

Abstract
The neurotoxicant 6-hydroxydopamine (6-OHDA) is used to investigate the cellular and molecular mechanisms underlying selective degeneration of dopaminergic neurons in Parkinson’s disease (PD). Oxidative stress and caspase activation contribute to the 6-OHDA-induced apoptotic cell death of dopaminergic neurons. In the present study, we sought to systematically characterize the key downstream signaling molecule involved in 6-OHDA-induced dopaminergic degeneration in cell culture and animal models of PD. Treatment of mesencephalic dopaminergic neuronal N27 cells with 6-OHDA (100 μM) for 24h significantly reduced mitochondrial activity and increased cytosolic cytochrome c, followed by sequential activation of caspase-9 and caspase-3. Co-treatment with the free radical scavenger MnTBAP (10 μM) significantly attenuated 6-OHDA-induced caspase activities. Interestingly, 6-OHDA induced proteolytic cleavage and activation of protein kinase C delta (PKCδ) was completely suppressed by treatment with a caspase-3-specific inhibitor, Z-DEVD-FMK (50 μM). Furthermore, expression of caspase-3 cleavage site-resistant mutant PKCδD327A and kinase dead PKCδK376R or siRNA-mediated knockdown of PKCδ protected against 6-OHDA-induced neuronal cell death, suggesting that caspase-3-dependent PKCδ promotes oxidative stress-induced dopaminergic degeneration. Suppression of PKCδ expression by siRNA also effectively protected N27 cells from 6-OHDA-induced apoptotic cell death. PKCδ cleavage was also observed in the substantia nigra of 6-OHDA-injected C57 black mice but not in control animals. Viral-mediated delivery of PKCδD327A protein protected against 6-OHDA-induced PKCδ activation in mouse substantia nigra. Collectively, these results strongly suggest that proteolytic activation of PKCδ is a key downstream event in dopaminergic degeneration, and these results may have important translational value for development of novel treatment strategies for PD.
Keywords: Oxidative Stress, 6-OHDA, PKC delta, Apoptosis, animal model, Parkinson’s disease
Introduction
Parkinson’s disease (PD) is the most common neurodegenerative movement disorder affecting more than 1% of the population over the age of 60. The significant loss of dopaminergic neurons (>70%) in the substantia nigra pars compacta is a hallmark of PD (Burke and Kholodilov, 1998; Orth and Tabrizi, 2003). Metabolic or neurotoxic insults often cause oxidative stress-mediated neuronal apoptosis and thereby contribute to the pathogenesis and progression of several forms of neurodegenerative diseases, including PD. Oxidative stress results from the imbalance of pro-oxidant/antioxidant homeostasis, leading to the generation of toxic reactive oxygen species (ROS). Oxidative stress often causes extensive damage to lipids, proteins, and DNA, resulting in cell death by a variety of different mechanisms including activation or inactivation of various apoptotic cell signaling molecules. Typically, unregulated ROS generation results in calcium dysregulation that leads to excitotoxic cell death or to mitochondrial dysfunction, resulting in activation of the apoptotic caspase cascade (Henchcliffe and Beal, 2008; Zhou et al., 2008; Naoi et al., 2009; Tsang and Chung, 2009; Cannon and Greenamyre, 2010; Kanthasamy et al., 2010).
MPTP, 6-OHDA and rotenone are neurotoxins commonly used to investigate the pathogenesis and progression of PD in cellular and animal models (Moore et al., 2005; Andrabi et al., 2008; Henchcliffe and Beal, 2008; Zhou et al., 2008; Lee et al., 2009; Naoi et al., 2009; Shin et al., 2009; Tsang and Chung, 2009; Beal, 2010; Cannon and Greenamyre, 2010; Horowitz and Greenamyre, 2010; Kanthasamy et al., 2010; Gao et al., 2011). Results from MPTP and 6-OHDA studies suggest that oxidative stress-mediated apoptotic cell death is a major contributing factor to the selective degeneration of dopaminergic neurons in the PD model. 6-OHDA has been shown to impair mitochondrial dysfunction and caspase activation in a variety of cell lines and primary neuronal cultures (Choi et al., 1999; Dodel et al., 1999; Elkon et al., 2004; Jordan et al., 2004; Liu et al., 2007; Puttonen et al., 2008; Kim et al., 2010; Kupershmidt et al., 2010; Yoon et al., 2010). However, the role of cell signaling molecules downstream of caspases in the degeneration of dopaminergic neurons in PD is yet to be identified.
Recently, we identified the novel PKC isoform protein kinase Cδ (PKCδ) is highly expressed in nigral dopaminergic neurons (Zhang et al., 2007b) and the kinase is a prominent endogenous substrate for caspase-3, as well as a key player in oxidative stress-induced neuronal apoptosis (Kanthasamy et al., 2003). The PKCδ is proteolytically cleaved by caspase-3 to yield a 38 kDa regulatory fragment and a 41 kDa persistently active catalytic fragment (Greenberg et al., 1998; Brodie and Blumberg, 2003). In the present study, we examined the role of PKCδ in 6-OHDA-induced dopaminergic cell death by using loss of PKCδ function dominant negative mutants and RNAi-mediated knockdown of PKCδ expression. Both cell culture and animal models were used to delineate the PKCδ signaling in 6-OHDA-triggered oxidative damage. We demonstrated that proteolytic cleavage of PKCδ by caspase-3 is a critical event in 6-OHDA-induced dopaminergic degeneration in cell culture and animal models of PD.
Materials and Methods
Materials
ATP, Protein-A-Sepharose, 6-hydroxydopamine, and β-actin mouse monoclonal antibody were obtained from Sigma, Z-Asp-Glu-Val-Asp-fluoromethyl ketone (Z-DEVD-FMK), Acetyl-Leu-Glu-His-Asp-7-amino-4-fluorcoumarin (Ac-LEHD-AFC) and acetyl-Asp-Glu-Val-Asp-7-amino-4-fluorcoumarin (Ac-DEVD-AFC), were obtained from MP Biochemicals (Irvine, CA). Anti-PKCδ antibody (rabbit polyclonal) was purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and the ECL chemiluminescence kit was purchased from Amersham Pharmacia Biotech (Piscataway, NJ). Sytox green nucleic acid fluorescence stain was purchased from Molecular Probes (Eugene, OR). Cell Death Detection ELISA Plus assay kit was purchased from Roche Molecular Biochemicals (Indianapolis, IN). [32P-]ATP was purchased from Perkin Elmer (Downers Grove, IL). Bradford protein assay kit was purchased from Bio-Rad (Hercules, CA). RPMI-1640 medium, horse serum, fetal bovine serum, L-glutamine, penicillin, and streptomycin were obtained from Invitrogen (Gaithersburg, MD). Plasmids for pPKCδK376R-green fluorescent protein (GFP) and pEGFP-N1 were kind gifts from Dr. Stuart Yuspa (National Cancer Institute, Bethesda, MD). PKCδD327A-GFP (PKCδ-CRM) construct was a kind gift from Dr. Mary Reyland at the University of Colorado (Boulder, CO).
Cell culture
The immortalized rat mesencephalic dopaminergic neuronal cell line (N27) was developed from the ventral mesencephalon, a region of the brain that is directly affected in PD (Zhou et al., 2000). N27 cells represent a homogenous population of tyrosine hydroxylase-positive (TH+) neurons with functional characteristics including dopamine synthesis and cellular signaling pathway (Zhang et al., 2007b, Jin et al., 2011a, Jin et al., 2011b). We and others have extensively used this cell model for studying neurodegenerative mechanisms in PD (Kaul et al., 2003; Kitazawa et al., 2003; Anantharam et al., 2004; Latchoumycandane et al., 2005; Kanthasamy et al., 2010). The cells were grown in RPMI 1640 medium containing 10% fetal bovine serum (FBS), 2 mM L-glutamine, 50 U penicillin and 50 μg/ml streptomycin (Kaul et al., 2003; Anantharam et al., 2004). Cells were maintained in a humidified atmosphere of 5% CO2 at 37°C. N27 cells of passage 2–4 were used for most of the experiments.
Primary mesencephalic neuronal culture
Primary mesencephalic neuronal cultures were prepared from the ventral mesencephalon from embryonic day 15–17 Sprague Dawley rats as described previously (Yang et al., 2004; Afeseh Ngwa et al., 2009; Song et al. 2010). Briefly, ventral mesencephalic brain regions were dissected out of the rat embryos and the tissues were maintained on ice-cold, calcium-free EBSS supplemented with gentamycin (50 mg/ml) and penicillin/streptomycin (200 U), and then dissociated in EBSS solution containing trypsin (0.25%) for 15 min. The dissociated cells were plated at equal density (0.5 × 106 cells) in 30 mm-diameter tissue culture wells precoated with poly-L-lysine (1 mg/ml). The primary cultures were maintained in a chemically defined, serum-free media consisting of neurobasal medium fortified with B-27 supplements, L-glutamine (500 μM), penicillin (100 U), and streptomycin (100 μg/ml) (Life Technologies). The cells were maintained in a CO2 incubator at 5% CO2 and 37°C for 24 h, and then treated with cytosine arabinoside (10 μM) for 24 h to inhibit glial cell proliferation. Half of the culture medium was replaced every 2 days. Approximately 6–7 day-old cultures were used for experiments.
