2.6. Statistical analyses
Results were presented in the means ± SD. P values were examined using student t test in the two-tail condition. P < 0.05 is considered as statistically significant.
3. Results
3.1. Reducing RIP140 or its nuclear export stimulus, PKCε, enhances adiponectin secretion
We previously found that RIP140 can be exported into the cytoplasm of adipocytes both in vitro and in vivo [7, 10]. In 3T3-L1 adipocytes, silencing RIP140, or more specifically knocking down its nuclear export stimulator PKCε to decrease cytoplasmic RIP140, enhanced basal adiponectin secretion without altering adiponectin mRNA levels (Fig. 1A). But we found no change in other adipokines such as leptin, resistin, IL-6 and TNFα (data not shown). To rule out potential effects on adiponectin translation or its protein stability, we performed metabolic labeling in the presence of a proteosome inhibitor, MG132, and a post-Golgi trafficking inhibitor, Brefeldin A to block protein degradation and post-Golgi secretion [23]. Neither general translation nor specific adiponectin translation was significantly altered, which ruled out the possibility that these treatments might affect adiponectin production (Fig. 1B). Adiponectin secretion is mainly controlled by post-translational modifications in ER and vesicle transport in trans-Golgi networks (TGN) [13, 24–26]. Disulfide bond formation of adiponectin molecules is also critical to their secretion. We performed Western blotting in a non-reducing condition to detect various forms of secreted adiponectin obtained from adipocyte cultures under RIP140- or PKCε–silencing. The results show that silencing of RIP140 or PKCε significantly enhanced the secretion of all forms of adiponectin (Fig. 1C), suggesting that RIP140 regulates adiponectin secretion in an unbiased manner.
Fig. 1.
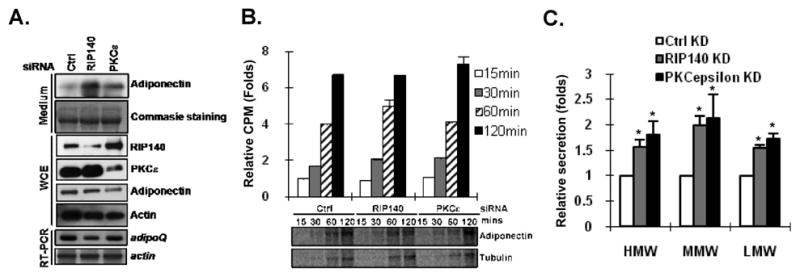
Targeting RIP140 or PKCε increases adiponectin secretion. (A) Adiponectin secretion pattern in ctrl-, RIP140- or PKCε-silenced 3T3-L1 adipocytes. Commasie staining shows loading control. WCE: whole cell extract. (B) Production of adiponectin and its mRNA. Differentiating adipocytes were transfected with indicated siRNA on day 5. On day 8, mature adipocytes were treated with MG132 and Brefeldin A in the presence of 35S-labeled methionine for indicated time. General (total cpm counts) and specific (determined by antibody against adiponectin and actin, respectively) translation was each examined. The general translation rate, per 100 μg whole cell lysate, in each experimental condition was represented as the relative fold of CPM using the CPM of control at 15 minutes as the value of 1. The differences among indicated knockdown at each time point examined are not statistically significant. (C) Secreted adiponectin profiles. Differentiating adipocytes were transfected with indicated siRNA on day 5. On day 8, mature adipocytes were cultured in serum-free medium for 6 h. Adiponectin profile was determined by western blotting in non-reducing condition. HMW: high molecular weight complex; MMW: medium molecular weight complex; LMW: low molecular weight complex. *: P < 0.05, compared to ctrl knockdown group (CtrlKD).
We then assessed the functional role of RIP140 in modulating adiponectin secretion using RIP140-null adipocytes. As shown in Fig. 2, RIP140-null adipocytes rescued with a wild type (Wt) RIP140 secreted less adiponectin as compared to the control vector (GFP). On the contrary, adiponectin secretion from RIP40-null adipocytes rescued with a mutant RIP140-GFP defected in its nuclear export (CN, a phospho-deficient form that loses cytoplasmic activity) was not affected as compared to the control group. Taken together, these results show that cytoplasmic RIP140 dampens adiponectin secretion in an unbiased manner, and targeting the nuclear export or RIP140 itself can elevate the secretion of adiponectin.
