

Abstract
The A/U-rich RNA binding protein tristetraprolin (TTP) is an mRNA destabilizing factor which plays a role in the regulated turnover of many transcripts encoding proteins involved in immune function and cell growth control. TTP also plays a role in stress-induced destabilization of mRNAs. Here we report the interaction of TTP with a component of the mTORC2 kinase, Protor-2 (PRR5-L, protein Q6MZQ0/FLJ14213/CAE45978). Protor-2 is structurally similar to human PRR5 and has been demonstrated to bind mTORC2 via Rictor and/or Sin1 and may signal downstream events promoting apoptosis. Protor-2 dissociates from mTORC2 upon hyperactivation of the kinase and is not required for mTORC2 integrity or activity. We identified Protor-2 in a yeast two-hybrid screen as a TTP interactor using the C-terminal mRNA decay domain of TTP as bait. The interaction of Protor-2 with TTP was also confirmed in vivo in co-immunoprecipitation experiments and Protor-2 was also detected in immunoprecipitates of rictor. Protor-2 was shown to stimulate TTP-mediated mRNA turnover of several TTP-associated mRNAs (TNF-α, GM-CSF, IL-3 and COX-2) in Jurkat cells when overexpressed while the half-lives of transcripts which do not decay via a TTP-mediated mechanism were unaffected. Knockdown of Protor-2 via RNAi inhibited TTP-mediated mRNA turnover of these TTP-associated mRNAs and inhibited association of TTP with cytoplasmic stress granules (SG) or mRNA processing bodies (P-bodies) following induction of the integrated stress response. These results suggest that Protor-2 associates with TTP to accelerate TTP-mediated mRNA turnover and functionally links the control of TTP regulated mRNA stability to mTORC2 activity.
Keywords: Protor-2, tristetraprolin, mTORC2, mRNA stability, stress
1. Introduction
The tandem zinc finger protein tristetraprolin (TTP; also known as Nup475, Tis11, or Zfp36) [1–3] is widely expressed and functions to regulate gene expression by binding to a conserved adenosine/uridine-rich element (ARE) within the 3' untranslated region of several mRNAs [4]. TTP is known to promote mRNA deadenylation [5] and 3' to 5' degradation of the mRNA body [6], consistent with its ability to recruit several factors involved in these processes such as Dcp1, Dcp2 and components of the exosome [7]. The critical role for TTP in the regulation of tumor necrosis factor-α is demonstrated by the proinflammatory phenotype of TTP−/− mice in which chronic overexpression of TNF-α in macrophages results in severe polyarthritis and cachexia [8]. The overexpression of TNF-α in these mice is due to the stabilization of TNF-α mRNA as a result of the lack of TTP function [9]. TTP has been implicated in the posttranscriptional regulation of granulocyte-macrophage colony-stimulating factor (GM-CSF), interleukin-2 (IL-2) and cyclooxygenase 2 (COX-2) [6]. It may also regulate its own expression by binding to an ARE within the 3' UTR of TTP mRNA [10]. The minimum binding site of TTP is the nonameric sequence UUAUUUAUU [11], and it is probable that additional targets of TTP containing this sequence remain to be discovered.
The p38 mitogen-activated protein kinase (MAPK) and its downstream kinase MK2 are known to activate TTP [12]. The two major sites of MK2-mediated phosphorylation of TTP identified in vitro and in vivo are serines 52 and 178 [13]. These phosphorylations result in the recruitment of 14-3-3 proteins, functional adaptors which specifically interact with particular serine- or threonine-phosphorylated proteins [14]. The recruitment of 14-3-3 proteins leads to exclusion of TTP from stress granules (SGs) and mRNA processing bodies (P-bodies), cytoplasmic structures at which translationally stalled transcripts accumulate and mRNAs are degraded, respectively, under conditions of cellular stress [12]. The phosphorylation of TTP and its exclusion from SGs and P-bodies are associated with stabilization of ARE-containing mRNAs [1, 12].
TOR (Target of Rapamycin) kinase is a highly conserved, central sensor of cell growth [15]. In humans, dysregulated mTOR signaling plays a role in cancer development and progression, as well as the response to mitogens, nutrients and chemotherapeutic agents [16]. Aberrant mTOR signaling has also been implicated in tuberous sclerosis complex and lymphangiomyelomatosis [17]. TOR is found, in yeast to humans, in at least two functionally and structurally distinct multiprotein complexes termed TOR complex 1 (TORC1) and TORC2 [18]. The mammalian TOR complex 1 (mTORC1) contains mTOR, mLST8 and Raptor and is rapamycin sensitive. MTORC2 consists of Rictor, mSIN1, mLST8, Protor and mTOR and is rapamycin insensitive [19].
Previous studies have provided evidence that the TOR signaling cascade controls mRNA turnover in yeast [20, 21]. It has been shown that blocking TOR signaling either through nutrient limitation or rapamycin treatment causes accelerated turnover of a subset of mRNAs while others appear unaffected. More recently, several factors which regulate mRNA decay have been shown to be potential substrates of mTOR [22]. Here we demonstrate that the mTORC2 component Protor-2 interacts with TTP to promote mRNA turnover of transcripts known to degrade via TTP. We also demonstrate that Protor-2 and TTP interact in vivo and that overexpression of Protor-1 accelerates the decay of TTP-associated transcripts. Moreover, knockdown of TTP expression inhibited TTP-mediated mRNA turnover and reduced the ability of TTP to associate with SGs and P-bodies following stress induction.