Animal studies
Six- to 8-week-old 26/C57/Bl mice weighing 25–30 g were housed in standard conditions: constant temperature (22 ± 1°C), humidity (relative, 30%), and a 12-h light/dark cycle. Mice were allowed free access to food and water. Use of the animals and protocol procedures were approved and supervised by the Institutional Animal Care and Use Committee (IACUC) at Iowa State University. C57 black mice were injected stereotaxically with 4 μl of 1 μg/l 6-OHDA (in 0.2% ascorbic acid) into the right substantia nigra (SN) and saline into the left SN at 10 angles of the following coordinates from the bregma: AP: −3.2; ML: 2.0; and DV: −4.7 as described previously (Filipov et al., 2002). The mice were sacrificed 7 days later; left and right striatum and SN were removed and processed for Western blot and immunoprecipitation kinase assays. For PKCδ-CRM lentiviral studies in animals, C57 black mice were injected stereotaxically with lenti-PKCδ-CRM (2 μl viral particles (5×109 gc/mL) at a rate of 0.2 μl/min using a microinjection pump into the right SN and lenti-LacZ into the left SN. After 6 days of lentiviral injection, the mice were re-injected with 4μg 6-OHDA. The mice were sacrificed 7-days later; left and right striatum and SN were removed and subjected to neurochemical, biochemical, and immunohistochemical analyses including western blot and immunoprecipitation kinase assays.
Assessment of cell viability
Cell viability was determined by trypan blue exclusion method and Sytox assay after exposing N27 cells to 100 μM 6-OHDA (Latchoumycandane et al., 2005). Sytox assay was determined by using a cell-impermeable nucleic acid fluorescence-based dye (Sytox), which enters into dead cells and intercalates with DNA to produce green fluorescence (Jin et al., 2011a). Sytox-stained cells were then observed using a Nikon TE 200 fluorescence microscope, and images were captured with a Spot RT camera.
Determination of mitochondrial activity
The N27 cells were incubated with 100 μM 6-OHDA for 12–24 h, and mitochondrial activity was determined by using MTT (3-[4,5-dimethylthiazol-3-yl]-2,5-diphenyl tetrazolium bromide) assay, which is based on the formation of formazan from tetrazolium inside the active mitochondria (Latchoumycandane et al., 2005). Briefly, N27 cells were plated in 24-well plates and treated with 6-OHDA. After treatment, the cells were washed once with PBS and then incubated in serum-free medium containing 0.25 mg/ml MTT for 2 h at 37°C. Formation of formazan was measured at 570 nm with reference wavelength at 630 nm using a SpectraMax microplate reader (Molecular Devices, Sunnyvale, CA).
Determination of cytosolic cytochrome c
The cytosolic cytochrome c levels were measured in cytosolic fractions obtained from control and 6-OHDA-treated N27 cells using an ELISA Kit as described previously (Anantharam et al., 2002). Briefly, N27 cells (~5 × 106 cells) were exposed to 100 μM 6-OHDA for 3 and 6 h. After treatment, cytosolic fractions were obtained from lysed cells and the level of cytosolic cytochrome c was quantified by ELISA assay using a SpectraMax Gemini XS microplate reader.
Determination of caspase-9 and -3 activities
The activities of caspase-9 and -3 were measured using caspase-9- and -3-specific fluorescence substrates, Ac-LEHD-AFC and Ac-DEVD-AFC, respectively as described previously (Kaul et al., 2003). Briefly, cells (1–2 × 105 cells/well) were subcultured in 24-well tissue culture plates and treated with 100 μM 6-OHDA for 3, 6, 12 and 24 h. Formation of 7-amino-4-methylcoumarine (AMC), resulting from caspase substrate cleavage, was measured using a SpectraMax Gemini XS microplate reader with excitation at 400 nm (slit width 10 nm) and emission at 505 nm (slit width 20 nm). Enzyme activities are expressed as fluorescence units/mg protein/h.
Determination of proteolytic activation of PKCδ
N27 cells (~1 × 107 cells) were exposed to 100 μM 6-OHDA for 12 and 24 h and cell lysates were prepared as described previously (Kaul et al., 2003). N27 cells were washed with phosphate buffered saline (PBS) and resuspended in homogenization buffer containing 25 mM HEPES, pH 7.5, 20 mM β-glycerophosphate, 0.1 mM sodium orthovanadate, 0.1% Triton X-100, 0.3 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5 mM DTT, 10 mM NaF and 4 μg/ml each of aprotonin and leupeptin. The cells were sonicated for 15 sec under ice-cold conditions, and then the samples from control and treated cells were centrifuged at 16,000 × g for 60 min at 4 °C. The supernatants were collected as cell lysates. Mouse brain nigral lysates from 6-OHDA treated animals were prepared as described previously (Zhang et al., 2007). Cell and mouse brain lysates containing equal protein were mixed with 2x gel loading buffer containing 10% SDS and 200 mM DTT, and placed in a boiling water bath for 5 min. Proteins were resolved by 12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and blotted onto nitrocellulose membrane (Bio-Rad Laboratories). The non-specific binding sites were blocked with 5% non-fat dry milk blocking solution (Amersham Pharmacia Biotech), and the membrane was then treated with anti-PKCδ (1:2000 dilution) antibody (Santa Cruz) or tyrosine hydroxylase antibody (1:2000) followed by secondary HRP-conjugated anti-rabbit or anti-mouse (1:2000) antibody. Antibody-bound proteins were detected by an enhanced chemiluminescence (ECL) system using a Kodak Imager (Kodak Image Station 2000R, Eastman Kodak Company, New Haven, CT). To confirm equal protein in each lane, membranes were reprobed with -actin (1:5000).
Determination of PKCδ activity
PKCδ enzymatic activity was measured using immunoprecipitation as described previously (Reyland et al., 1999; Anantharam et al., 2002). The proteins were immunoprecipitated from cell and mouse brain lysates overnight at 4 °C using anti-PKCδ antibody. After immunoprecipitation, 25μl of samples containing PKCδ bound to Sepharose A beads were incubated with 25μl of reaction buffer containing 0.4 mg histone H1 and 5 μCi of [γ-32P] ATP (4,500 Ci/mM) for 10 min at 30 °C. The reaction was terminated after mixing with SDS gel loading buffer (2x) and boiling for 5 min. The samples were separated on a 15% SDS-PAGE, and histone-phosphorylated bands were detected using Phosphoimager (Personal Molecular Imager FX, Bio-Rad Laboratories, Hercules, CA) and quantified using Quantity One 4.2.0 Software (Bio-Rad Laboratories).
Lentiviral-mediated transduction of caspase resistant PKCδD327A and kinase inactive PKCδK376R mutant proteins
We used the ViraPower Lentiviral Expression System (Invitrogen) to stably transduce a caspase-resistant mutant of PKCδ (PKCδD327A, lysine to arginine mutation at position 327) into N27 cells or primary mesencephalic neurons. Initially, the cDNA coding for PKCδD327A (herein referred to as plenti6/PKCδ-CRM) was transferred from pEGFP-N1 vector to plenti6/V5-D-TOPO expression vector using standard cloning procedures. The PCR primers were: forward, 5′CACCATGGCACCCTTCCTGCTC3′ and reverse, 5′AATGTCCAGGAATTGCTCAAAC 3′. The cloning and preparation of lentivirus coding for pLenti6PKCδK376R–DN have been described previously (Kitazawa et al., 2005). Standard expression procedures were used to produce lentiviruses. Briefly, the plenti6/PKCδ-CRM constructs were transfected into human 293FT cells using Lipofectamine 2000 transfection reagent as per the manufacturer’s protocol (Invitrogen). The lentivirus in the medium was collected by centrifuging at 1500 × g for 15 min 48–72 h post-transfection. Lentivirus containing plenti/LacZ was also produced to serve as a vector control. Lentiviruses containing either plenti/PKCδD327A-CRM or plenti/PKCδK376R-DN and polybrene (6 μg/ml) were added into cultured N27 cells. Stable cell lines were established by selection in 10 μg/ml Blasticidin 48 h after transfection. Colonies were isolated, then replated and grown to confluence in T75 flasks. Subsequently, the stable cell lines were maintained in 5 μg/ml blasticidin. PKCδ-CRM-V5-, PKCδ-DN-V5- or LacZ-V5-expressing N27 cells were identified by immunostaining of the V5 epitope present in the C-terminus.