Fig. 2.
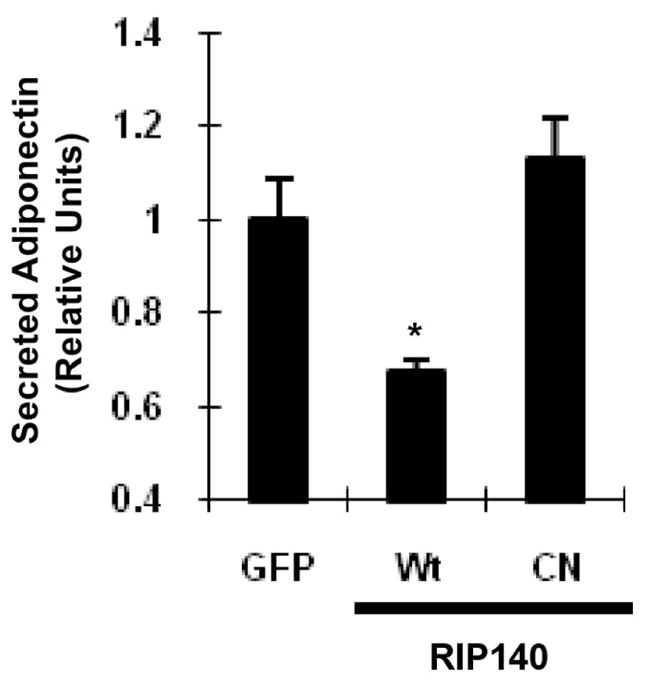
Cytoplasmic RIP140 reduces adiponectin secretion in RIP140-null adipocytes. The effect of various forms of RIP140 on adiponectin secretion in RIP140-null adipocytes rescued with RIP140 expression vectors. Wt: wild type RIP140; CN: phosphor-defective mutant RIP140. All values represent the means ± SD., n=3; *: P < 0.05, compared to ctrl group.
3.2. Adiponectin secretion is regulated by AS160 activity
AS160 is a Rab GTPase-activating protein (GAP) whose activity can be diminished by Akt-mediated phosphorylation upon insulin binding with insulin receptor [27]. Rab GTPases are important in vesicle secretion in many steps including recycling, trafficking, tethering and membrane docking. The increased activity of AS160 reduces GLUT4 vesicle secretion by decreasing GTP-bound form of Rab proteins [27]. In addition, insulin also promotes the secretion of adiponectin vesicle, but the mechanism is unclear [28]. Recently, we found that, in adipocytes, AS160 activity can be maintained by interacting with RIP140 in the cytoplasm [7]. A recent study demonstrated that Rab11 regulates adiponectin vesicle trafficking and suspected that AS160 might modulate Rab11 activity by interacting with Rab11-interacting protein, Rip11 (also see Discussion) [28]. We then determined the functional role for AS160 in adiponectin secretion using a gain-of-function approach. Figs. 3A and 3B show that over-expression of the wild type (Wt) AS160, or its constitutively active form (4P) [27], dramatically reduced basal and insulin-stimulated adiponectin secretion. Of note, constitutively active AS160 (4P) almost blocked the secretion of adiponectin completely. Besides, both over-expressions of the wild type (Wt) AS160 and constitutively active form (4P) blunted insulin’s effect on promoting adiponectin secretion. Altogether, the data support the notion of negative regulation of adiponectin secretion by AS160.
Fig. 3.