2. Materials and Methods
2.1. Plasmids, cell lines and reagents
The coding region of Protor-2 was subcloned from an EST clone obtained from the German Resource Center for Genome Research (DKFZp686N03132) and cloned in-frame with a C-terminal myc-epitope tag into pTRACER (Invitrogen, Carlsbad, CA). HeLa and Jurkat cell lines were obtained from ATCC. The generation of tetracycline-inducible shRNA TSCsiJurkat (T-RExJurkat, Invitrogen) cells was as described in [23]. Actinomycin D (Sigma) was dissolved in water and used at a final concentration of 10 µg/ml for actinomycin D-chase analyses as described [24]. FCCP (carbonyl cyanide p-trifluoromethoxyphenylhydrazone) (Sigma) was added to media at a final concentration of 1 µM unless otherwise indicated. The TTP (CARP-3) antibody was generously provided by Dr. William Rigby (Dartmouth University). Antibodies to Protor-1, Protor-2 (FLJ14213), TIA-1, Dcp1 and actin were obtained from Abcam. Rictor antibodies were from Bethyl Laboratories. siRNA transfection targeting Protor-2 (sc-96853) or Rictor (siGENOME™ SMARTpool siRNA) were performed using synthetic oligonucleotides obtained from Santa Cruz Biotechnology and Dharmacon, respectively. A siRNA with a scrambled sequence was used as a negative-targeting control. Transfections were performed using FuGene 6 transfection reagent as directed by the manufacturer (Roche, Indianapolis, IN).
2.2. Yeast two-hybrid screen
The yeast two-hybrid screen to isolate TTP interacting screen was performed as previously described [25]. Several N-terminal and RNA-binding zinc-finger domain fragments of the human TTP cDNA were cloned into pGB12 in frame with the Gal4 DNA-binding domain (Gal4DBD). Most of these fusions were found to autoactivate Gal4UAS based reporters and thus unable to be utilized for screening purposes. A portion of the C-terminal half of TTP (C-terminal mRNA decay domain, amino acids 174–326) was cloned into pGB12 and successfully used to screen several libraries. This construct was used to transform AH109 (Mat a, trp-901, leu2–3, 112, ura3–52, his3–200, gal4d, gal80d, LYS2::GAL1TATA-HIS3, GAL2UAS-GAL2TATA-ADE2, URA3::MEL1UAS-MEL1TATA-LacZ, MEL1) to obtain a strain which expressed the GAL4DBD-C-term-TTP fusion. We confirmed that this fusion protein alone did not result in any significant reporter activation. This stain was used to screen mammary gland, prostate and placenta MATCHMAKER cDNA libraries in pACT2 (BD Biosciences, Clontech). Additionally, we screened a library constructed in pACT2 containing cDNAs reverse-transcribed from mRNA isolated from Jurkat cells stimulated with TNF-α. Transformants were selected by plating on appropriate dropout media. Plasmids were recovered from HIS+ and β-gal+ colonies and sequenced and subsequently retested by retransformation into AH109. Growth under selective conditions was confirmed to required both the DBD and AD-fusion proteins. Liquid β-gal assays were performed as previously described [25].
2.3. RNA and Protein analysis
Following transfection with siRNAs cells were treated with the indicated agents and RNA extracted using TRIZOL (Invitrogen). 2 µg of total RNA was used for reverse transcription using random primers and MMLV-RT (Promega, Madison, WI). Real time qRT-PCR was performed a previously described [26] using SYBRgreen technology using 2xTAq-Master mix (Promega) in a cycler (EP Mastercycler realplex2 optical module, Eppendorf AG, Germany). For all amplicons, an annealing temperature of 60°C was used. Primer sequences are available upon request. Relative changes in mRNA amounts were calculated based on the ΔC1 method, as described by Livak and Schmittgen [27]. For immunoblotting, total protein extracts were analyzed as previously described [28]. Immunoprecipitations were performed as described [24], briefly 250 µg aliquots of lysates from the indicated cell lines were immunoprecipitated with either the CARP-3 antibody or antibodies specific for the Protor isoforms and subjected to immunoblot analyses for total TTP or Protor levels.
2.4. Immunofluorescence
HeLa cells were grown on coverslips and treated with FCCP (1 µM) 48 hrs after transfection. Fixed cells were processed for immunostaining as previously described [29] using the antibodies described above. All image acquisition and analysis was performed using standard settings on a microscope (E600; Nikon) equipped with a camera (model C474-95; Hamamatsu), a Lucia software module (Laboratory Imaging, Ltd.) and a 60× objective (Plan Fluor; Nikon).
2.5. Statistics
Significant differences were determined between groups via student’s t test and ANOVA models. P values < 0.05 were considered significant. For mRNA half-life (t1/2) determinations values were calculated following plotting decay rates on a logarithmic scale.
3. Results
3.1. Protor-2 interacts with TTP in a yeast two-hybrid assay
To begin to identify new TTP binding proteins, we carried out a yeast two-hybrid screen using the C-terminal mRNA decay domain of human TTP (aa 174–326, C-term TTP) as bait screened against several Gal4-activation domain (Gal4AD) libraries, in addition to one constructed from cDNAs generated from mRNAs isolated from Jurkat cells (TNF-stimulated). A fusion of either the full-length, N-terminal mRNA decay domain (aa 1–100) or RNA-binding domain (aa 100–173) of the human TTP sequence to the Gal4DBD was found to activate Gal4UAS-based reporters alone. Screening with the C-term TTP as bait we identified a number of known TTP interactors, including components of the mRNA turnover machinery, hDcp1 and hXrn1, as well as additional potential interactors shown in table 1. Of these, Protor-2 was identified in approximately 4% of the total clones recovered from the screens. Other proline rich domain-containing proteins were not identified in the screens including the isoform Protor-1. Recently, Protor-2 has been shown to be a component of the mTORC2 complex and dissociates from hyperactivated mTORC2 and promotes apoptosis [23]. We also identified the DExH box RNA helicase RHAU as a C-terminal domain TTP interactor. Our previous studies have defined a role for regulated mRNA stability in controlling the expression of two critical determinants involved in mediating AKT-dependent rapamycin hypersensitivity of tumor cells [24], thus we investigated the Protor-2 interaction with TTP in greater detail.
Table 1.