PKCδ siRNA transfections
PKCδ-siRNA was prepared by an in vitro transcription method as described previously (Yang et al., 2004). Transfection of N27 cells was carried out using a commercially available TKO transfection reagent (Mirus Corporation, Madison, WI). Briefly, diluted TKO reagent was mixed with either 25 nM PKCδ siRNA duplex or 25 nM non-specific siRNA to form a lipid-siRNA complex; this complex was then added to the N27 cells (50–70% confluent). After 24 h, N27 cells were treated with 100 μM 6-OHDA for an additional 24 h, then used for DNA fragmentation assay. The siRNA-transfected cells were viewed under a Nikon TE 200 fluorescence microscope and images were captured with a Spot RT camera.
Determination of DNA fragmentation by ELISA
Extent of DNA fragmentation was measured using Cell Death Detection ELISA Plus Assay Kits, as per manufacturer’s protocol (Roche Molecular Biochemicals, Indianapolis, IN). This is a highly sensitive assay for the detection of early apoptotic events that measures the amount of histone-associated low molecular weight DNA in the cytoplasm of cells. Briefly, N27 cells were exposed to 100 μM 6-OHDA with or without 50 μM Z-DEVD-FMK (caspase-3-specific inhibitor) for 24 h. The fragmented DNA was quantified colorimetrically using a SpectraMax microplate reader (Anantharam et al., 2002).
Statistical analysis
All the data were analyzed using Prism 4.0 Software (GraphPad, San Diego, CA). Data were first analyzed using one-way ANOVA. Then Dunnett’s post hoc test was performed to compare the differences between control and treated samples. P-values <0.05 were considered significant.
Results
6-OHDA induces neuronal cell death
Quantification of cell viability or cell death is an important parameter in assessing neurotoxin-induced cytotoxic cell death. The dopaminergic neuronal cells derived from mesencephalic regions are more vulnerable to neurotoxin- and oxidative-stress-induced cell death. In the present study, cell viability was determined by trypan blue exclusion method and cell death was measured by Sytox green cell death assay. N27 cells treated with 100 μM 6-OHDA showed decreased cell viability (50% reduction) at 24 h as compared to control cells (Fig. 1A). Sytox green dye enters the cells via damaged plasma membrane, intercalates with DNA and emits green fluorescence. After 24 h treatment with 100 μM 6-OHDA, Sytox green-positive cells were viewed under a Nikon TE 200 fluorescence microscope, and images were captured with a Spot RT camera. The Sytox assay revealed 50% cell death as compared to control groups (Fig. 1B), confirming the results obtained with trypan blue exclusion assay. Together, these results suggest that exposure to 100 μM 6-OHDA for 24 h causes significant cell death in dopaminergic neuronal cells.
Fig. 1.

Dopaminergic neuronal cytotoxicity induced by the neurotoxin 6-OHDA. (A) Cell viability was measured by the trypan blue exclusion method after exposure to 100 μM 6-OHDA for 24 h. The data represent mean ± SEM for at least two separate experiments in triplicate. ** indicates a significant difference between the means at p<0.01. (B) Cell death was also assessed qualitatively using Sytox green nucleic acid stain. N27 cells were exposed to 100 μM 6-OHDA for 24 h with 1 μM Sytox green, and then observed under fluorescence microscopy with excitation of 485 nm and emission of 538 nm. The Sytox green-positive cells represent dead cells after exposure to 6-OHDA.
6-OHDA alters mitochondrial activity and release of cytochrome c
It is well known that mitochondria play a significant role in apoptosis. In response to a variety of stress stimuli, mitochondrial proteins, such as cytochrome c, are released into the cytosol, to further activate the downstream apoptotic caspase cascade (Susin et al., 1999). In the present study, mitochondrial activity was assessed by the intake and conversion of tetrazolium salts into formazan by intact mitochondria inside the cell. 6-OHDA treatment significantly decreased the mitochondrial activity as compared to control cells in a time-dependent manner (Fig. 2A). Furthermore, 6-OHDA also induced the release of cytochrome c from the mitochondria into the cytosol, as determined by ELISA. Cytochrome c release was observed as early as 3 h after 6-OHDA treatment, and continued to increase steadily up to 6 h (Fig. 2B). Exposure to 100 μM 6-OHDA for 3 and 6 h increased cytosolic cytochrome c levels by 80% and 200%, respectively, as compared with corresponding control cells, suggesting that mitochondrial dysfunction contributes to 6-OHDA-induced cell death.
Fig. 2.

6-OHDA impairs mitochondrial activity and induces cytochrome c release in dopaminergic neurons. (A) Relative mitochondrial activity was determined using MTT; the assay is based on the formation of formazan from tetrazolium inside the active mitochondria. The mitochondrial activity decreased significantly after exposure to 6-OHDA for 12 and 24 h (**p<0.01; n=6). (B) Cytosolic cytochrome c release was determined using an ELISA kit; the levels in the cytosolic fraction increased significantly after 3 and 6 h treatments with 6-OHDA (*p<0.05; **p<0.01; n=8).
6-OHDA activates caspase-3 and -9 in a time-dependent manner
Cytochrome c released from mitochondria is known to activate multiple caspases that play an important role in the execution of apoptosis in neuronal and non-neuronal cells. Exposure to 100 μM 6-OHDA induced increases in caspase-9 and -3 enzyme activities up to 12 h in a time-dependent manner, as compared to control (Fig. 3). The activity of caspase-9 increased by 75% and 65% at 6 h and 12 h, respectively, as compared to untreated controls (Fig. 3A). The activity of caspase-3 increased by 150% and 200% at 6 h and 12 h, respectively (Fig. 3B). These data indicate that caspase-9 activity peaks at 6 h or earlier and precedes caspase-3 activity, which peaks at 12 h or later. Co-treatment with 10 μM MnTBAP, a SOD mimetic, significantly inhibited 6-OHDA-induced increases in caspase-3 activity when compared with 6-OHDA-treated cells alone (Fig. 3C). MnTBAP inhibited caspase-3 activity by 71% and 74% at 6 h and12 h, respectively, when compared to 6-OHDA treated cells. This suggests that free radicals are critically involved in 6-OHDA-induced caspase-3 activation.
Fig. 3.
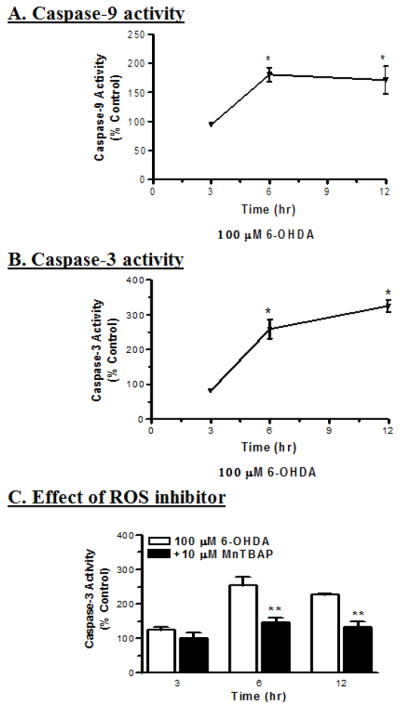
6-OHDA induces ROS-mediated activation of caspases in dopaminergic neuronal cells. The activities of caspase-9 (A) and caspase-3 (B) were determined using fluorogenic substrates, Ac-LEHD-AMC and Ac-DEVD-AMC, respectively, with excitation of 380 nm and emission of 460 nm. Significant increases in the activities of caspase-9 (n=6; *p<0.05) and caspase-3 (n=6; *p<0.05) were observed after exposure to 6-OHDA for 6 and 12 h. (C) Co-treatment with 10 μM MnTBAP (free radical scavenger) protected against 6-OHDA-induced changes in caspase-3 activities, with significant differences between time points (n=6; **p<0.01).
Caspase-3 mediates proteolytic activation of PKCδ in 6-OHDA-treated neuronal cells
In the present study, exposure to 100 μM 6-OHDA for 12 and 24 h resulted in concomitant increases in the magnitude of 41 kDa catalytic and 38 kDa regulatory PKCδ fragments (Fig. 4A), as compared to control, in a time-dependent manner. Co-treatment with 50 μM Z-DEVD-FMK, a caspase-3 specific inhibitor, almost completely blocked the 6-OHDA-induced PKCδ cleavage at both time points of 12 h and 24 h, suggesting that PKCδ cleavage is mediated via caspase-3 (Fig. 4A).
Fig. 4.

6-OHDA induces caspase-3-mediated proteolytic activation of PKCδ. (A) Proteolytic activation of PKCδ was determined in N27 cells and the cells overexpressing cleavage-resistant PKCδD327A protein after exposure to 6-OHDA with or without Z-DEVD-FMK (caspase-3 inhibitor) by Western blot. The results show that 6-OHDA induces caspase-3-mediated PKCδ cleavage; the induction was blocked by the caspase-3 inhibitor and by overexpression of PKCδ cleavage resistant protein (CRM). (B) PKCδ kinase activity was determined by immunoprecipitation followed by [32P]-phosphorylation, and then scanned with a phosphoimager. The data were analyzed using Quantity One Software. The results indicate that 6-OHDA induces PKCδ kinase activity (*p<0.05); that activity was completely inhibited by Z-DEVD-FMK.