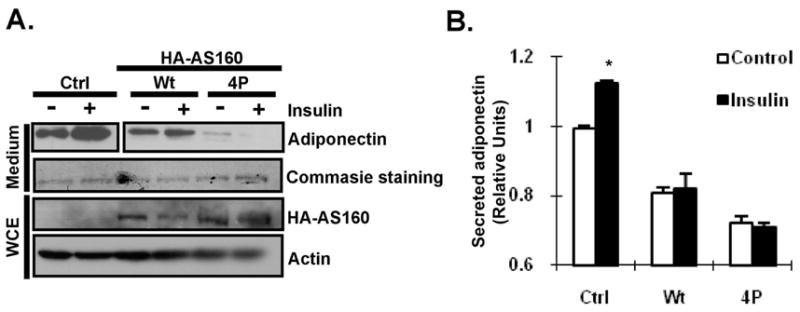
AS160 modulates adiponectin secretion. (A) Western blot detection of adiponectin secretion from adipocytes transfected with control vector (Ctrl) or indicated AS160 vectors. Differentiating 3T3-L1 adipocytes were electroporated with indicated vector on day 6. After two days, mature adipocytes were cultured in serum-free medium with or without 100nM Insulin for 6 h. Adiponectin profile was determined by western blotting in non-reducing condition. Wt: wild type; 4P: constitutive active form of AS160. Commasie staining shows loading control. (B) ELISA detection of secreted adiponectin from transfected adipocytes was examined by ELISA. All values represent the means ± SD., n=3; *: P < 0.05 compared to control treatment.
3.3. Blocking RIP140-AS160 interaction enhances adiponectin secretion
RIP140 interacts with AS160 through its amino terminal domain to prevent AKT-mediated inactivation [7]. The results described above suggest that AS160 can negatively regulate adiponectin vesicle secretion and cytoplasmic RIP140 may retard adiponectin secretion through interacting with AS160. Because RD1 is the main interacting domain for RIP140 to interact with AS160 [7], over-expression of this domain might compete the interaction between AS160 and the full length RIP140. We then examined if over-expressing RIP140’s RD1 (repressive domain 1) can enhance adiponectin secretion. Interestingly, expressing RD1 in 3T3-L1 adipocytes substantially elevated adiponetin secretion (Fig. 4). This result further supports the hypothesis that cytoplasmic RIP140 might dampen adiponectin secretion via interacting with AS160 and targeting the interaction of RIP140 with AS160 might elevate adiponectin vesicle trafficking.
Fig. 4.
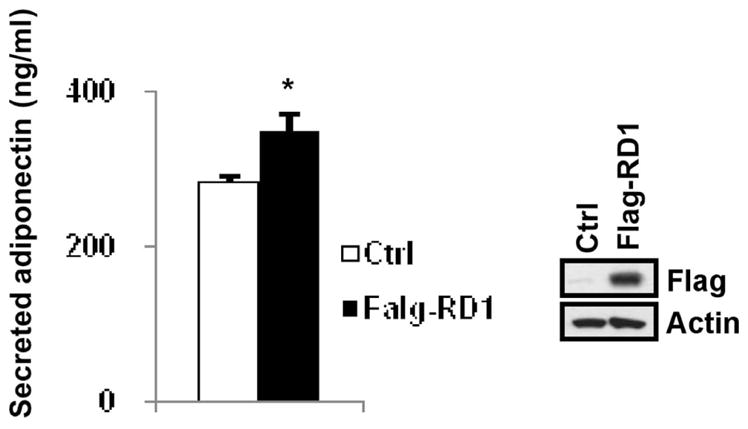
Over-expression of the amino terminus (RD-1) of RIP140 elevates adiponectin secretion. Left: Secreted adiponectin from transfected adipocytes was examined by ELISA. Control vector or flag-RD1 vector was electroporated into day 6 differentiating 3T3-L1 adipocytes. After two days, adipocytes were cultured in serum-free medium for 6 h. The adiponectin levels of culture supernatants were measured by ELISA. All values represent the means ± SD., n=3; *: P < 0.05 compared to control group. Right: immunoblotting of whole cell lysates for indicated antibodies to determine the efficiency of over-expression.