Genetic interactors of C-term TTP in Y2H.
| Gene | Accession No. | Cellular location | Description |
|---|---|---|---|
| Protor-2 | NM_024841 | cytoplasmic | PRR5-Like ORF, mTORC2 component |
| DNAJB1 | NP_006136 | nuclear | Heat shock protein (Hsp40) |
| hDcp1 | NM_018403 | cytoplasmic | mRNA decapping enzyme 1 |
| hXrn1 | NM_019001 | Nuclear/cytoplasmic | 5’ to 3’ RNA exonuclease |
| RHAU | NM_020865 | Nuclear/cytoplasmic | DExH RNA helicase |
3.2. Mapping of the regions involved in Protor-2 binding to TTP
We next determined which regions of TTP were required for efficient Protor-2 binding as determined by the yeast two-hybrid assay. We generated a set of deletion mutants, shown in figure 1, of both the C-terminal mRNA decay domain of TTP and Protor-2 fused to the Gal4-DNA binding domain (DBD) or Gal4-activation domains (AD), respectively. These constructs were transformed into the yeast two hybrid strain AH109 and the relative strength of the interactions were determined by liquid β-galactosidase assays. As shown, a region within the C-terminal half of Protor-2 (aa 293–368) was required for specific binding with the C-term TTP-AD fusion. This region is distinct from the conserved regions within the Protor isoforms described in its initial characterization as a component of the mTORC2 complex [30]. Deletion mutants of the C-term TTP sequences demonstrated that a region from amino acids 205 to 326 was required for the strong interaction with Protor-2. Fusions of only the interacting domains within Protor-2 and the C-term TTP sequences to the Gal4-AD or DBD, respectively were sufficient to mediate a robust interaction and led to high levels of β-gal activity. These data suggested that a domain within the C-terminal half of Protor-2 and the C-terminal mRNA decay domain of TTP mediate the interaction of these two proteins.
Figure 1.
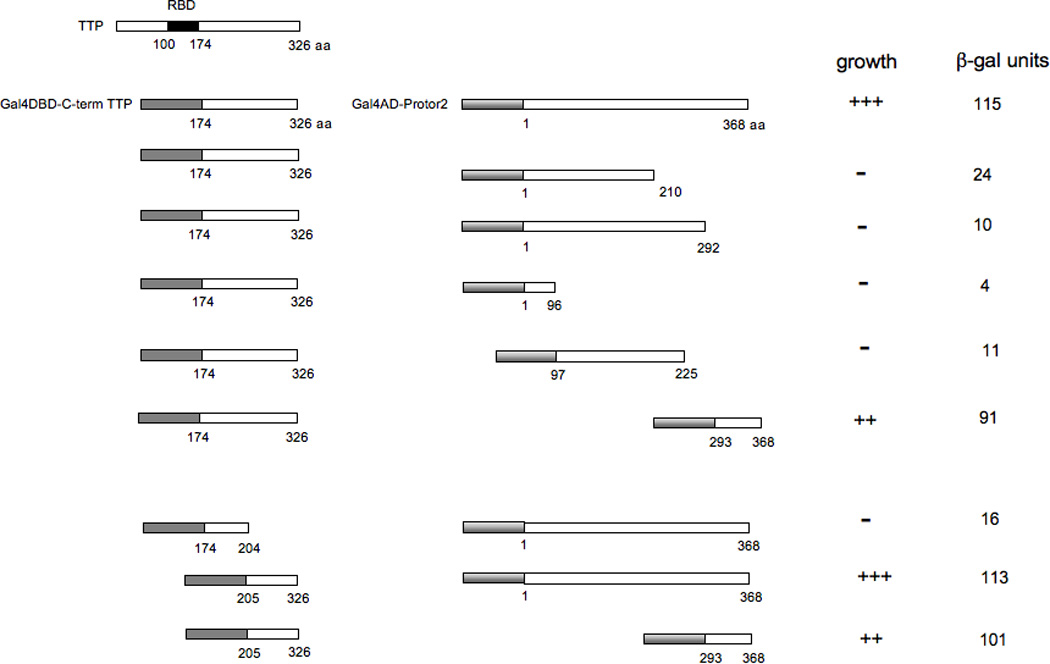
Y2H mapping of interacting regions of Protor-2 and TTP. The indicated deletion mutants of Gal4DBD-TTP or Gal4AD-Protor-2 were cotransfected into AH109 cells and plated onto selective media in the absence of histidine to determine whether an interaction between the proteins was detectable via activation of the HIS3 reporter (+++, positive growth; ++, moderate growth; −, no growth). Colonies which grew were assayed for β-gal activity as described (XX).
3.3. Protor-2 associates with TTP in vivo
To further confirm a direct association of Protor-2 with TTP in cells, we conducted co-immunoprecipitation experiments. As shown in figure 2, endogenous Protor-2 from Jurkat cells was detectable in immunoprecipitates of TTP in uninduced cells. However, the amount of Protor-2 found in immunoprecipitates of TTP was markedly increased in TSC1/2 deficient cells (TSCsiJurkat) while the total levels of Protor-2 and TTP were comparable in the cell extracts. TORC2 activity in these cells is markedly increased upon TSC1/2 knockdown via the tetracycline-induced expression of shRNAs targeting TSC1 and TSC2 (kindly provided by Dr. Michael Hall, Biozentrum, Basel, Switzerland) [23]. We also determined whether Protor-2 differentially associated with mTORC2 via rictor binding as had previously been reported [23]. As also shown in figure 2, high levels of endogenous Protor-2 was found in immunoprecipitates of rictor in unstimulated Jurkat cells and diminished significantly upon activation of mTORC2 in TSC1/2 knockdown cells. These data suggest that endogenous Protor-2 and TTP associate in vivo and that during activation of mTORC2 Protor-2 is released from rictor and binds TTP.
Figure 2.