A dramatic increase in PKCδ enzymatic activity, as determined by immunoprecipitation kinase assays, suggested that an increase in the PKCδ kinase activity parallels the proteolytic cleavage products (Fig. 4B). A 24 h treatment with 100 μM 6-OHDA induced a ten-fold increase in PKCδ kinase activity. The kinase activity assay was performed in the absence of lipids to determine the activity contributed solely by the proteolytically-cleaved PKCδ catalytic fragment. The increase in PKCδ kinase activity was almost completely blocked in cells co-treated with 50 μM Z-DEVD-FMK, suggesting that caspase-3 mediates 6-OHDA-induced activation of PKCδ.
Suppression of 6-OHDA-induced DNA fragmentation by caspase-3 inhibitor or free radical scavenger
Chromatin condensation and DNA fragmentation are key markers of apoptosis and have been shown to increase in dopaminergic neuronal cells upon treatment with dopaminergic toxins (Anantharam et al., 2002; Kitazawa et al., 2003). DNA fragmentation was measured quantitatively by ELISA (Roche Molecular Biochemicals), as previously described (Kitazawa et al., 2002). Exposure to 100 μM 6-OHDA significantly increased DNA fragmentation by 70% at 24 h, as compared to control (Fig. 5). However, co-treatment with 50 μM Z-DEVD-FMK or 10 μM MnTBAP significantly blocked 6-OHDA-induced DNA fragmentation by 95% and 92%, respectively (Fig. 5), suggesting that both generation of free radicals and activation of caspase-3 are essential for 6-OHDA-induced apoptosis.
Fig. 5.

ROS and caspase-3 mediate 6-OHDA-induced DNA fragmentation. The cells were cultured and treated with 100 μM 6-OHDA for 24 h in the presence or absence of a caspase-3 inhibitor, 50 μM Z-DEVD-FMK, or a free radical scavenger, 10 μM MnTBAP. DNA fragmentation was quantified using ELISA. The data are represented as the mean ± SEM of six individual measurements. *p<0.05 indicates significant differences between 6-OHDA-treated cells and control and inhibitor- or scavenger-treated cells.
Caspase cleavage-resistant mutant PKCδD327A-CRM protects against 6-OHDA-induced PKCδ cleavage, apoptotic cell death and TH neuronal cell loss
To determine the effect of caspase-3 on 6-OHDA-induced cleavage of PKCδ, N27 cells were engineered to stably express caspase cleavage-resistant PKCδ mutant (PKCδD327A-CRM) and then challenged with 6-OHDA. Figure 6A shows N27 cells stably expressing PKCδ-CRM-V5 fusion protein immunostained with V5 antibody. Exposure to 100 μM 6-OHDA failed to induce proteolytic cleavage of PKCδ in PKCδ-CRM transfected cells for up to 24 h as compared to 6-OHDA-treated naïve N27 cells (Fig. 4A). In a similar fashion, treatment with 6-OHDA also failed to induce a significant increase in DNA fragmentation in N27 cells stably expressing PKCδ-CRM compared to LacZ control N27 cells (Fig. 6B).
Fig. 6.

Overexpression of cleavage-resistant PKCδD327A mutant (PKCδ-CRM) blocks 6-OHDA-induced apoptosis. (A) Schematic of plasmid constructs: PKCδ-CRMV5 construct encodes cleavage resistant mutant of PKCδ-CRM with V5 tag, transcript mRNA is transcribed under the 5′ human cytomegalovirus (CMV) immediate early promoter, and the mRNA is stabilized with the 3′ SV40 mRNA polyadenylation signal (pA). Fluorescent images showing the stable expression of V5 tag in N27 cells expressing the PKCδ-CRM-V5 tag (200X magnification). (B) PKCδ-CRM blocks 6-OHDA-induced DNA fragmentation. The N27 cells stably expressing vector (LacZ) or PKCδ-CRM-V5 protein were treated with 100 μM 6-OHDA for 24 h, then DNA fragmentation was measured by ELISA. The data represent mean ± SEM of six individual measurements. The asterisk (*p<0.05) indicates a significant difference between the 100 μM 6-OHDA-treated PKCδ-CRM-V5 mutant cells and the 100 μM 6-OHDA-treated LacZ vector control cells.
To determine whether PKCδ-CRM can also protect against 6-OHDA-induced dopaminergic cell death in primary cultures, rat mesencephalic dopaminergic primary neurons were transiently transfected with plasmids PKCδD327A-CRM-V5 and Lac-V5 using a nucleofector device. The transfection efficiency was determined to be greater than 80% in both PKCδ-CRM and LacZ-transfected primary neurons as determined by V5-immunostaining. Twenty-four h after transfection, the primary neurons were treated with 30 μM 6-OHDA for 3 h. After treatment, the primary neurons were fixed and immunostained for tyrosine hydroxylase (TH). Immunostaining experiments revealed that 6-OHDA induced a significant loss in the number of TH+ neurons in LacZ-transfected primary neurons as compared to untreated LacZ-transfected primary neurons (Fig. 7B). However, 6-OHDA failed to induce a significant loss in the number of TH+ neurons in PKCδ-CRM-transfected primary neurons as compared to untreated PKCδ-CRM-transfected primary neurons (Fig. 7B). LacZ vector transfected primary neurons were affected by 6-OHDA while PKCδ-CRM-transfected primary neurons remained intact (Fig. 7A), indicating PKCδ-CRM offered significant protection against apoptotic cell death and TH+ neuronal cell loss.
Fig. 7.

Expression of PKCδ-CRM blocks 6-OHDA-induced primary dopaminergic neuronal cell death. (A) Primary dopaminergic neuronal cells from E16 embryos were collected and transfected with PKCδ-CRM. The primary neurons were exposed to 30 μM 6-OHDA for 3 h and then stained for tyrosine hydroxylase. PKCδ-CRM-expressing primary neurons were protected from 6-OHDA-induced cell death as compared to 6-OHDA-treated primary neurons. (B) The TH-positive neurons were counted by fluorescence microscopy using Metamorph Software. PKCδ-CRM-expressing primary dopaminergic neurons blocked 90% of cell death, compared to non-transfected cells treated with 6-OHDA (*p<0.05).
Loss-of-function-PKCδK376R-DN mutant rescues neuronal cells against 6-OHDA-induced apoptotic cell death
To determine whether enzyme activity of cleaved PKCδ also is critical for 6-OHDA-induced cell death, we engineered N27 cells to stably express loss-of-function kinase dead PKCδ mutant (PKCδK376R-DN) in which lysine at position 376 in the catalytic site is mutated to arginine, rendering the kinase inactive. We then challenged the cells with 6-OHDA. Figure 8A shows N27 cells stably expressing PKCδ-DN-V5 fusion protein immunostained with V5 antibody. Exposure to 100 μM 6-OHDA resulted in a 75% increase in DNA fragmentation in N27 cells stably expressing LacZ vector as compared to untreated LacZ-expressing cells. However, 6-OHDA treatment failed to induce a significant increase in DNA fragmentation in N27 cells stably expressing PKCδ-DN compared to untreated PKCδ-DN expressing cells (Fig. 8B). These results strongly suggest that PKCδ kinase activity is essential for 6-OHDA-induced apoptotic cell death in dopaminergic neuronal clonal cells.
Fig. 8.

Overexpression of catalytically inactive PKCδK376R protein (PKCδ-DN) attenuates 6-OHDA-induced apoptosis. (A) Fluorescent images showing the stable expression of PKCδ-DN-V5 (left panel), and phase contrast image (right panel) in N27 cells. (B) DNA fragmentation assay shows significant protection against 6-OHDA in the cells overexpressing catalytically inactive kinase dead PKCδ-DN mutant as compared to wild-type (LacZ) cells. *p<0.05 indicates a significant difference between the 100 μM 6-OHDA-treated PKCδ-DN mutant cells and the 100 μM 6-OHDA-treated LacZ vector control cells.
RNAi-mediated knockdown of PKCδ rescues neuronal cells against 6-OHDA-induced DNA fragmentation
To further substantiate the functional role of PKCδ in 6-OHDA-induced neuronal cell death, we examined the effect of siRNA targeted to PKCδ on 6-OHDA-induced DNA fragmentation. We recently developed siRNAs targeted against the coding region of PKCδ that specifically suppress PKCδ expression without producing any cytotoxic effect in dopaminergic cells (Yang et al., 2004). PKCδ-siRNA dramatically suppressed PKCδ protein expression, as demonstrated by secondary staining with Alexa 488 in Cy3-labeled PKCδ-siRNA (siRNA PKCδ-4) transfected N27 cells, compared to non-specific siRNA (siRNA-NS) transfected N27 cells (Fig. 9A). SiRNA transfection studies revealed that exposure to 100 μM 6-OHDA for 24 h resulted in a significant increase in DNA fragmentation in non-specific siRNA transfected N27 cells, whereas 6-OHDA failed to induce a significant increase in DNA fragmentation in PKCδ siRNA-transfected cells (Fig. 9B), thus confirming the pivotal role of PKCδ in 6-OHDA-induced dopaminergic cell death.