3.4. Targeting cytoplasmic RIP140 is beneficial for glucose metabolism in C2C12 muscle cells and hepatocytes in an adiponectin-dependent manner
Adiponectin is an important insulin sensitizer to promote glucose uptake in muscle cells and to reduce glucose production in hepatocytes through an AMPK-dependent signaling cascade. The net effect of elevation of circulating adiponectin is better glucose tolerance [13]. To further examine if targeting RIP140 or its nuclear export trigger, PKCε, can be beneficial for glucose metabolism in muscle cells, we determined the effect of conditioned media collected from RIP140- or PKCε-silencing 3T3-L1 adipocytes on glucose uptake ability of C2C12 muscle cells. Indeed, the conditioned media from both RIP140- and PKCε-knocked down adipocytes increased both basal and insulin-stimulated glucose uptake (Fig. 5A). Besides, the phosphorylation status of AMP-activated protein kinase (AMPK) in C2C12 cells was also elevated by conditioned media from both RIP140- and PKCε-knocked down adipocytes (Fig. 5B). Finally, the anti-adiponectin antibody substantially blocked the effect of conditioned media from both RIP140- and PKCε-knocked down adipocytes on elevation of glucose uptake ability (Fig. 5C), supporting that the beneficial effect on glucose uptake ability in C2C12 muscle cells was likely due to the secreted adiponectin in the conditioned media.
Fig. 5.

Targeting RIP140 or PKCε in adipocytes enhances functional adiponectin secretion to promote glucose uptake in muscle cells. (A) The effect of conditioned media from ctrl-, RIP140- or PKCε-silenced adipocytes on glucose uptake in C2C12 cells in the absence (control) or presence of insulin. All values represent the means ± SD., n=3; *: P < 0.05 compared to control group in Ctrl conditioned medium. **: P < 0.05 compared to insulin group in Ctrl conditioned medium. (B) The effect of conditioned media in (A) on AMPK activation in C2C12 cells. The ratio of pAMPK/AMPK was quantified. (C) Anti-adiponectin antibody (anti-adipoQ) neutralized the effect of conditioned media on C2C12 glucose uptake in the presence of insulin
In addition to the effect on C2C12 muscle cells, we further investigated the effect of adipocyte-conditioned media from RIP140- or PKCε-silencing adipocytes on gluconeogenesis ability in hepatocytes. As shown in Fig. 6A, conditioned media from both RIP140- and PKCε-knocked down adipocytes significantly reduced glucose production in HepG2 hepatocytes. In the presence of insulin, there is no further reduction of glucose production in HepG2 cells. As same as the effects on C2C12 muscle cells, these conditioned media promoted the level of phosphorylated AMPK in HepG2 cells. Most importantly, the anti-adiponectin antibody effectively neutralized the effect of conditioned media on glucose production in the absence of insulin (Fig 6C).
Fig. 6.

Targeting RIP140 or PKCε in adipocytes enhances functional adiponectin secretion to reduce gluconeogenesis in hepatocytes. (A) The effect of conditioned media from ctrl-, RIP140- or PKCε-silenced adipocytes on gluconeogenesis in HepG2 cells in the absence (control) or presence of insulin. All values represent the means ± SD., n=3; *: P < 0.05 compared to control group in Ctrl conditioned medium. (B) The effect of conditioned media on AMPK activation in HepG2 cells. The ratio of pAMPK/AMPK was quantified. (C) Anti-adiponectin antibody (anti-adipoQ) neutralized the effect of conditioned media on glucose production ability of HepG2 in the absence of insulin. *: P < 0.05 compared to IgG group in conditioned medium from indicated silencing group.
All together, the data demonstrate that cytoplasmic RIP140 may play a role in regulating adiponectin secretion in adipocytes, and the elevated secretion of adiponectin is functional in promoting glucose uptake in muscle cells and reducing glucose production in hepatocytes.
4. Discussion
We have previously shown that HFD increases the levels of RIP140 in the cytoplasm of epididymal adipocytes, and proposed that the cytoplasmic accumulation of RIP140 may provide a disease marker [7]. The cytoplasmic RIP140 interacts with AS160 to retard GLUT4 vesicle trafficking, and with perilipin A on lipid droplets to promote lipolysis [7, 19]. The current study reports a new function of cytoplasmic RIP140 in adipocytes, that is to negatively regulate adiponectin secretion. Because adiponectin can target multiple cell/tissue types for different functions, its role in maintaining metabolic homeostasis on a systemic level has been intensively studied; but regulation of its secretion is poorly understood. The regulatory role of cytoplasmic RIP140 in adiponectin secretion significantly expands the scope of the biological activity of cytoplasmic RIP140, and suggests that targeting the functions of cytoplasmic RIP140 may provide a potential therapeutic strategy in managing metabolic diseases.