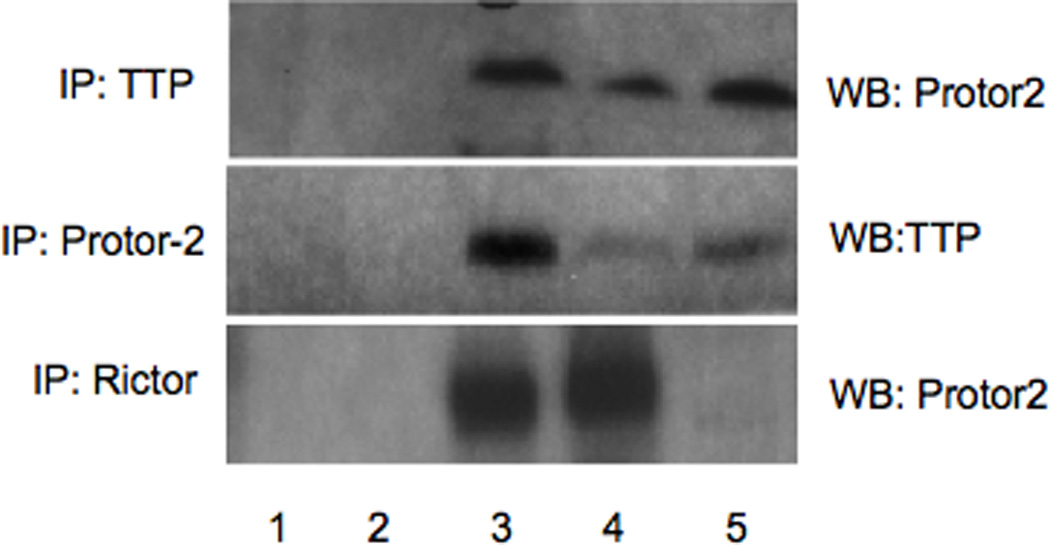
Protor-2 interacts with TTP in vivo. TTP, Protor-2 or Rictor was immunoprecipitated from Jurkat or TSCsiJurkat cells as indicated. Western blots were subsequently performed on the immunoprecipitates for the indicated proteins. Lane 1, beads without antibody; lane 2, immunoprecipitation using irrelevant antibody (actin Ab), lane 3, input uninduced Jurkat cell lysate, lane 4, immunoprecipitation antibody, lane 5, TSCsiJurkat cell lysate with immunoprecipitation antibody.
3.4. Overexpression of Protor-2 leads to accelerated mRNA turnover of TTP-associated transcripts
To determine whether the association of Protor-2 with TTP had functional significance, in terms of TTP-mediated regulation of mRNA stability, we initially tested whether overexpression of Protor-2 affected mRNA turnover in Jurkat cells. We stably transfected cells with myc epitope-tagged Protor-2 and determined the relative half-lives of several TTP-associated mRNA including TNF-α, GM-CSF, IL-3 and COX-2 [6] in standard ActD-half-life experiments. We also determined the half-lives of non-ARE containing mRNAs, tubulin and GAPDH in these cells. As shown in figure 3, cells overexpressing Protor-2 (Fig. 3A) displayed increased mRNA turnover of all the TTP regulated transcripts examined (Fig. 3B). For example, the TNF-α mRNA which has a relatively short native half-life of 30 minutes in control cells, decayed with a half-life of only 10 minutes in cells overexpressing Protor-2. The relative stabilities of tubulin and GAPDH mRNA were unaffected by Protor-2 overexpression. Moreover, we found that the TNF-α, GM-CSF, IL-3 and COX-2 transcripts also had relatively shorter half-lives in TSC1/2 deficient cells as compared to control cells while the half-lives of tubulin and GAPDH mRNA was unaltered (Fig. 3C). Treatment of cells with tetracycline to induce TSC1/2 knockdown had no effect on the half-lives of these mRNAs (data not shown). These data suggest that Protor-2 is involved in the regulation of mRNA stability of transcripts associated with TTP.
Figure 3.
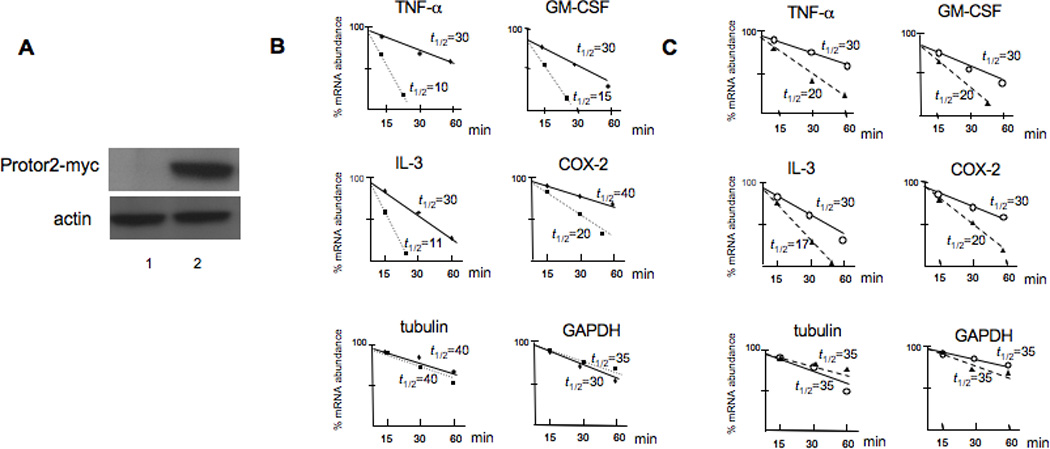
Overexpression of Protor-2 promotes TTP-associated mRNA turnover. A. Expression of myc-tagged Protor-2 in Jurkat cells. Lane 1, JurkatPuro control transfectants, Lane 2, JurkatProtor2 overexpression clone. Western blot was probed with a-myc epotope antibodies (4E11) and actin. B. Relative mRNA turnover and half-lives (t1/2) of the indicated transcripts from JurkatPuro (shaded circles) or JurkatProtor2 (shaded squares) as determined by ActD-mRNA half-life experiments followed by real time qRT-PCR of the indicated transcripts. C. Relative mRNA turnover of the indicated mRNAs in JurkatZeo (open circles) versus TSCsiJurkat (shaded triangles) cells following tet-induced suppression of TSC1/TSC2.