Fig. 9.

Suppression of PKCδ by siRNA protects N27 cells from 6-OHDA-induced apoptotic cell death. (A) PKCδ siRNA suppresses PKCδ protein expression. N27 cells were transfected with cy3-labeled PKCδ siRNA (25 nM) or cy3-labeled non-specific (NS) siRNA; PKCδ expression was observed following immunostaining using anti-PKCδ antibody and Alexa 488 secondary antibody. The nuclei were visualized by Hoechst 33342 counterstaining. Merged images show expression of PKCδ (green), cy3-labeled siRNA or siRNA–NS 9 (red) and nuclear staining (blue). The cells were observed under a fluorescence microscope. (B) PKCδ siRNA attenuates 6-OHDA-induced DNA fragmentation. The N27 cells were transfected with siRNA-δ-4 (25 nM) or non-specific siRNA for 24 h; cells were then treated with 100 μM 6-OHDA for an additional 24 h, and then DNA fragmentation was quantified. The data represent the mean ± SEM from six individual measurements. * indicates differences between 6-OHDA-treated non-transfected and NS siRNA cells and the untreated control cells (p<0.05). ## indicates the differences between 6-OHDA-treated siRNA cells and 6-OHDA-treated non-transfected and NS siRNA cells (p<0.01).
Proteolytic cleavage of PKCδ in the substantia nigra of 6-OHDA injected animals
To determine if 6-OHDA-induced proteolytic activation of PKCδ observed in N27 cells (Fig. 4) also occurs in vivo, we examined the proteolytic activation of PKCδ in an animal PD model. Adult C57 black male mice were stereotaxically injected with 4 ug 6-OHDA in the right substantia nigra (SN), and saline was injected into the left SN as described in the Methods section. After 7 days animals were sacrificed and then nigral tissues were dissected out using a tissue punch. Western blot analysis of nigral tissue lysate revealed the proteolytic cleavage of native PKCδ (72–74 kDa) into a PKCδ catalytic fragment (41 kDa) and a 38 kDa regulatory fragment in the 6-OHDA-injected side (Fig. 10A). No cleavage was observed in saline-injected substantia nigra. Tyrosine hydroxylase (TH) immunoblot of each sample was used as a marker of nigral tissue, and β-actin bands represented equal protein loading. These results indicate that the 6-OHDA-induced PKCδ proteolytic cleavage is not limited to cell culture models, but also occurs in mouse nigral tissues following 6-OHDA treatment. The proteolytic cleavage of PKCδ was also followed by a dramatic increase in the PKCδ enzymatic activity, as determined by immunoprecipitation kinase assays. This suggests that an increase in the PKCδ kinase activity parallels the proteolytic cleavage products observed in 6-OHDA-injected substantia nigra as compared to saline-injected substantia nigra (Fig. 10B). The kinase activity assay was performed in the absence of lipids to determine the activity contributed solely by the proteolytically cleaved PKCδ catalytic fragment. These data suggest that proteolytic cleavage of PKCδ may play a critical role in the 6-OHDA-induced dopaminergic cell death observed in animal models of PD.
Fig. 10.

6-OHDA induces proteolytic activation of PKCδ in mouse substantia nigra. (A) Mice were stereotaxically injected with 6-OHDA, and proteolytic cleavage of PKCδ in mouse substantia nigra was analyzed using Western blot. The results showed that 6-OHDA induces PKCδ cleavage. (B) PKCδ enzyme activity was determined by immunoprecipitation kinase assay with [32P]-phosphorylation, and then scanned using a phosphoimager. The densitometric analysis of the [32P]-phosphorylated 32 kDa histone band is shown. The data represent mean ± SEM from 3 independent images. *** indicates a significant difference between the 10 μM 6-OHDA-injected and saline-injected substantia nigra (p<0.001).
Caspase cleavage-resistant mutant PKCδD327A-CRM protects against 6-OHDA-induced PKCδ cleavage and activation in mouse substantia nigra
Since PKCδD327A-CRM protected against 6-OHDA-induced proteolytic activation in N27 cells, we next determined whether PKCδD327A-CRM protects against 6-OHDA-induced proteolytic activation of PKCδ in mouse brain substantia nigra. Adult C57 black male mice were stereotaxically injected with lentivirus coding for PKCδD327A-CRM in the left SN and LacZ in the right SN 6 days prior to injection of 4 μg 6-OHDA in the right SN and saline into the left SN. Six days later, 12 animals were sacrificed, nigral tissues were dissected out, and Western blot and kinase assays were performed. Western blot analysis of nigral tissue lysate revealed the proteolytic cleavage of native PKCδ in the LacZ and 6-OHDA-injected side (Fig. 11A), whereas proteolytic cleavage of PKCδ was significantly blocked in the PKCδD37A-CRM- and 6-OHDA-injected side. β-actin bands represented equal protein loading. These results indicate that the caspase-cleavage resistant PKCδD37A-CRM significantly prevented 6-OHDA-induced PKCδ proteolytic cleavage in a dominant-negative manner in mouse nigral tissues similar to that observed in cell cultures studies. The proteolytic cleavage of PKCδ was also followed by a dramatic increase in the PKCδ enzymatic activity in the LacZ-injected side, whereas PKCδ enzymatic activity was significantly blocked in the PKCδD37A-CRM-injected side following 6-OHDA treatment, as assessed by immunoprecipitation kinase assays (Fig. 11B). This suggests that PKCδD37A-CRM also attenuates 6-OHDA-induced increases in PKCδ kinase activity in mouse substantia nigra similar to that observed in N27 cells.
Fig. 11.

Caspase-3 cleavage-resistant PKCδD327A CRM mutant prevents 6-OHDA-induced proteolytic activation of PKCδ in mouse substantia nigra. (A) Mice were stereotaxically injected with either LacZ or PKCδD327A CRM and 6-OHDA, and proteolytic cleavage of PKCδ in mouse SN was measured, as described in the Methods. SN samples were analyzed using Western blot and immunoprecipitation kinase assays. The results showed that 6-OHDA induces PKCδ cleavage. (B) PKCδ enzyme activity was determined by immunoprecipitation kinase assay with [32P]-phosphorylation, and then scanned using a phosphoimager. The densitometric analysis of the [32P]-phosphorylated 32 kDa histone band is shown. The data represent mean ± SEM from 3 independent images. *** indicates a significant difference between the 10 μM 6-OHDA and LacZ-injected and 6-OHDA and PKCδD327A CRM-injected SN (p<0.001).
Discussion
We reported that PKCδ is an oxidative stress sensitive kinase activated by pro-oxidants such as hydrogen peroxide or neurotoxic agents such as manganese and pesticides in neuronal culture models (Kaul et al., 2003; Kitazawa et al., 2004; Yang et al., 2004; Latchoumycandane et al., 2005). In this report, we extended our previous studies to investigate the role of proteolytically activated PKCδ in mediating dopaminergic cell death in cell culture and animal PD models using 6-OHDA. The 6-OHDA-treatment induced apoptosis in mesencephalic dopaminergic neuronal cells (N27) via free radical generation, mitochondrial dysfunction, cytochrome c release, activation of caspase-9 and caspase-3, proteolytic activation of PKCδ and DNA fragmentation. PKCδ siRNA and loss-of-function PKCδ mutants (PKCδD327A and PKCδK376R) all rescued 6-OHDA-induced cell death and TH+ neuronal cell loss in both N27 cells and in primary mesencephalic neurons. Loss-of-function PKCδD327A mutant also rescued 6-OHDA-induced proteolytic activation in the mouse brain SN.
Oxidative stress results from an imbalance between ROS generation, antioxidant levels and/or the depletion of enzymatic or non-enzymatic scavengers of ROS. The neurotoxin 6-OHDA, a hydroxylated analogue of dopamine, enters the cells via the dopamine transporter. Inside the cell, Fe2+ and H2O2 enhance the formation of 6-OHDA by non-enzymatic hydroxylation of dopamine (Linert et al., 1996). It has been reported that alterations in mitochondrial membrane permeability result in the release of cytochrome c into the cytosol (Fiskum et al., 2003; Polster and Fiskum, 2004). In this study, N27 cells exposed to 6-OHDA induced cytotoxic cell death in a time- and dose-dependent manner. Based on cytotoxicity experiments, as well as a review of literature, we used 100 μM 6-OHDA for all subsequent studies (Choi et al., 2000). In this study, we show that treatment with 100 μM 6-OHDA induces altered mitochondrial activity, as determined by the MTT assay. These alterations in mitochondrial activity were followed by an increase in cytosolic cytochrome c levels. Once cytochrome c is released from the mitochondria, it forms a complex with apoptosis protease- activating factor-1 (Apaf-1) and activates caspase-9, which in turn activates caspase-3 (Zhou and Tang, 2002; Yuyama et al., 2003; Polster and Fiskum, 2004). We demonstrated that co-treatment with MnTBAP almost completely blocked 6-OHDA-induced increases in DNA fragmentation, suggesting that ROS serves as an initiator of 6-OHDA-induced apoptotic cell death.