Although both adiponectin release and GLUT4 trafficking can be stimulated by insulin, the two proteins are localized in two different types of vesicles [18]. But both adiponectin and GLUT4 vesicles reside in the trans-Golgi network (TGN) and the recycling endosomes [26]. Secretion of these vesicles is regulated by Rab GTPases in multiple steps such as trafficking, docking, tethering and fusion of the vesicles [27, 29]. AS160 is a RIP140 interacting GAP that can inactivate several Rab GTPases [27]. RIP140 can maintain AS160 activity through a direct interaction and preventing its Akt-mediated inactivation. This would presumably affect the trafficking of multiple types of vesicles. This would also suggest a possible mechanism for the action of insulin in enhancing both adiponectin release and GLUT4 trafficking, and substantiates the significance of the molecular interaction between RIP140 and AS160. Rab5 and Rab11 are the only two Rab GTPases shown to be involved in adiponectin vesicles trafficking to modulate adiponectin release [26, 28]. However, neither Rab5 nor Rab11 activities can be directly modulated by AS160 in vitro [27]. Therefore, the detailed mechanism by which AS160 regulates adiponectin vesicles, such as via regulating Rab5 or Rab11, or other unknown factors, remains to be examined. To this end, a previous study has suggested that AS160 may modulate Rab11 by interacting with its interacting proteins such as Rip11 [27, 30], which would be a potential topic of future studies.
Hightlights.
This study reports a new functional role for cytoplasmic RIP140 in adipocyte in regulating adiponectin secretion. This is via its interaction with AS160, a known RIP140-interacting protein. The study suggests that targeting cytoplasmic RIP140 may be a potentially effective therapeutic strategy to improve adiponectin secretion and possibly to manage metabolic disorders.
Acknowledgments
This study was supported in part by NIH grants DK60521, DK060521-07S1, DA11190, DK54733, DK054733-09S1, K02-DA13926 and the Distinguished McKnight University Professorship to L.-N. W. P.-C. H. is supported by predoctoral fellowship of American Heart Association. We thank Gustav Lienhard for providing AS160 constructs, Malcolm Parker for providing RIP140-null MEF cells. We thank Shawna D. Persaud, C.-H. Hung, Y.-S. Chuang, A. Smith, F. A. Beyan and S.-C. Luo for technical help. We also thank Dr. Timothy Walseth for providing essential equipment.
Abbreviations
- RIP140
Receptor-interacting protein 140
- AS160
Akt substrate 160
- HFD
High-fat diet
- PKCε
Protein kinase C epsilon
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Herman MA, Kahn BB. J Clin Invest. 2006;116:1767–1775. doi: 10.1172/JCI29027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guilherme A, Virbasius JV, Puri V, Czech MP. Nat Rev Mol Cell Biol. 2008;9:367–377. doi: 10.1038/nrm2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schenk S, Saberi M, Olefsky JM. J Clin Invest. 2008;118:2992–3002. doi: 10.1172/JCI34260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wellen KE, Hotamisligil GS. J Clin Invest. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mostaqul Huq MD, Gupta P, Wei LN. Curr Med Chem. 2008;15:386–392. doi: 10.2174/092986708783497382. [DOI] [PubMed] [Google Scholar]