3.5. Knockdown of Protor-2 inhibits TTP-mediated mRNA turnover during stress
We then determined whether inhibition of Protor-2 via RNAi affected the relative mRNA stabilities of the transcripts we had previously examined following stress-induced destabilization. TTP has been shown to participate in the regulated destabilization of mRNAs following the induction of cellular stress [31]. We transiently transfected Jurkat cells with non-targeting siRNA (scrambled, scr) or siRNA targeting Protor-2 and examined the half-lives of the TNF-α, GM-CSF, IL-3 and COX-2 mRNAs as before, following treatment with the mitochondrial inhibitor FCCP. As shown in figure 4A, transfection of these cells with siRNA targeting Protor-2 resulted in greater then 95% inhibition of expression as compared to controls and was specific in that actin expression was unaffected. As shown in figure 4B, inhibition of Protor-2 expression resulted in significant increases in mRNA half-life for these transcripts. The TNF-α mRNA half-life increased from 15 min in FCCP-treated cells to over 25 min in cells in which Protor-2 was knocked down following FCCP treatment. Similarly, the GM-CSF mRNA half-life increased from 15 min to 30 min, the IL-3 mRNA half-life increased similarly from 15 min to 30 min and the COX-2 mRNA half-live increased from 10 min to 25 min. The half-lives of the tubulin and GAPDH mRNAs were unchanged irrespective of FCCP treatment or Protor-2 knockdown. These results indicate that Protor-2 knockdown results in inhibition of TTP-mediated mRNA destabilization during stress.
Figure 4.
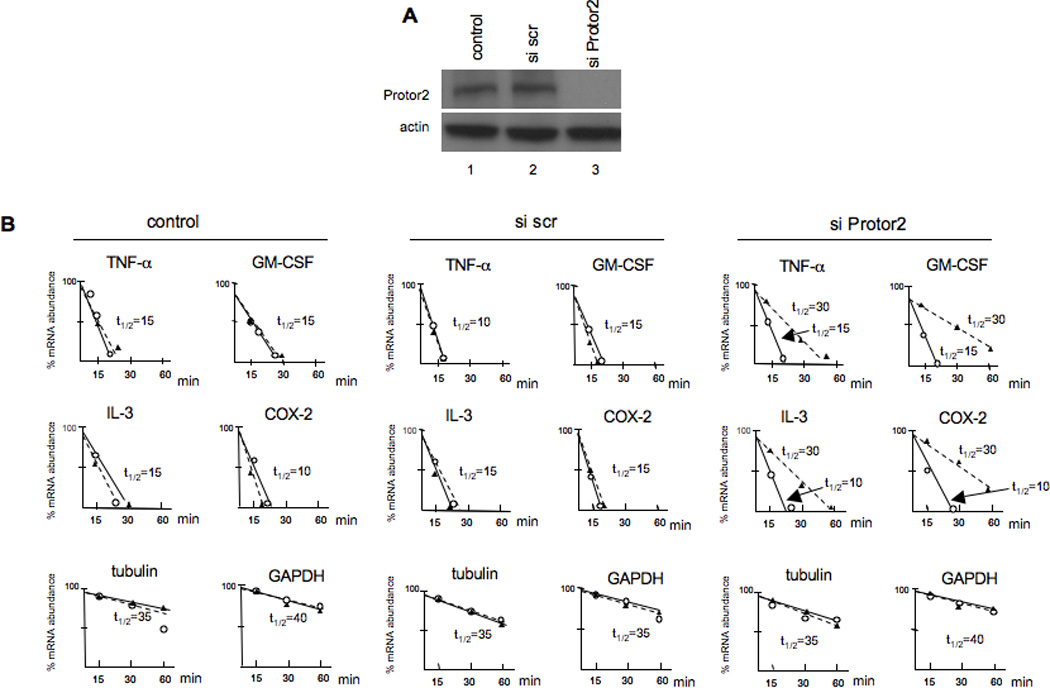
Effect of Protor-2 knockdown on TPP-associated mRNAs during stress-induced destabilization. A. Jurkat cells were transfected with control, non-targeting scrambled sequence or Protor-2-targeting siRNAs and immunoblotted for the indicated proteins. B. Relative mRNA turnover and half-lives (t1/2) of Jurkat cells treated with FCCP (10 µM) and transfected with control, scrambled sequence or Protor-2-targeting siRNAs as indicated.
3.6. Knockdown of Protor-2 inhibits TTP association with stress granules or P bodies during the integrated stress response
Recent data indicate that TTP is found in discrete cytoplasmic foci in cells subjected to environmental stress [1]. Stress granules represent one type of these structures in which mRNAs are translationally arrested initiated via phosphorylation of the translation initiation factor eIF2α [32, 33]. Processing bodies (P-bodies) are similar structures, however current data suggests that these are exclusively sites of mRNA degradation [34]. TTP has been found in both SGs and P-bodies upon energy starvation following exposure to the mitochondrial inhibitor FCCP [12]. Thus, we determined whether knockdown of Protor-2 affected the ability of TTP to localize to SGs or P-bodies during the starvation response. As shown in figure 5, TTP was found to localize in both SGs and P-bodies following FCCP exposure in HeLa cells, consistent with prior reports [1, 12]. However, in cells which had been transfected with siRNAs targeting Protor-2, resulting in greater then 95% inhibition of Protor-2 expression (see supplementary fig. 1), TTP did not localize into discrete SGs or P-bodies and was found diffusely localized to the cytoplasm. These data indicate that Protor-2 is required for TTP localization to SGs and P-bodies following exposure to FCCP.
Figure 5.