We found that exposure of N27 cells to 6-OHDA resulted in increases in both caspase-9 and caspase-3 activities, indicating that caspases play a role in 6-OHDA-induced dopaminergic cell death. Co-treatment with 10 μM MnTBAP almost completely blocked 6-OHDA-induced increases in caspase-3 activity, indicating that ROS mediates caspase-3 activation. These results are in agreement with those from recent studies by our lab and from others in which both caspase-9 and caspase-3 activation was observed in neuronal cultures after MPP+(1-methyl-4-phenylpyridinium) treatment (Kaul et al., 2003; Yang et al., 2004). Our results are also in agreement with previous studies, which have reported oxidative stress, alterations in mitochondrial function, cytochrome c release, and caspase-9 and caspase-3 activation during 6-OHDA-induced cell death in the PC12 pheochromocytoma cell line (Elkon et al., 2004), SH-SY5Y human neuroblastoma cells (Jordan et al., 2004), MN9D murine dopaminergic cells (Choi et al., 1999) and cerebellar granule cells (Dodel et al., 1999).
Events that occur downstream of caspase-3 activation are still unclear. PKCδ has been reported to be an endogenous substrate for caspase-3 (Reyland et al., 1999; Anantharam et al., 2002; Kaul et al., 2003; Kitazawa et al., 2003), and activated caspase-3 cleaves PKCδ, yielding a 38 kDa regulatory fragment and a 41 kDa persistently active catalytic fragment (Greenberg et al., 1998; Brodie and Blumberg, 2003). Recently, we established that caspase-3-mediated proteolytic activation of PKCδ is an important downstream event in apoptotic cell death of dopaminergic neurons. In this study, we demonstrate that 6-OHDA induces caspase-3-dependent proteolytic cleavage of PKCδ, and causes increases in PKCδ kinase activity in N27 cells in a time-dependent manner. This suggests that 6-OHDA-treated N27 cells have higher levels of persistently active catalytic PKCδ fragments. Furthermore, we demonstrated that N27 cells and primary mesencephalic neurons engineered to express PKC-CRM or catalytically inactive PKCδK376R protein (PKCδ-DN) were both resistant to 6-OHDA-induced cell death. The protective effect of PKCδ-siRNA further confirmed the proapoptotic function of the kinase. These in vitro results strongly implicate PKCδ in oxidative damage of dopaminergic neurons. Furthermore, 6-OHDA also induced proteolytic cleavage and activation in mouse substantia nigra, suggesting that PKCδ may mediate 6-OHDA-induced neurodegeneration in animals. Loss-of-function PKCδD327A mutant also significantly attenuated 6-OHDA-induced proteolytic activation in mouse brain SN, providing in vivo evidence for our hypothesis. While we were completing these studies, another research group reported that PKCδ is activated in adrenal chromaffin cells (Hanrott et al., 2006) and in a rat model (Hanrott et al., 2008). However, our systematic investigation using dominant negative mutants, CRM mutants and siRNA provides clear evidence for proapoptotic function of PKCδ in dopaminergic neurodegeneration.
The role of PKCδ in nigral neurons is beginning to emerge. We recently reported that PKCδ is highly expressed in nigral dopaminergic neurons (Zhang et al., 2007b). In addition, we demonstrated that PKCδ also negatively regulates tyrosine hydroxylase activity and dopamine synthesis by enhancing protein phosphatase-2A activity in dopaminergic neurons (Zhang et al., 2007b). Recently, we have also showed that PKCδ contributes manganese-induced tyrosine hydroxylase activity (Zhang et al., 2011). Previously, we demonstrated -synuclein interacts with proapoptotic proteins PKCδ and BAD to protect dopaminergic neuronal cells against MPP+-induced apoptotic cell death (Kaul et al., 2005a). We also showed that tyrosine phosphorylation regulates the proteolytic activation of PKCδ in dopaminergic neuronal cells (Kaul et al., 2005b). Furthermore, we reported that α-synuclein negatively regulates protein kinase Cδ expression to suppress apoptosis in dopaminergic neurons by reducing p300 histone acetyl-transferase activity (Jin et al., 2011b). Very recently, we have implicated transcriptional regulation of PKCδ in oxidative stress-induced neuronal cell death (Jin et al., 2011b). With respect to environmental neurotoxic chemical exposure and pathogenesis of PD, we showed that exposure to the transition metal manganese, pesticides and proteasome inhibitors can trigger a PKCδ-dependent apoptotic cascade in dopaminergic neuronal cells. Collectively, our findings demonstrate that PKCδ plays a critical role in mediating nigral dopaminergic neurodegeneration during neurotoxic insults. These results also suggest that PKC may serve as a potential therapeutic target for development of novel neuroprotective strategies for treatment of PD. In this regard, we extended our mechanistic findings into translational research, in which we showed that the pharmacological inhibitor rottlerin protected against MPTP-induced motor deficits, dopamine depletion and TH neuronal cell loss in an animal model of PD (Zhang et al., 2007a).
In summary, the present study demonstrates that the oxidative stressor 6-OHDA activates the PKCδ-dependent apoptotic pathway in both cell culture and animal models of PD. The use of loss-of-function PKCδ mutants, as well as selective targeting of the proapoptotic kinase PKCδ by siRNA, clearly establishes PKCδ as a key downstream mediator of 6-OHDA-induced apoptosis. These findings indicate that further exploration of PKCδ signaling in the nigral dopaminergic system may have a significant therapeutic implication in the treatment of PD related neurological disorders.
Highlights.
Dopaminergic neurotoxicant 6-hydroxydopamine (6-OHDA) activates the mitochondrial-apoptotic signaling pathway.
Proteolytic activation of PKCδ is a key downstream event of 6-OHDA-induced neurotoxicity.
Expression of cleavage resistant PKCδD327A, kinase dead PKCδK376R or PKCδ siRNA protects against 6-OHDA-induced dopaminergic cell death.
Viral-mediated delivery of PKCδD327A attenuates 6-OHDA induced PKCδ proteolytic activation in mouse substantia nigra.
The study strongly suggests that PKCδ proteolytic activation is a key downstream event in dopaminergic degeneration.
Acknowledgments
This study was supported by grants from the National Institute of Health NS065167, NS38644 and ES10586. The W. Eugene and Linda Lloyd Endowed Chair and Professorship to AGK is also acknowledged. The authors also acknowledge Faneng Sun for technical assistance and Ms. Mary Ann deVries for the assistance in the preparation of this manuscript.