- 6.Leonardsson G, Steel JH, Christian M, Pocock V, Milligan S, Bell J, So PW, Medina-Gomez G, Vidal-Puig A, White R, Parker MG. Proc Natl Acad Sci U S A. 2004;101:8437–8442. doi: 10.1073/pnas.0401013101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ho PC, Lin YW, Tsui YC, Gupta P, Wei LN. Cell Metab. 2009;10:516–523. doi: 10.1016/j.cmet.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Powelka AM, Seth A, Virbasius JV, Kiskinis E, Nicoloro SM, Guilherme A, Tang X, Straubhaar J, Cherniack AD, Parker MG, Czech MP. J Clin Invest. 2006;116:125–136. doi: 10.1172/JCI26040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mostaqul Huq MD, Gupta P, Tsai NP, White R, Parker MG, Wei LN. EMBO J. 2006;25:5094–5104. doi: 10.1038/sj.emboj.7601389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gupta P, Ho PC, Huq MD, Khan AA, Tsai NP, Wei LN. PLoS One. 2008;3:e2658. doi: 10.1371/journal.pone.0002658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huq MD, Tsai NP, Lin YP, Higgins L, Wei LN. Nat Chem Biol. 2007;3:161–165. doi: 10.1038/nchembio861. [DOI] [PubMed] [Google Scholar]
- 12.Dridi S, Taouis M. J Nutr Biochem. 2009;20:831–839. doi: 10.1016/j.jnutbio.2009.06.003. [DOI] [PubMed] [Google Scholar]
- 13.Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. J Clin Invest. 2006;116:1784–1792. doi: 10.1172/JCI29126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Folco EJ, Rocha VZ, Lopez-Ilasaca M, Libby P. J Biol Chem. 2009;284:25569–25575. doi: 10.1074/jbc.M109.019786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li S, Shin HJ, Ding EL, van Dam RM. JAMA. 2009;302:179–188. doi: 10.1001/jama.2009.976. [DOI] [PubMed] [Google Scholar]
- 16.Antoniades C, Antonopoulos AS, Tousoulis D, Stefanadis C. Obes Rev. 2009;10:269–279. doi: 10.1111/j.1467-789X.2009.00571.x. [DOI] [PubMed] [Google Scholar]
- 17.Shibata R, Ouchi N, Murohara T. Circ J. 2009;73:608–614. doi: 10.1253/circj.cj-09-0057. [DOI] [PubMed] [Google Scholar]
- 18.Bogan JS, Lodish HF. J Cell Biol. 1999;146:609–620. doi: 10.1083/jcb.146.3.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ho PC, Chuang YS, Hung CH, Wei LN. Cell Signal. 2011;23:1396–1403. doi: 10.1016/j.cellsig.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ho PC, Gupta P, Tsui YC, Ha SG, Huq M, Wei LN. Cell Signal. 2008;20:1911–1919. doi: 10.1016/j.cellsig.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tulipano G, Spano P, Cocchi D. Mol Cell Endocrinol. 2008;292:42–49. doi: 10.1016/j.mce.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 22.Rasouli N, Yao-Borengasser A, Varma V, Spencer HJ, McGehee RE, Jr, Peterson CA, Mehta JL, Kern PA. Arterioscler Thromb Vasc Biol. 2009;29:1328–1335. doi: 10.1161/ATVBAHA.109.186957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bedi D, Clarke KJ, Dennis JC, Zhong Q, Brunson BL, Morrison EE, Judd RL. Biochem Biophys Res Commun. 2006;345:332–339. doi: 10.1016/j.bbrc.2006.04.098. [DOI] [PubMed] [Google Scholar]
- 24.Wang Y, Lam KS, Yau MH, Xu A. Biochem J. 2008;409:623–633. doi: 10.1042/BJ20071492. [DOI] [PubMed] [Google Scholar]
- 25.Xie L, Boyle D, Sanford D, Scherer PE, Pessin JE, Mora S. J Biol Chem. 2006;281:7253–7259. doi: 10.1074/jbc.M511313200. [DOI] [PubMed] [Google Scholar]
- 26.Xie L, O’Reilly CP, Chapes SK, Mora S. Biochim Biophys Acta. 2008;1782:99–108. doi: 10.1016/j.bbadis.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sakamoto K, Holman GD. Am J Physiol Endocrinol Metab. 2008;295:E29–37. doi: 10.1152/ajpendo.90331.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clarke M, Ewart MA, Santy LC, Prekeris R, Gould GW. Biochem Biophys Res Commun. 2006;342:1361–1367. doi: 10.1016/j.bbrc.2006.02.102. [DOI] [PubMed] [Google Scholar]
- 29.Stenmark H. Nat Rev Mol Cell Biol. 2009;10:513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- 30.Welsh GI, Leney SE, Lloyd-Lewis B, Wherlock M, Lindsay AJ, McCaffrey MW, Tavare JM. J Cell Sci. 2007;120:4197–4208. doi: 10.1242/jcs.007310. [DOI] [PubMed] [Google Scholar]