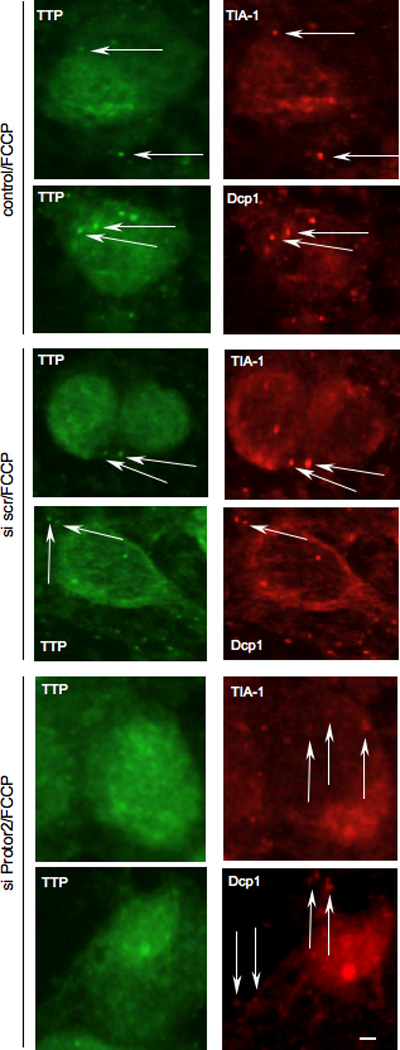
Inhibition of Protor-2 expression reduces TTP association with stress granules and P-bodies. HeLa cells were treated with control, non-targeting or Protor-2-targeting siRNA and treated with FCCP (1 µM) to induce SG and P-bodies formation. Subcellular localization of TTP and stress granule (TIA-1) and P-body (Dcp1) markers (right panels, red, as indicated) were determined by dual label immunofluorescence using CARP-3, an affinity-purified antibody reactive with TTP (left panels, green). Arrows point out SGs and P-bodies. Bar size is 10 µm.
4. Discussion
Our previous data have demonstrated that TTP plays an important role in the regulation of mRNA stability in response to mTOR inhibition [24]. Here we demonstrate that TTP function is linked to TOR activity via an mTORC2 component Protor-2. Protor-2 was identified as an interactor with TTP in a yeast two-hybrid screen and the two proteins were subsequently shown to be co-immunoprecipitatable in whole cell extracts. Binding of Protor-2 to TTP was shown to require sequences within the C-terminal half of both proteins and binding was induced in mTORC2 activated cells, consistent with previous reports describing the liberation of Protor-2 from the mTORC2 complex following hyperactivation [23]. Forced ectopic expression of Protor-2 resulted in increased mRNA turnover of transcripts known to be regulated by TTP. Additionally, knockdown of Protor-2 reduced mRNA degradation of TTP-associated messages and inhibited TTP association with SGs and P-bodies following stress induction. These data are most consistent with a model of mTORC2 activity dependent TTP function in which, upon activation of mTORC2, Protor-2 is released from the complex and subsequently associates with TTP to promote decay of TTP-regulated mRNAs. In support of this model, we also tested the notion of whether the mitochondrial oxidative phosphorylation uncoupler FCCP would induce mTORC2 activity. As shown in supplementary figure 2, FCCP exposure induced phospho-S473-AKT accumulation in a rictor-dependent fashion. These data are consistent with reports demonstrating that several inhibitors of mitochondrial function can induce AKT activity via the accumulation of reactive oxygen species or inactivation of PTEN [35, 36]. Additionally, we have demonstrated that during other cellular stress responses such as heat shock, the mTORC2/AKT pathway is activated [37].
TTP is known to play a role in the regulated mRNA turnover of several mRNA following T lymphocyte activation [2]. TPA or LPS mediated activation of T cells results in induction of immediate, early genes the transcripts for several of which contain AREs which bind TTP. Current models suggest that upon cell stimulation, a class of TTP-regulatable mRNAs are induced coordinately with TTP expression. TTP then binds to these mRNAs and selectively mediates transcript destabilization resulting in a rapid cessation of expression of these factors [6]. Many of these mRNAs encode pro-inflammatory proteins whose overexpression may be deleterious to the cell and thus, are regulated in this manner post-transcriptionally. The role of mTORC2 activity in the regulation of immediate, early gene expression is not known, however our data are indicative that it may play a role in the regulation of TTP-mediated mRNA destabilization. It is tempting to speculate that mTORC2 is activated following T cell stimulation and liberates Protor-2 which promotes TTP-mediated mRNA turnover.
The role of TTP in the regulation of mRNA turnover in response to cell stress is well known [6]. However, the molecular events regulating TTP activity have only recently been described [12, 38]. The expression of ARE-containing mRNAs requires the activation of signaling cascades which ablate this decay program. The p38 MAPK/MK2 cascade phosphorylates TTP at two residues (serines 52 and 178) which induces adaptor 14-3-3 protein binding and sequesters TTP within the cytoplasm of the cell [12]. 14-3-3 protein binding also results in the exclusion of TTP from cytoplasmic stress granules (SGs) and mRNA processing bodies (P-bodies), sites of storage of translationally arrested mRNAs and mRNA degradation, respectively [1, 12]. The observation that knockdown of Protor-2 inhibits TTP association with these subcellular structures, suggests a role for Protor-2 in trafficking TTP-bound mRNA substrates to SGs and/or P-bodies. The mechanism of Protor-2 promotion of TTP mediated decay may involve increased binding affinities of TTP to its mRNA substrates, regulating subcellular localization or controlling subsequent steps in the decay process such as mRNA deadenylation or decapping. It is also of interest whether other TIS11 family members such as BRF1 and BRF2, can bind Protor-2 and are potentially subject to mTORC2 regulation.
Recently Kedar et al., [39] have reported TTP interactors derived from large-scale yeast two-hybrid screening efforts. As in our experiments, full-length TTP was found to be autoactivating, as were baits containing N-terminal fragments of the protein. While there was no overlap in the reported focused list of TTP interacting proteins with those identified in this study, the identification of proline-rich domain containing proteins was in common. In our screens, we also identified components of the mRNA decay machinery which appear to be able to interact with the C-terminal mRNA decay domain of TTP. Other studies have suggested that the N-terminal decay domain may function as a binding platform for mRNA decay enzymes [7], however our results suggest that the C-terminal domain is also able to interact with components of the decay machinery. Additionally, it is also possible that the C-terminal domain may interact with other factors involved in mRNA remodeling or in the localization of mRNAs to cytoplasmic processing-bodies as previously suggested [7]. Our finding that the RNA-helicase RHAU can bind the C-terminal domain of TTP supports this [40].