Abbreviations
- 6-OHDA
6-hydroxydopamine
- PD
Parkinson’s disease
- SN
substantia nigra
- PKCδ
protein kinase C delta
- ROS
reactive oxygen species
- TH
tyrosine hydroxylase
- MTT
3-(4,5-Dimethylthiazol-3-yl)-2,5-diphenyl tetrazolium bromide
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MPP+
1-methyl-4-phenylpyridinium
- AMC
7-amino-4-methylcoumarine
- Z-DEVD-FMK
Z-Asp-Glu-Val-Asp-fluoromethyl ketone
- Ac-LEHD-AFC
Acetyl-Leu-Glu-His-Asp-7-amino-4-fluorcoumarin
- Ac-DEVD-AFC
Acetyl-Asp-Glu-Val-Asp-7-amino-4-fluorcoumarin
Footnotes
Conflict of interest statement
All authors declare that there is no conflict of interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Afeseh Ngwa H, Kanthasamy A, Anantharam V, Song C, Witte T, Houk R, Kanthasamy AG. Vanadium induces dopaminergic neurotoxicity via protein kinase Cdelta dependent oxidative signaling mechanisms: relevance to etiopathogenesis of Parkinson’s disease. Toxicol Appl Pharmacol. 2009;240:273–285. doi: 10.1016/j.taap.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharam V, Kitazawa M, Latchoumycandane C, Kanthasamy A, Kanthasamy AG. Blockade of PKCdelta proteolytic activation by loss of function mutants rescues mesencephalic dopaminergic neurons from methylcyclopentadienyl manganese tricarbonyl (MMT)-induced apoptotic cell death. Ann N Y Acad Sci. 2004;1035:271–289. doi: 10.1196/annals.1332.017. [DOI] [PubMed] [Google Scholar]
- Anantharam V, Kitazawa M, Wagner J, Kaul S, Kanthasamy AG. Caspase-3-dependent proteolytic cleavage of protein kinase Cdelta is essential for oxidative stress-mediated dopaminergic cell death after exposure to methylcyclopentadienyl manganese tricarbonyl. J Neurosci. 2002;22:1738–1751. doi: 10.1523/JNEUROSCI.22-05-01738.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi SA, Dawson TM, Dawson VL. Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann N Y Acad Sci. 2008;1147:233–241. doi: 10.1196/annals.1427.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF. Parkinson’s disease: a model dilemma. Nature. 2010;466:S8–10. doi: 10.1038/466S8a. [DOI] [PubMed] [Google Scholar]
- Brodie C, Blumberg PM. Regulation of cell apoptosis by protein kinase c delta. Apoptosis. 2003;8:19–27. doi: 10.1023/a:1021640817208. [DOI] [PubMed] [Google Scholar]
- Burke RE, Kholodilov NG. Programmed cell death: does it play a role in Parkinson’s disease? Ann Neurol. 1998;44:S126–133. doi: 10.1002/ana.410440719. [DOI] [PubMed] [Google Scholar]
- Cannon JR, Greenamyre JT. Neurotoxic in vivo models of Parkinson’s disease recent advances. Prog Brain Res. 2010;184:17–33. doi: 10.1016/S0079-6123(10)84002-6. [DOI] [PubMed] [Google Scholar]
- Choi HY, Song JH, Park DK, Ross GM. The effects of ascorbic acid on dopamine-induced death of PC12 cells are dependent on exposure kinetics. Neurosci Lett. 2000;296:81–84. doi: 10.1016/s0304-3940(00)01647-5. [DOI] [PubMed] [Google Scholar]
- Choi WS, Yoon SY, Oh TH, Choi EJ, O’Malley KL, Oh YJ. Two distinct mechanisms are involved in 6-hydroxydopamine- and MPP+-induced dopaminergic neuronal cell death: role of caspases, ROS, and JNK. J Neurosci Res. 1999;57:86–94. doi: 10.1002/(SICI)1097-4547(19990701)57:1<86::AID-JNR9>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Dodel RC, Du Y, Bales KR, Ling Z, Carvey PM, Paul SM. Caspase-3-like proteases and 6-hydroxydopamine induced neuronal cell death. Brain Res Mol Brain Res. 1999;64:141–148. doi: 10.1016/s0169-328x(98)00318-0. [DOI] [PubMed] [Google Scholar]
- Elkon H, Melamed E, Offen D. Oxidative stress, induced by 6-hydroxydopamine, reduces proteasome activities in PC12 cells: implications for the pathogenesis of Parkinson’s disease. J Mol Neurosci. 2004;24:387–400. doi: 10.1385/JMN:24:3:387. [DOI] [PubMed] [Google Scholar]
- Filipov NM, Cao L, Seegal RF, Lawrence DA. Compromised peripheral immunity of mice injected intrastriatally with six-hydroxydopamine. J Neuroimmunol. 2002;132:129–139. doi: 10.1016/s0165-5728(02)00321-1. [DOI] [PubMed] [Google Scholar]
- Fiskum G, Starkov A, Polster BM, Chinopoulos C. Mitochondrial mechanisms of neural cell death and neuroprotective interventions in Parkinson’s disease. Ann N Y Acad Sci. 2003;991:111–119. doi: 10.1111/j.1749-6632.2003.tb07469.x. [DOI] [PubMed] [Google Scholar]
- Gao HM, Zhang F, Zhou H, Kam W, Wilson B, Hong JS. Neuroinflammation and alpha-Synuclein Dysfunction Potentiate Each Other Driving Chronic Progression of Neurodegeneration in a Mouse Model of Parkinson’s Disease. Environ Health Perspect. 2011;119:807–14. doi: 10.1289/ehp.1003013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SS, Jie O, Zhao X, Wang JF. Role of PKC and tyrosine kinase in ethanol-mediated inhibition of LPS-inducible nitric oxide synthase. Alcohol. 1998;16:167–175. doi: 10.1016/s0741-8329(97)00187-0. [DOI] [PubMed] [Google Scholar]
- Hanrott K, Gudmunsen L, O’Neill MJ, Wonnacott S. 6-hydroxydopamine-induced apoptosis is mediated via extracellular auto-oxidation and caspase 3-dependent activation of protein kinase Cdelta. J Biol Chem. 2006;281:5373–5382. doi: 10.1074/jbc.M511560200. [DOI] [PubMed] [Google Scholar]
- Hanrott K, Murray TK, Orfali Z, Ward M, Finlay C, O’Neill MJ, Wonnacott S. Differential activation of PKC delta in the substantia nigra of rats following striatal or nigral 6-hydroxydopamine lesions. Eur J Neurosci. 2008;27:1086–1096. doi: 10.1111/j.1460-9568.2008.06097.x. [DOI] [PubMed] [Google Scholar]
- Henchcliffe C, Beal MF. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neurol. 2008;4:600–609. doi: 10.1038/ncpneuro0924. [DOI] [PubMed] [Google Scholar]
- Horowitz MP, Greenamyre JT. Gene-environment interactions in Parkinson’s disease: the importance of animal modeling. Clin Pharmacol Ther. 2010;88:467–474. doi: 10.1038/clpt.2010.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, Kanthasamy A, Anantharam V, Rana A, Kanthasamy AG. Transcriptional regulation of Pro-apoptotic protein kinase C-delta: Implications for oxidative stress-induced neuronal cell death. J Biol Chem. 2011a;286:19840–59. doi: 10.1074/jbc.M110.203687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H, Kanthasamy A, Ghosh A, Yang Y, Anantharam V, Kanthasamy AG. Alpha-synuclein negatively regulates protein kinase Cdelta expression to suppress apoptosis in dopaminergic neurons by reducing p300 histone acetyltransferase activity. J Neurosci. 2011b;31:2035–2051. doi: 10.1523/JNEUROSCI.5634-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan J, Galindo MF, Tornero D, Gonzalez-Garcia C, Cena V. Bcl-x L blocks mitochondrial multiple conductance channel activation and inhibits 6-OHDA-induced death in SH-SY5Y cells. J Neurochem. 2004;89:124–133. doi: 10.1046/j.1471-4159.2003.02299.x. [DOI] [PubMed] [Google Scholar]
- Kanthasamy A, Jin H, Mehrotra S, Mishra R, Rana A. Novel cell death signaling pathways in neurotoxicity models of dopaminergic degeneration: relevance to oxidative stress and neuroinflammation in Parkinson’s disease. Neurotoxicology. 2010;31:555–561. doi: 10.1016/j.neuro.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanthasamy AG, Kitazawa M, Kaul S, Yang Y, Lahiri DK, Anantharam V, Kanthasamy A. Proteolytic activation of proapoptotic kinase PKCdelta is regulated by overexpression of Bcl-2: implications for oxidative stress and environmental factors in Parkinson’s disease. Ann N Y Acad Sci. 2003;1010:683–686. doi: 10.1196/annals.1299.125. [DOI] [PubMed] [Google Scholar]
- Kaul S, Anantharam V, Kanthasamy A, Kanthasamy AG. Wild-type alpha-synuclein interacts with pro-apoptotic proteins PKCdelta and BAD to protect dopaminergic neuronal cells against MPP+-induced apoptotic cell death. Brain Res Mol Brain Res. 2005a;139:137–152. doi: 10.1016/j.molbrainres.2005.05.022. [DOI] [PubMed] [Google Scholar]
- Kaul S, Anantharam V, Yang Y, Choi CJ, Kanthasamy A, Kanthasamy AG. Tyrosine phosphorylation regulates the proteolytic activation of protein kinase Cdelta in dopaminergic neuronal cells. J Biol Chem. 2005b;280:28721–28730. doi: 10.1074/jbc.M501092200. [DOI] [PubMed] [Google Scholar]