The regulatory subunits of several multisubunit kinase complexes have been reported as having kinase-independent functions [41]. These include the p85α regulatory subunit of PI3K, which has been shown to potentiate JNK signaling under certain conditions [42]. It has also been proposed that the glucose regulatable yeast Snf1 kinase complex may be targeted to specific intracellular locations via interaction with regulatory subunits [43]. There is also evidence that the regulatory subunits of casein kinase II have functions independent of the holoenzyme involving activation of c-Raf, c-Mos or Chk1 [41]. It is certainly possible that many regulatory subunits or closely associated factors of specific kinase complexes have evolved kinase-independent functions and our studies with Protor-2 support this. In fact, the Rictor and Sin1 mTORC2 regulatory subunits are known to have TOR-independent functions [44, 45].
In conclusion, this work presents experimental evidence that mTORC2 activity may be functionally linked to regulated mRNA turnover. Recently Protor-2 has been implicated in the promotion of apoptosis [23]. The regulated destabilization of antiapoptotic mRNAs known to contain AREs, such as Bcl-2, would be consistent with the function of Protor-2 we have observed and its role in apoptosis.
Highlights.
> The mTORC2 kinase component Protor-2 is found to interact with the mRNA destabilizing factor tristetraprolin. > Modulation of Protor-2 levels specifically alters the stability of mRNAs known to be degraded by tristetraprolin. > Inhibition of Protor-2 expression blocks the association of tristetraprolin with stress granules or processing bodies during stress. > We conclude that Protor-2 regulates tristetraprolin mediated mRNA turnover and may link mTORC2 signaling to regulated mRNA stability.
Supplementary Material
Supplementary figure 1. siRNA-mediated inhibition of Protor-2 expression. HeLa cells were transfected with control, non-targeting scrambled (scr) or Protor-2 targeting siRNA as indicated (10 nM, 24 hr) and lysates immunoblotted for Protor-2 and actin.
Supplementary figure 2. Knockdown of Rictor expression via siRNAs and induction of mTORC2 activity by FCCP. A, As in supplementary figure 1, except Rictor-targeting siRNAs were used and lysates immunobloted for Rictor. B. FCCP activates mTORC2 in a Rictor-dependent manner. HeLa cells were treated with FCCP (1 µM) following Rictor knockdown as indicated and lysates immunoblotted for phospho-S473-AKT, Rictor and actin. As can be seen, FCCP treatment resulted in the accumulation of S473-phosphorylated AKT which was markedly abrogated in Rictor siRNA transfected cells.
Acknowledgments
We thank Drs. Michael Hall and William Rigby for providing cell lines and antibodies, Dr. Robert Nishimura for critical reading of the manuscript and Ardella Sherwood for excellent administrative assistance. This work was supported, in part, by grants from the NIH (RO1CA109312) and the US Department of Veterans Affairs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Stoecklin G, Anderson P. Genes Dev. 2007;21:627–631. doi: 10.1101/gad.1538807. [DOI] [PubMed] [Google Scholar]
- 2.Carrick D, Lai W, Blackshear P. Arthritis Res Ther. 2004;6:248–264. doi: 10.1186/ar1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson P, Phillips K, Stoecklin G, Kedersha N. J Leukoc Biol. 2004;76:42–47. doi: 10.1189/jlb.1103536. [DOI] [PubMed] [Google Scholar]
- 4.Chen C-YA, Shyu A-B. Trends in Biochemical Sciences. 1995;20:465–470. doi: 10.1016/s0968-0004(00)89102-1. [DOI] [PubMed] [Google Scholar]
- 5.Lai WS, Kennington EA, Blackshear PJ. Mol. Cell. Biol. 2003;23:3798–3812. doi: 10.1128/MCB.23.11.3798-3812.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blackshear PJ. Biochem. Soc. Trans. 2002;30:945–952. doi: 10.1042/bst0300945. [DOI] [PubMed] [Google Scholar]
- 7.Lykke-Andersen J, Wagner E. Genes Dev. 2005;19:351–361. doi: 10.1101/gad.1282305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, Schenkman DI, Gilkeson GS, Broxmeyer HE, Haynes BF, Blackshear PJ. Immunity. 1996;4:445–454. doi: 10.1016/s1074-7613(00)80411-2. [DOI] [PubMed] [Google Scholar]
- 9.Lai WS, Carballo E, Strum JR, Kennington EA, Phillips RS, Blackshear PJ. Mol. Cell. Biol. 1999;19:4311–4323. doi: 10.1128/mcb.19.6.4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tchen CR, Brook M, Saklatvala J, Clark AR. J. Biol. Chem. 2004;279:32393–32400. doi: 10.1074/jbc.M402059200. [DOI] [PubMed] [Google Scholar]
- 11.Pagano JM, Farley BM, McCoig LM, Ryder SP. J. Biol. Chem. 2007;282:8883–8894. doi: 10.1074/jbc.M700079200. [DOI] [PubMed] [Google Scholar]
- 12.Stoecklin G, Stubbs T, Kedersha N, Wax S, Rigby W, Blackwell T, Anderson P. EMBO J. 2004;23:1313–1324. doi: 10.1038/sj.emboj.7600163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chrestensen CA, Schroeder MJ, Shabanowitz J, Hunt DF, Pelo JW, Worthington MT, Sturgill TW. J. Biol. Chem. 2004;279:10176–10184. doi: 10.1074/jbc.M310486200. [DOI] [PubMed] [Google Scholar]
- 14.Hermeking H, Benzinger A. Seminars in Cancer Biology. 2006;16:183–192. doi: 10.1016/j.semcancer.2006.03.002. [DOI] [PubMed] [Google Scholar]
- 15.Soulard A, Hall MN. Cell. 2007;129:434.e431–434.e432. doi: 10.1016/j.cell.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 16.Reiling JH, Sabatini DM. Oncogene. 25:6373–6383. doi: 10.1038/sj.onc.1209889. [DOI] [PubMed] [Google Scholar]