- Kaul S, Kanthasamy A, Kitazawa M, Anantharam V, Kanthasamy AG. Caspase-3 dependent proteolytic activation of protein kinase C delta mediates and regulates 1-methyl-4-phenylpyridinium (MPP+)-induced apoptotic cell death in dopaminergic cells: relevance to oxidative stress in dopaminergic degeneration. Eur J Neurosci. 2003;18:1387–1401. doi: 10.1046/j.1460-9568.2003.02864.x. [DOI] [PubMed] [Google Scholar]
- Kim SY, Woo MS, Park JS, Hyun JW, Kim YS, Kim HS. The neuroprotective role of tissue inhibitor of metalloproteinase-2 in MPP+- or 6-OHDA-treated SK-N-BE(2)C and SH-SY5Y human neuroblastoma cells. Neurosci Lett. 2010;468:136–140. doi: 10.1016/j.neulet.2009.10.084. [DOI] [PubMed] [Google Scholar]
- Kitazawa M, Anantharam V, Kanthasamy A, Kanthasamy AG. Dieldrin promotes proteolytic cleavage of poly(ADP-ribose) polymerase and apoptosis in dopaminergic cells: protective effect of mitochondrial anti-apoptotic protein Bcl-2. Neurotoxicology. 2004;25:589–598. doi: 10.1016/j.neuro.2003.09.014. [DOI] [PubMed] [Google Scholar]
- Kitazawa M, Anantharam V, Kanthasamy AG. Dieldrin induces apoptosis by promoting caspase-3-dependent proteolytic cleavage of protein kinase Cdelta in dopaminergic cells: relevance to oxidative stress and dopaminergic degeneration. Neuroscience. 2003;119:945–964. doi: 10.1016/s0306-4522(03)00226-4. [DOI] [PubMed] [Google Scholar]
- Kitazawa M, Anantharam V, Yang Y, Hirata Y, Kanthasamy A, Kanthasamy AG. Activation of protein kinase C delta by proteolytic cleavage contributes to manganese-induced apoptosis in dopaminergic cells: protective role of Bcl-2. Biochem Pharmacol. 2005;69:133–146. doi: 10.1016/j.bcp.2004.08.035. [DOI] [PubMed] [Google Scholar]
- Kitazawa M, Wagner JR, Kirby ML, Anantharam V, Kanthasamy AG. Oxidative stress and mitochondrial-mediated apoptosis in dopaminergic cells exposed to methylcyclopentadienyl manganese tricarbonyl. J Pharmacol Exp Ther. 2002;302:26–35. doi: 10.1124/jpet.302.1.26. [DOI] [PubMed] [Google Scholar]
- Kupershmidt L, Okun Z, Amit T, Mandel S, Saltsman I, Mahammed A, Bar-Am O, Gross Z, Youdim MB. Metallocorroles as cytoprotective agents against oxidative and nitrative stress in cellular models of neurodegeneration. J Neurochem. 2010;113:363–373. doi: 10.1111/j.1471-4159.2010.06619.x. [DOI] [PubMed] [Google Scholar]
- Latchoumycandane C, Anantharam V, Kitazawa M, Yang Y, Kanthasamy A, Kanthasamy AG. Protein kinase Cdelta is a key downstream mediator of manganese-induced apoptosis in dopaminergic neuronal cells. J Pharmacol Exp Ther. 2005;313:46–55. doi: 10.1124/jpet.104.078469. [DOI] [PubMed] [Google Scholar]
- Lee JK, Tran T, Tansey MG. Neuroinflammation in Parkinson’s disease. J Neuroimmune Pharmacol. 2009;4:419–429. doi: 10.1007/s11481-009-9176-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linert W, Herlinger E, Jameson RF, Kienzl E, Jellinger K, Youdim MB. Dopamine, 6-hydroxydopamine, iron, and dioxygen--their mutual interactions and possible implication in the development of Parkinson’s disease. Biochim Biophys Acta. 1996;1316:160–168. doi: 10.1016/0925-4439(96)00020-8. [DOI] [PubMed] [Google Scholar]
- Liu WG, Wang XJ, Lu GQ, Li B, Wang G, Chen SD. Dopaminergic regeneration by neurturin-overexpressing c17.2 neural stem cells in a rat model of Parkinson’s disease. Mol Neurodegener. 2007;2:19. doi: 10.1186/1750-1326-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson’s disease. Annu Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- Naoi M, Maruyama W, Yi H, Inaba K, Akao Y, Shamoto-Nagai M. Mitochondria in neurodegenerative disorders: regulation of the redox state and death signaling leading to neuronal death and survival. J Neural Transm. 2009;116:1371–1381. doi: 10.1007/s00702-009-0309-7. [DOI] [PubMed] [Google Scholar]
- Orth M, Tabrizi SJ. Models of Parkinson’s disease. Mov Disord. 2003;18:729–737. doi: 10.1002/mds.10447. [DOI] [PubMed] [Google Scholar]
- Polster BM, Fiskum G. Mitochondrial mechanisms of neural cell apoptosis. J Neurochem. 2004;90:1281–1289. doi: 10.1111/j.1471-4159.2004.02572.x. [DOI] [PubMed] [Google Scholar]
- Puttonen KA, Lehtonen S, Lampela P, Mannisto PT, Raasmaja A. Different viabilities and toxicity types after 6-OHDA and Ara-C exposure evaluated by four assays in five cell lines. Toxicol In Vitro. 2008;22:182–189. doi: 10.1016/j.tiv.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Reyland ME, Anderson SM, Matassa AA, Barzen KA, Quissell DO. Protein kinase C delta is essential for etoposide-induced apoptosis in salivary gland acinar cells. J Biol Chem. 1999;274:19115–19123. doi: 10.1074/jbc.274.27.19115. [DOI] [PubMed] [Google Scholar]
- Shin JH, Dawson VL, Dawson TM. SnapShot: pathogenesis of Parkinson’s disease. Cell. 2009;139:440, e441–442. doi: 10.1016/j.cell.2009.09.026. [DOI] [PubMed] [Google Scholar]
- Song C, Kanthasamy A, Anantharam V, Sun F, Kanthasamy AG. Environmental neurotoxic pesticide increases histone acetylation to promote apoptosis in dopaminergic neuronal cells: relevance to epigenetic mechanisms of neurodegeneration. Mol Pharmacol. 2010;77:621–632. doi: 10.1124/mol.109.062174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Tsang AH, Chung KK. Oxidative and nitrosative stress in Parkinson’s disease. Biochim Biophys Acta. 2009;1792:643–650. doi: 10.1016/j.bbadis.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Wang B, Luo F, Ge X, Fu A, Han J. Effect of 6-OHDA lesions of the dopaminergic mesolimbic system on drug priming induced reinstatement of extinguished morphine CPP in rats. Beijing Da Xue Xue Bao. 2003;35:449–452. [PubMed] [Google Scholar]
- Yang Y, Kaul S, Zhang D, Anantharam V, Kanthasamy AG. Suppression of caspase-3-dependent proteolytic activation of protein kinase C delta by small interfering RNA prevents MPP+-induced dopaminergic degeneration. Mol Cell Neurosci. 2004;25:406–421. doi: 10.1016/j.mcn.2003.11.011. [DOI] [PubMed] [Google Scholar]
- Yoon J, Bang SH, Park JS, Chang ST, Kim YH, Min J. Increased in vitro lysosomal function in oxidative stress-induced cell lines. Appl Biochem Biotechnol. 2010;163:1002–1011. doi: 10.1007/s12010-010-9104-z. [DOI] [PubMed] [Google Scholar]
- Yuyama K, Yamamoto H, Nishizaki I, Kato T, Sora I, Yamamoto T. Caspase-independent cell death by low concentrations of nitric oxide in PC12 cells: involvement of cytochrome C oxidase inhibition and the production of reactive oxygen species in mitochondria. J Neurosci Res. 2003;73:351–363. doi: 10.1002/jnr.10669. [DOI] [PubMed] [Google Scholar]
- Zhang D, Anantharam V, Kanthasamy A, Kanthasamy AG. Neuroprotective effect of protein kinase C delta inhibitor rottlerin in cell culture and animal models of Parkinson’s disease. J Pharmacol Exp Ther. 2007a;322:913–922. doi: 10.1124/jpet.107.124669. [DOI] [PubMed] [Google Scholar]
- Zhang D, Kanthasamy A, Yang Y, Anantharam V. Protein kinase C delta negatively regulates tyrosine hydroxylase activity and dopamine synthesis by enhancing protein phosphatase-2A activity in dopaminergic neurons. J Neurosci. 2007b;27:5349–5362. doi: 10.1523/JNEUROSCI.4107-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Kanthasamy A, Anantharam V, Kanthasamy A. Effects of manganese on tyrosine hydroxylase (TH) activity and TH-phosphorylation in a dopaminergic neural cell line. Toxicol Appl Pharmacol. 2011;254:65–71. doi: 10.1016/j.taap.2010.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou C, Huang Y, Przedborski S. Oxidative stress in Parkinson’s disease: a mechanism of pathogenic and therapeutic significance. Ann N Y Acad Sci. 2008;1147:93–104. doi: 10.1196/annals.1427.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Tang XC. Huperzine A attenuates apoptosis and mitochondria-dependent caspase-3 in rat cortical neurons. FEBS Lett. 2002;526:21–25. doi: 10.1016/s0014-5793(02)03107-1. [DOI] [PubMed] [Google Scholar]
- Zhou W, Hurlbert MS, Schaack J, Prasad KN, Freed CR. Overexpression of human alpha-synuclein causes dopamine neuron death in rat primary culture and immortalized mesencephalon-derived cells. Brain Res. 2000;866:33–43. doi: 10.1016/s0006-8993(00)02215-0. [DOI] [PubMed] [Google Scholar]