- 17.Guertin D, Sabatini D. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 18.Sabatini DM. Nat Rev Cancer. 2006;6:729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 19.Sturgill TW, Hall MN. Nat Cell Biol. 2007;9:1221–1222. doi: 10.1038/ncb1107-1221. [DOI] [PubMed] [Google Scholar]
- 20.Albig AR, Decker CJ. Mol. Biol. Cell. 2001;12:3428–3438. doi: 10.1091/mbc.12.11.3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Foat BC, Houshmandi SS, Olivas WM, Bussemaker HJ. Proc Natl Acad Sci U S A. 2005;102:17675–17680. doi: 10.1073/pnas.0503803102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hsu PP, Kang SA, Rameseder J, Zhang Y, Ottina KA, Lim D, Peterson TR, Choi Y, Gray NS, Yaffe MB, Marto JA, Sabatini DM. Science. 2011;332:1317–1322. doi: 10.1126/science.1199498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thedieck K, Polak P, Kim M, Molle K, Cohen A, Jeno P, Arrieumerlou C, Hall M. PLoS ONE. 2007;2:e1217. doi: 10.1371/journal.pone.0001217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marderosian M, Sharma A, Funk AP, Vartanian R, Masri J, Jo OD, Gera JF. Oncogene. 2006;25:6277–6290. doi: 10.1038/sj.onc.1209645. [DOI] [PubMed] [Google Scholar]
- 25.Gera J, Hazbun T, Fields S. Methods Enzymol. 2002;350:499–512. doi: 10.1016/s0076-6879(02)50981-2. [DOI] [PubMed] [Google Scholar]
- 26.Sharma A, Masri J, Jo OD, Bernath A, Martin J, Funk A, Gera J. J. Biol. Chem. 2007;282:9505–9516. doi: 10.1074/jbc.M608874200. [DOI] [PubMed] [Google Scholar]
- 27.Livak KJ, Schmittgen TD. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 28.Masri J, Bernath A, Martin J, Jo OD, Vartanian R, Funk A, Gera J. Cancer Res. 2007;67:11712–11720. doi: 10.1158/0008-5472.CAN-07-2223. [DOI] [PubMed] [Google Scholar]
- 29.Farina KL, Huttelmaier S, Musunuru K, Darnell R, Singer RH. J. Cell Biol. 2003;160:77–87. doi: 10.1083/jcb.200206003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pearce LR, Huang X, Boudeau J, Pawlowski R, Wullschleger S, Deak M, Ibrahim AF, Gourlay R, Magnuson MA, Alessi DR. Biochem J. 2007;405:513–522. doi: 10.1042/BJ20070540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abdelmohsen K, Kuwano Y, Kim HH, Gorospe M. Biol Chem. 2008;389:243–255. doi: 10.1515/BC.2008.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Anderson P, Kedersha N. J. Cell Biol. 2006;172:803–808. doi: 10.1083/jcb.200512082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kedersha N, Anderson P. Biochem. Soc. Trans. 2002;30:963–969. doi: 10.1042/bst0300963. [DOI] [PubMed] [Google Scholar]
- 34.Parker R, Sheth U. Molecular Cell. 2007;25:635–646. doi: 10.1016/j.molcel.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 35.Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. J Cell Sci. 2007;120:4155–4166. doi: 10.1242/jcs.011163. [DOI] [PubMed] [Google Scholar]
- 36.Pelicano H, Xu RH, Du M, Feng L, Sasaki R, Carew JS, Hu Y, Ramdas L, Hu L, Keating MJ, Zhang W, Plunkett W, Huang P. J Cell Biol. 2006;175:913–923. doi: 10.1083/jcb.200512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin J, Masri J, Bernath A, Nishimura RN, Gera J. Biochem Biophys Res Commun. 2008;372:578–583. doi: 10.1016/j.bbrc.2008.05.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun L, Stoecklin G, Van Way S, Hinkovska-Galcheva V, Guo R-F, Anderson P, Shanley TP. J. Biol. Chem. 2007;282:3766–3777. doi: 10.1074/jbc.M607347200. [DOI] [PubMed] [Google Scholar]
- 39.Kedar VP, Darby MK, Williams JG, Blackshear PJ. PLoS One. 2010;5:e9588. doi: 10.1371/journal.pone.0009588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chalupnikova K, Lattmann S, Selak N, Iwamoto F, Fujiki Y, Nagamine Y. J Biol Chem. 2008;283:35186–35198. doi: 10.1074/jbc.M804857200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bibby A, Litchfield D. Int J Biol Sci. 2005;1:67–79. doi: 10.7150/ijbs.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Taniguchi CM, Aleman JO, Ueki K, Luo J, Asano T, Kaneto H, Stephanopoulos G, Cantley LC, Kahn CR. Mol. Cell. Biol. 2007;27:2830–2840. doi: 10.1128/MCB.00079-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ludin K, Jiang R, Carlson M. Proceedings of the National Academy of Sciences. 1998;95:6245–6250. doi: 10.1073/pnas.95.11.6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghosh D, Srivastava GP, Xu D, Schulz LC, Roberts RM. Proc Natl Acad Sci U S A. 2008;105:11673–11678. doi: 10.1073/pnas.0803182105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McDonald PC, Oloumi A, Mills J, Dobreva I, Maidan M, Gray V, Wederell ED, Bally MB, Foster LJ, Dedhar S. Cancer Res. 2008;68:1618–1624. doi: 10.1158/0008-5472.CAN-07-5869. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure 1. siRNA-mediated inhibition of Protor-2 expression. HeLa cells were transfected with control, non-targeting scrambled (scr) or Protor-2 targeting siRNA as indicated (10 nM, 24 hr) and lysates immunoblotted for Protor-2 and actin.
Supplementary figure 2. Knockdown of Rictor expression via siRNAs and induction of mTORC2 activity by FCCP. A, As in supplementary figure 1, except Rictor-targeting siRNAs were used and lysates immunobloted for Rictor. B. FCCP activates mTORC2 in a Rictor-dependent manner. HeLa cells were treated with FCCP (1 µM) following Rictor knockdown as indicated and lysates immunoblotted for phospho-S473-AKT, Rictor and actin. As can be seen, FCCP treatment resulted in the accumulation of S473-phosphorylated AKT which was markedly abrogated in Rictor siRNA transfected cells.